1. Introduction
The human skin is a vital barrier that safeguards the body from external environmental fluctuations [
1,
2]. In addition, the immune system [
3,
4] functions as an additional protective barrier, guarding against foreign agents of varying scales that traverse the skin barrier and infiltrate the body. However, atopic dermatitis is an allergic ailment in which the equilibrium between the barrier function of the skin and the immune system, functioning as a shield against external elements, is compromised [
5,
6]. Among the etiological factors contributing to atopic dermatitis, instances of chemical irritation linked to the nature of dyes used in textiles and physical skin irritation from interactions between textile surfaces (accumulation of lint, electrostatic charges, yarn twist count, etc.) and the skin have been identified as exacerbating factors [
7,
8]. Furthermore, the friction engendered between clothing and the skin can cause a degradation of the protective barrier function of the skin [
9]. This reduction in barrier efficacy heightens the sensitivity of the skin even to minor irritations, where the act of scratching to alleviate itchiness can exacerbate dermatitis symptoms. Considerable efforts have been devoted to fabricating fibers characterized by diminished lint propensity and a sleek texture, particularly for garments in direct contact with the skin [
10,
11]. Notably, silk, a natural [
12,
13] and biomedical [
14,
15] fiber, is an exemplar with a velvety fiber surface devoid of lint, offering a tactile experience that is gentle on the skin with minimal friction and hypoallergenic properties [
8,
11]. The primary objective of this study was to engineer synthetic fibers that elicit reduced skin irritation by attenuating the friction between the fiber and the skin. The present hypothesis postulates that a smooth fiber surface akin to that of silk could potentially abate friction-induced skin irritation. Certain organisms regulate friction through their microstructural adaptations to survive arduous environmental conditions [
16]. For instance, the boa snake exhibits an epidermal surface characterized by a reticular pattern with micrometer-scale pores [
17]. This distinctive epidermal topography affords a coefficient of friction that is lower than that of a polished metal surface [
18]. This feature presumably mitigates the friction forces encountered when traversing surfaces. This exemplifies how surface microstructures influence frictional behavior. Concerted efforts are underway to harness insights from biological surface adaptations, regulate material friction, and mitigate challenges related to material degradation, structural impairment, and energy dissipation. Previous studies have underscored the potential for mitigating frictional forces by manipulating material surface geometry and reducing the contact area between interacting solids [
19,
20,
21,
22]. Although Amonton’s law [
23], a cornerstone of frictional phenomena, posits that frictional force remains uninfluenced by the area of a sliding interface (contact surface), empirical findings have revealed that friction can be effectively reduced through modifications in the extent of contact between two solid surfaces [
19,
20,
21,
22]. Thus, the present study focused on modifying the surface morphology of a synthetic polymer widely used for textile constituents.
Here, we focus on polyurethane and its cross-linked materials, which can be applied to a variety of material forms, including films and foams, and is also known as one of the materials used to make synthetic fibers for clothing [
24,
25,
26,
27]. Polyurethane has been industrialized and mass-produced and continues to be used in a wide range of fields as a matrix for polyurethane foams, coatings, and composite materials, and is known as a fundamental polymeric material for building human society [
28]. Fibers made from polyurethane are elastomers that exhibit excellent elastic behavior and can form fabrics with excellent flexibility and stretchability [
29,
30]. For this reason, polyurethane fibers constitute one of the essential fibers used in underwear and other clothing that adheres closely to the human skin and in clothing for sports applications, where it is necessary to maintain skin–clothing adhesion even when the body is exercised vigorously [
30,
31]. Polyurethanes (polymers containing carbamate bonds from diisocyanates and diols) or polyurethane–ureas (polymers containing carbamate and urea bonds from diisocyanates, diols, and diamines) are even today well-known polymer materials and have recently been used as the basic backbone of polymers in research on the creation of functional elastomer materials [
32]. Many studies have been conducted on the synthesis of thermoplastic elastomers [
33,
34] and liquid crystalline elastomers [
35,
36,
37] that exhibit excellent mechanical properties. Research and development are being carried out to exploit these polyurethane elastomers’ potential and add new functions by changing monomer components. Thus, while polyurethane or polyurethane–urea systems have been known for a long time and are in stable supply, they are also recognized as polymeric material systems of interest, where research on the creation of new materials based on their polymer skeletons is actively being conducted.
