
Polymerization Initiated by Graphite Intercalation Compounds Revisited: One-Pot Synthesis of Amphiphilic Pentablock Copolymers †



Abstract
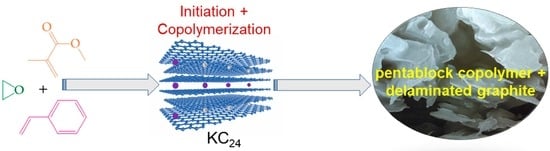
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
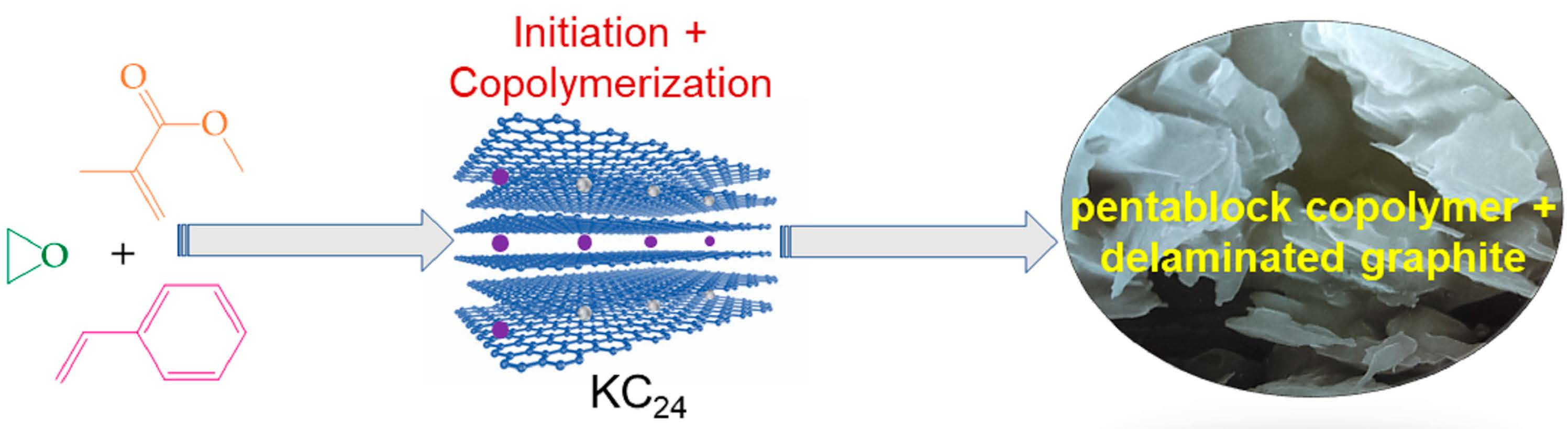
2.2.1. Initiator Synthesis
2.2.2. Copolymerization