In this study, we attempted to create new materials based on the hydrophilic moiety and cross-linked structure of a polyurethane-urea system by introducing a hydrophilic moiety and a cross-linked structure and by utilizing the swelling state (hydrogel state) of the cross-linked structure. This structure was subjected to controlled swelling in a mixture of water and other hydrophilic solvents, including ethanol. A freeze-drying process was subsequently implemented, resulting in a dehydrated fiber surface endowed with assorted surface configurations, including elevations and undulations. This study was aimed at obtaining cross-linked hydrophilic fibers and changing the physical cross-link structure of these fibers by achieving a hydrogel state [
38,
39,
40] in various solvents. The fibers could be easily permeated by substances and freely change their shape, leading to a change in surface morphology. By engineering diverse surface profiles for these fibers, this study deciphered the intricate interplay between fiber surface morphology and the resulting frictional attributes and skin irritation potential. This investigation serves as a foundation for the development of fibers with reduced skin-related irritations. The engineering of fibers that demonstrate minimal frictional interaction with the skin holds promise for mitigating discomfort from clothing-induced irritation, which is a significant contributor to pruritus. The present investigation encompassed the fabrication of polyurethane–urea fibers through the desiccation of cross-linked hydrogels and the ensuing surface topography and resulting tensile strength characteristics of the fibers. In the context of the present study, it should be noted that although previous studies have been conducted on the creation of porous and high-strength liquid crystalline hydrogels through freeze-drying [
41], as well as the surface porous structuring of hydrogel scaffold materials through freeze-drying [
42], the present study was aimed at achieving solvent-induced internal structural changes in hydrogels under mild conditions. To accomplish this goal, a (freeze-)drying process using a mixed-solvent system was employed, with the term “drying” used in the case of organic solvents. This study attempted to control the properties of the fibers obtained after drying by improving the fabrication process through changing the solvent composition to one that can maintain the swelling of the gel state of the polyurethane–urea fibers with the solvent. It showed the possibility of controlling the gel fiber’s surface morphology and fiber strength after drying using a simple method.
2. Materials and Methods
1.3-Bis(isocyanatomethyl)cyclohexane (BICH) and dibutyl tin dilaurate (DTDL) were procured from Tokyo Chemical Industry Co., Ltd., Tokyo, Japan, while polyethylene glycol (PEG) with a molecular weight of 1500 was obtained from Sigma-Aldrich Japan (Merck KGaA, Darmstadt, Germany). The solvents were sourced from Wako Pure Chemical Industries, Ltd., Tokyo, Japan, and utilized without further purification. Milli-Q water obtained from an Elix UV 3 Milli-Q integral water purification system (Nihon Millipore K.K., Tokyo, Japan) was employed as pure water.
To initiate, a diisocyanate prepolymer solution was prepared as described [
37]. Under a nitrogen atmosphere, a 50 mL round-bottom flask housed 0.741 mL (4.20 mmol) of BICH, 3.0 g (2.00 mmol) of PEG, 0.027 g (42 μmol) of DTDL, and 6.0 mL of anhydrous DMF. The mixture was heated and stirred at 60 °C for 2 h under a nitrogen atmosphere, followed by 1 h at room temperature to conclude the reaction. Subsequently, the reaction was allowed to stand at room temperature for 1 additional hour to achieve termination, resulting in the formation of a prepolymer solution. Thereafter, 0.5 mL of the prepared prepolymer solution was loaded into a syringe SS-02LZ with 2.5 mL capacity (Terumo Corporation, Tokyo, Japan) fitted with a non-beveled needle NN-2238N, 22G (length 38 mm, diameter 0.70 mm, Terumo Corporation, Tokyo, Japan) and positioned on a turntable electric T-AuN (AS ONE CORPORATION, Osaka, Japan) set at 44 rpm. The solution was transferred into a 20cc vial containing a 10 mL DMF/diethyl ether mixed solvent (1/6
v/v) with 40 vol% triethylenetetramine (TETA), employing a syringe pump YSP-101 standard type (YMC Co., Ltd., Kyoto, Japan) set at a flow rate of 0.5 mL/min. Post extrusion, the sample was immersed in the multifunctional cross-linker solvent for 1 day.
To eliminate unreacted monomers and the cross-linker TETA from the resulting polyurethane–urea gel fibers, the fibers were initially immersed in a DMF/diethyl ether mixture (1/10 v/v) for 2 days, with solution replacement every alternate day. Subsequently, they were subjected to 2 h in diethyl ether (with hourly solvent exchange). Following solvent immersion and washing steps, the samples were subjected to vacuum drying at 70 °C for 6 h.
The dried fibers were reswelled in 12 solvent systems, encompassing various combinations of pure water, ethanol, and acetone (described below). Following 3 days of reswelling with daily solution renewal, the samples were frozen in a –12 °C freezer for approximately 1 day, followed by freeze-drying in a freeze-dryer FDM-1000 (Tokyo Rikakikai Co., Ltd., Tokyo, Japan) for 2 h.
In this study, we employed a Leica ML9300 polarized optical microscope (Meiji Techno Co., Ltd., Saitama, Japan) with crossed polarizers to conduct polarized light microscopy observations on composite hydrogels. For SEM image measurements, a JSM-6700FN scanning electron microscope (JEOL Ltd., Tokyo, Japan) operating at 1.0 keV was utilized. The freeze-dried xerogel samples were carefully positioned on a conductive tape situated on the brass SEM stage. Prior to imaging, a 10 nm thick layer of Pt was applied to the samples using a sputtering technique to enhance their electrical conductivity.
ATR–FTIR spectra were recorded using an FTIR6600 spectrometer (JASCO Corporation, Tokyo, Japan) in conjunction with a single-bounce diamond-attenuated total reflectance (ATR) accessory.
The degree of swelling for fibers in each solvent system was calculated using the following formula: degree of swelling (q), q = (swollen weight of a fiber)/(dried weight of a fiber). Surface morphology of the freeze-dried samples was examined using electron microscopy, optical microscopy, and polarized light microscopy. Additionally, tensile testing was performed to assess material strength and elongation. The functional groups within the samples were investigated through the attenuated total reflectance Fourier-transform infrared spectroscopy (ATR–FTIR) measurements by use of an FTIR6600 spectrometer (JASCO Corporation, Tokyo, Japan) in conjunction with a single-bounce diamond-attenuated total reflectance accessory. A preliminary tactile experiment was conducted by comparing the tactile perception of samples before and after re-swelling, focusing on samples with distinct surface topographies.
Samples re-swelled and freeze-dried in each solvent were cut to 30 mm, with cross-sectional areas calculated from sample diameter measurements acquired using a digital multimeter. Samples were securely placed in a force tester, with the test area set at 20 mm from the edge of the sample. A constant speed of 40 mm/min was employed for sample elongation testing.
3. Results
The synthesis of polyurethane–urea fibers proceeded via the synthetic pathway illustrated in
Figure 1. Fiber formation and cross-linking were simultaneously achieved by injecting a solution of diisocyanate chain-extended with polyethylene glycol into a solvent mixture containing triethylenetetramine (TETA), a cross-linking agent, using a nonbeveled injection needle attached to a syringe. The obtained polyurethane–urea fiber gel was immersed in diethyl ether to remove unreacted reagents and subjected to subsequent purification, followed by vacuum heating at 70 °C for 6 h. The dried fibers were reswollen using a solvent system of 12 solvent compositions comprising pure water, ethanol, and acetone (
Table 1). The resulting swollen fibers were subjected to freeze-drying to obtain the fiber samples (
Figure 2).
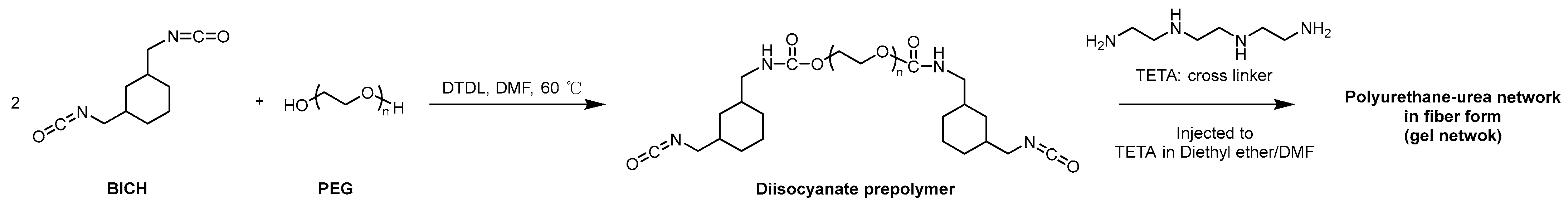
ATR–FTIR spectroscopy was employed to analyze the dried fiber samples.
Figure 3a showed that all the samples, including the preswollen sample, exhibited infrared absorption peaks within the range of approximately 3450–3000 cm⁻
1 attributable to the –NH stretching vibration, an infrared absorption peak near 1530 cm⁻
1 attributable to the –NH bending vibration (δNH, Amide II), and an infrared absorption peak at 1100 cm⁻
1 corresponding to ether bonds [
43,
44]. Similar spectra were recorded for all the polyurethane–urea samples swollen in the 12 solvent systems (
Figure 3b–d). This confirmed the presence of polyurethane–urea in the samples. The FTIR spectra did not show new peaks or shifts in the peaks, indicating the presence of characteristic intermolecular interactions between the polymer chains that constitute the fiber owing to differences in solvent systems.
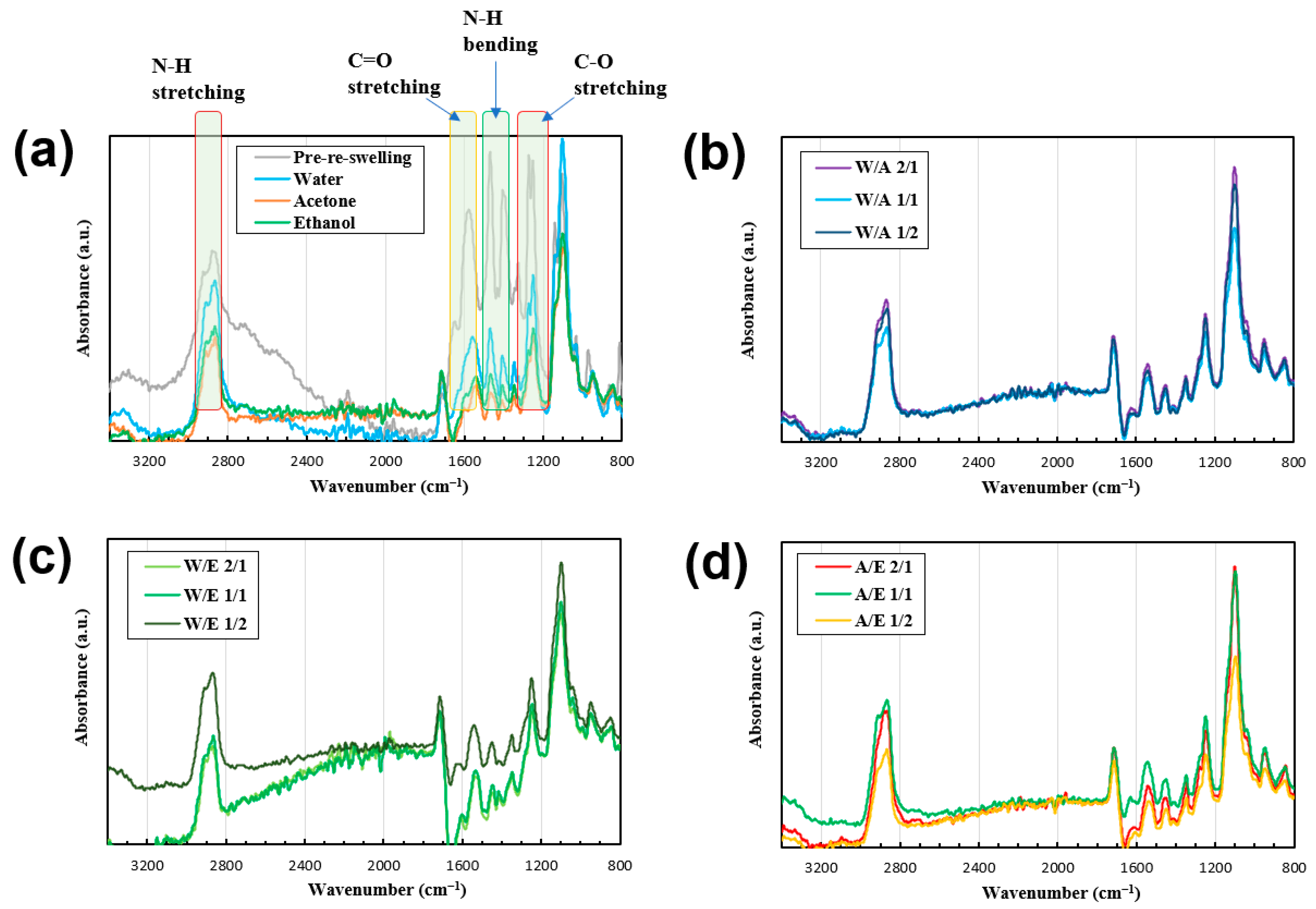
The fiber sample shapes were observed via optical microscopy (
Figure 4).
Figure 4 suggests that by subjecting the samples to reswelling and subsequent freeze-drying, the samples could be endowed with diverse shapes. Moreover,
Figure 4 indicates that under crossed-polarizer observation conditions, all the samples exhibited coloration, suggesting that the network in the gel-formed fibers exhibited an oriented configuration [
45].
SEM was employed to examine the microscopic surface morphology (
Figure 5). The surface of the pre-reswelling sample (
Figure 5a), which underwent neither reswelling nor freeze-drying, exhibited cavities, whereas the cavity-free regions appeared uniformly shaped. Conversely, the samples shown in
Figure 5b,c, subjected to reswelling and freeze-drying, exhibited topographies with convex and concave structures, such as spherical protuberances and fissures. The samples shown in
Figure 5d–g exhibited undulating ridges, whereas those shown in
Figure 5h–k exhibited perforations. Finally, the samples shown in
Figure 5l and m exhibited scale-like formations.
To investigate the swelling behavior of the polyurethane–urea gel fiber in mixed-solvent environments, the swelling degree in each solvent system was evaluated. Notably, the swelling degree in pure water was approximately five and six times that in acetone and ethanol, respectively. In the solvent mixture of pure water and ethanol, the swelling degree proportionally increased with an increase in the ethanol content. Contrarily, in the solvent mixture of pure water and acetone, the swelling degree decreased as the acetone content increased. The degree of swelling of the fiber in the solvent mixture of acetone and ethanol was low compared with that in the other two solvent systems. The degree of swelling in the acetone and ethanol mixture was lower than that in the other two solvent mixtures. Interestingly, the degree of swelling in the pure water/ethanol 1/2 and pure water/acetone 2/1 exceeded that of pure water.
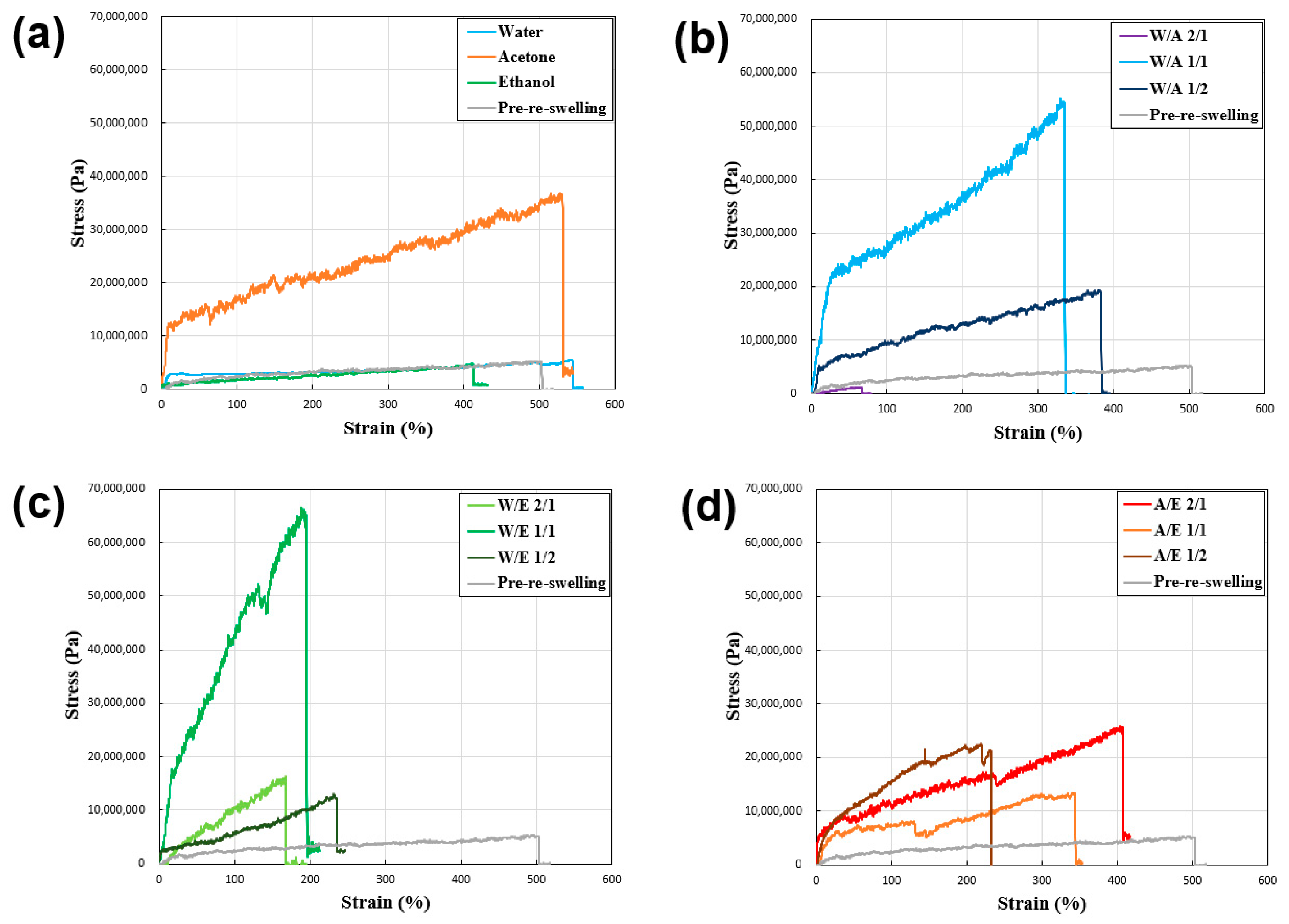
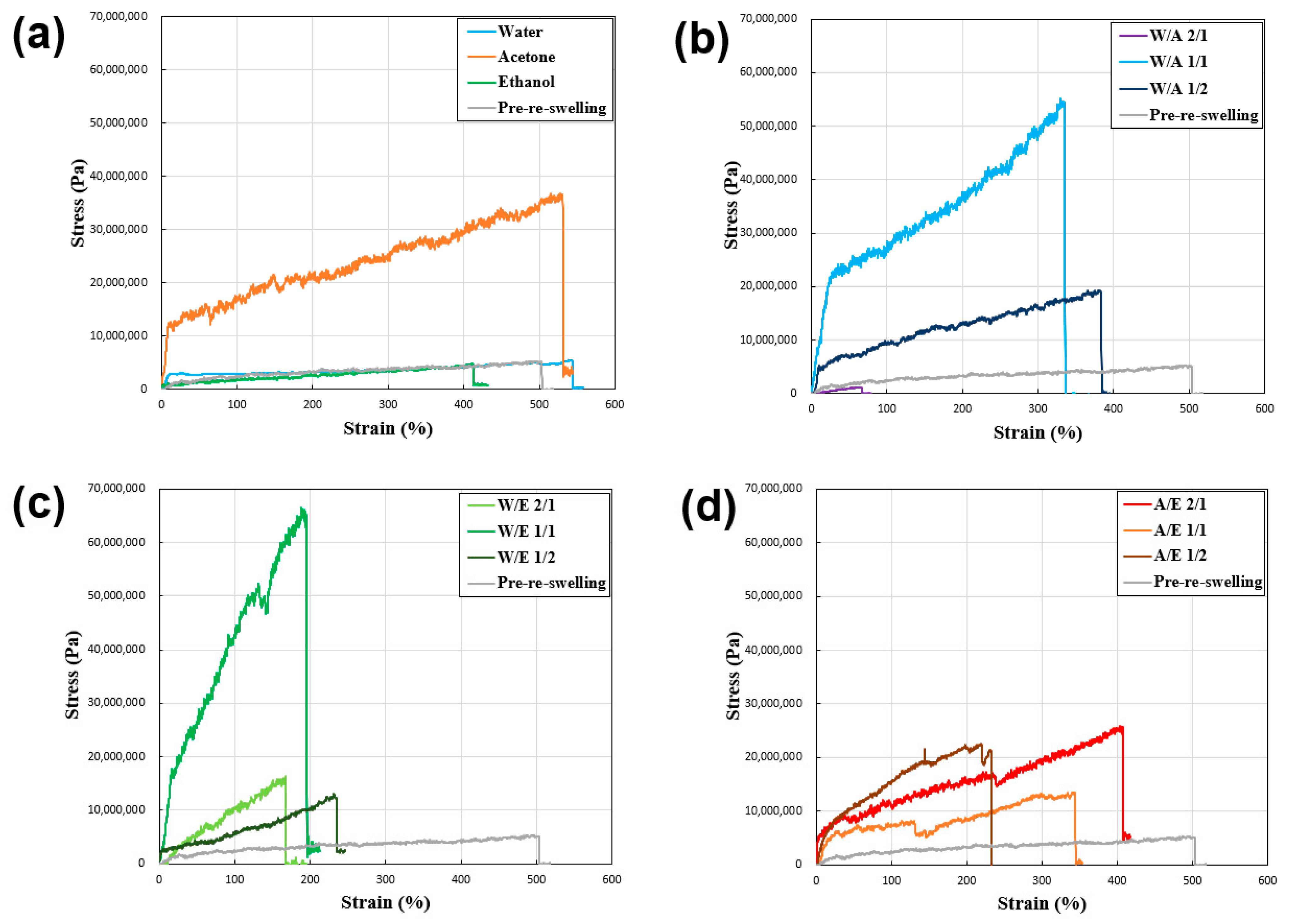
Figure 6 and
Table 1 show the results of tensile tests conducted on desiccated polyurethane–urea fibers to elucidate the inter-relation between the fiber strength and solvent composition.
Table 1 shows that all the samples exhibited an increase in tensile strength and a decrease in tensile strain except for the sample reswollen with water and acetone only. This showed that the swelling and freeze-drying processes using various hydrophilic solvent mixtures on the polyurethane–urea fibers resulted in different fiber strengths. The reasons for this are subsequently discussed, as well as the correlation between the degree of swelling and fiber strength.
Finally, we performed a tactile assessment of the fibers. The pre-reswollen acetone/ethanol 1/1 fiber sample retained a feeble and undulating texture when glided between the fingertips (
Figure 7). When squeezed, it elicited a rigid and uneven sensation. Oppositely, the post-reswelling acetone/ethanol 1/1 fiber sample offered a smooth and consistent feel when slid between the fingers. Furthermore, when handled, the pre- and post-reswollen samples exhibited a transition from a firm, knotted texture to a smooth, supple, and firm texture. The preliminary sensory tests revealed that the reswelling and freeze-drying of the hydrogel-forming polyurethane–urea fibers, coupled with alterations in their surface morphology, could potentially impact the tactile qualities of the fibers. In the future, we intend to fabricate an array of fibers with controlled surface profiles using the present approach and evaluate their relationship with skin irritation via sensory testing.
4. Discussion
Through the SEM images shown in
Figure 5, we postulate that the process of reswelling and freeze-drying the gelatinous samples facilitated the generation of distinct structures, including bumps and wrinkles, on the previously uniform surface. Furthermore, by varying the reswelling solvents used on the gelatinous samples, the surface morphology could be manipulated on a micron-scale level. This was considered to be caused by the different degrees of aggregation between the polymer chains due to the different solvation and drying processes between the solvent and polymer chains in the different solvent systems and the reconstruction of the physical cross-link structure as described below.
As shown in
Table 1, comparing the pre-reswelling to post-reswelling states, the alteration in the reswelling fiber signified a transformation in the mesh structure of the gel fibers. This suggested a reconstruction of the physically cross-linked network in the gel due to interactions based on hydrogen bonding and hydrophobic interactions in the gel, mediated by the hydrophilic solvent. Specifically, the progression of reswelling (dilution) and drying (concentration) led to a partial disruption and subsequent reformation of the physically cross-linked architecture. Consequently, the fibers with elevated q values demonstrated a reduction in physical cross-linking, whereas those with decreased swelling potentially exhibited an increased degree of physical cross-linking. This indicated that the substantial increase in the swelling ratio observed with pure water, which is approximately 5–6 times that of other pure solvents, may be attributed to the engagement of H₂O in solvent–solute interactions in urethane bonds, thereby reducing the influence of hydrogen-bond-based physical cross-linking.
Furthermore, the comparatively low q values of acetone and ethanol were probably due to their hydrophobicity, which could have resulted in a reduction in the hydrogen-bond-based physical cross-linking, as observed for water. The diminished q values of the mixed-solvent systems of acetone and ethanol might be attributed to the inherently low swelling capacities of these solvents when used independently. Moreover, a reason for the higher q values of W/A 2/1 and W/E 1/2, compared with those of W alone, could be the affinity of hydrophobic acetone and ethanol for the hydrophobic segments (cyclohexane units) in the polyurethane matrix [
25,
26]. This compatibility could have disrupted the hydrophobic interactions and, thus, the associated physical cross-linking between these segments, leading to a reduction in the overall cross-linking in the gel, resulting in increased swelling. These findings showed that controlling the swelling behavior involved the utilization of mixed-solvent systems rather than single hydrophilic solvents. This approach effectively controlled the swelling behavior of the polyurethane–urea fiber system, as demonstrated by the results.
Based on the tensile test results of the reswelling–(freeze-)dried samples (
Figure 6), when comparing the samples not subjected to reswelling with those reswollen using water and ethanol, it was observed that the tensile strength of the samples reswollen with acetone and the mixed solvent was higher. The results showed that, for the majority of mixed systems, the tensile strength generally improved compared with that of the pre-reswelling samples, although a decrease in tensile strain was observed. In fiber systems undergoing gel-state transition, the relationship between tensile strength and strain remains unclear. However, the favorable outcomes demonstrated in acetone and certain mixed-solvent systems suggest the potential for solvents with hydrophobic and hydrophilic properties to promote the successful reconstruction of physical cross-linking.
Mechanical strength (specific strength) generally tends to increase with an increase in material density [
46], and this trend also applies to polymer materials [
47]. Within gels, a high cross-link density at low swelling ratios generally leads to a high concentration of polymer chains per unit volume, resulting in increased mechanical strength [
38]. However, in the present context, although the reswelling–(freeze-)drying process enhanced the tensile strength, it should not be inferred that low q values inherently translate to increased tensile strength. Among the examined cases, although acetone (q = 4.2), ethanol (q = 3.6), A/E 1/1 (q = 3.7), and A/E 1/2 (q = 5.9) exhibited higher tensile strengths in acetone (9 MPa), other samples such as W/A 1/1 (q = 17.0) and W/E 1/1 (q = 18.2) exhibited lower tensile strengths (53 and 65 MPa, respectively). Thus, the enhanced tensile strength in most of the present samples was believed to have resulted from the successful reconstitution of physically cross-linked networks mediated by solvent interactions. However, instances such as W/A 1/2, which exhibited poor reconstruction, leading to lower tensile strength, highlight the importance of considering cases where solvent interactions might have adverse effects, underscoring the need to anticipate such scenarios. The tensile test results of the reswelling–freeze-dried samples (
Figure 6) indicate the potential for adjusting the tensile strength by reswelling the samples. The adjustment was attributed to the successful reconstitution of the physically cross-linked network via solvent interactions, although caution must be exercised in cases where such interactions might have unfavorable consequences.
Herein, polyurethane–urea fibers with different surface morphologies were obtained by controlling the swelling and drying processes of cross-linked materials. The present polyurethane–urea fibers exhibited various surface morphologies and tunable-tensile-strength fibers with surface morphology in the order of micrometers by changing the reswelling conditions in which physical recross-linking occurs. Although the feel of the fiber was preliminarily evaluated in preparation for the sensory test [
48], the correlation between the results of sensory tests, such as feeling, and the results of friction evaluation of the state of a single yarn should be studied in the future.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}