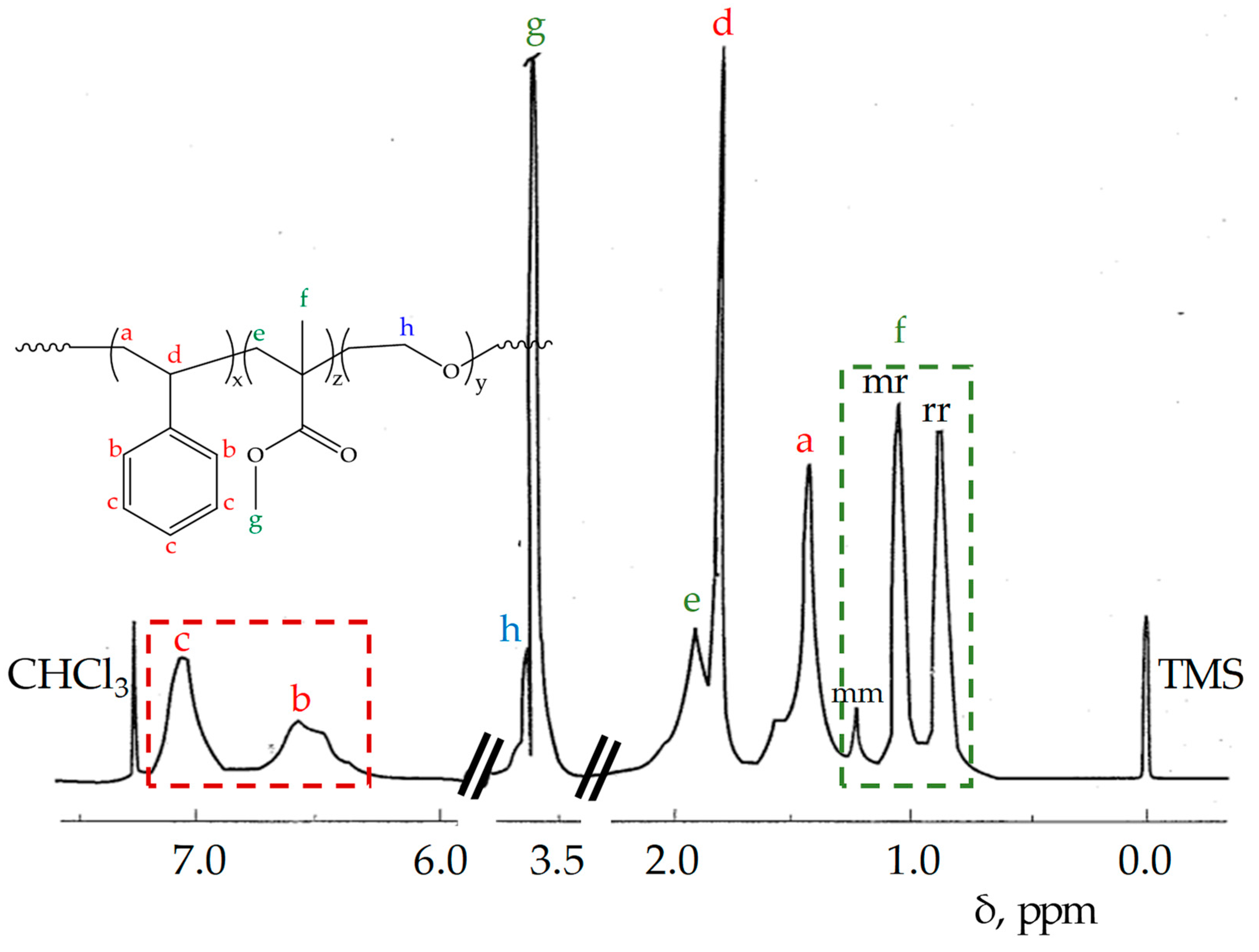
2.2.3. Polymer Characterization
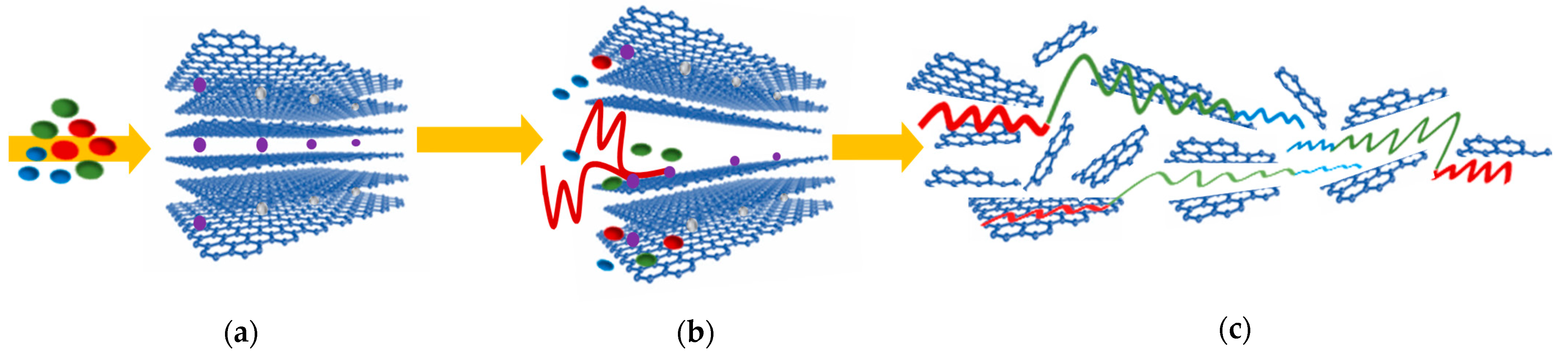
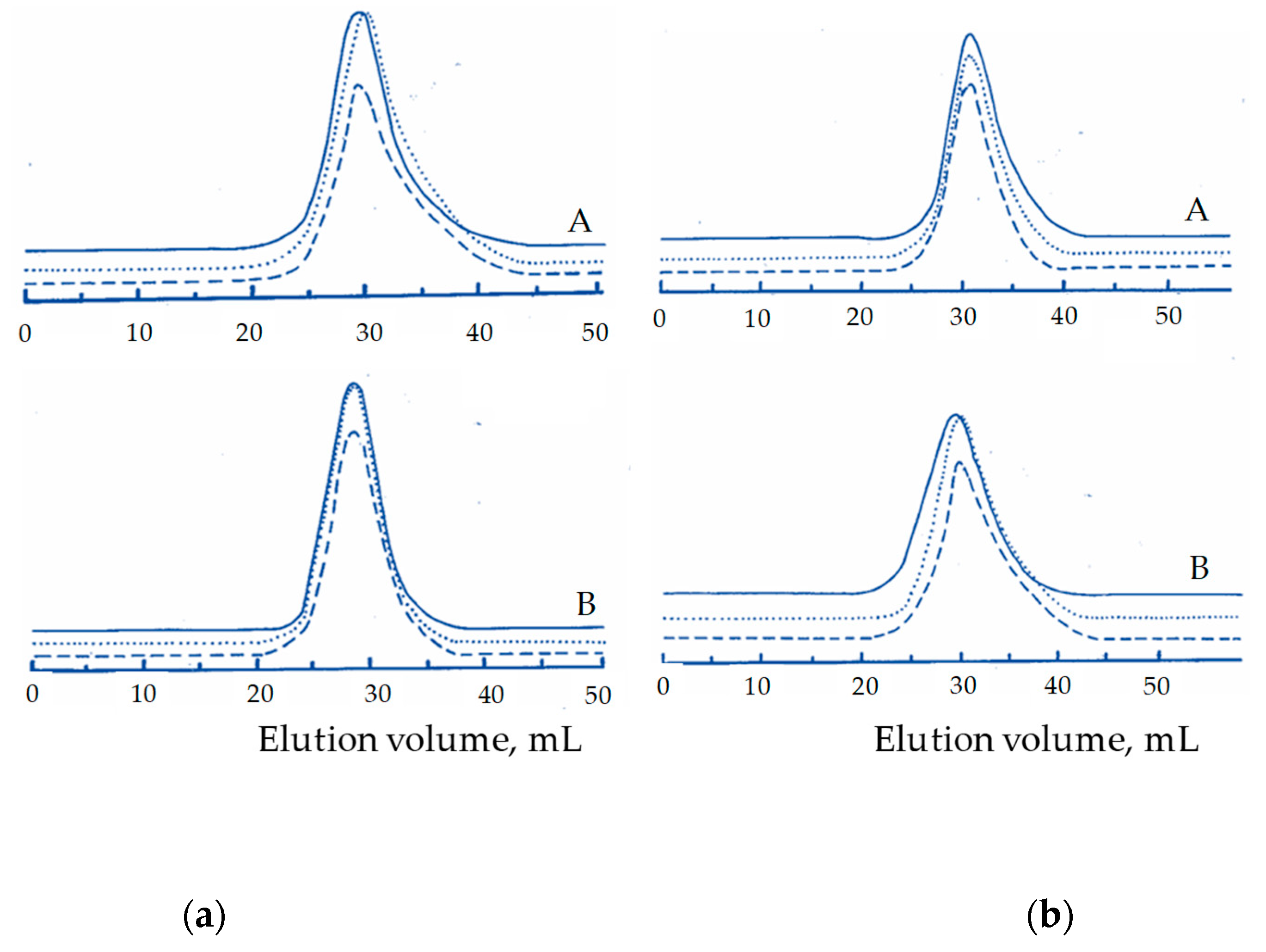
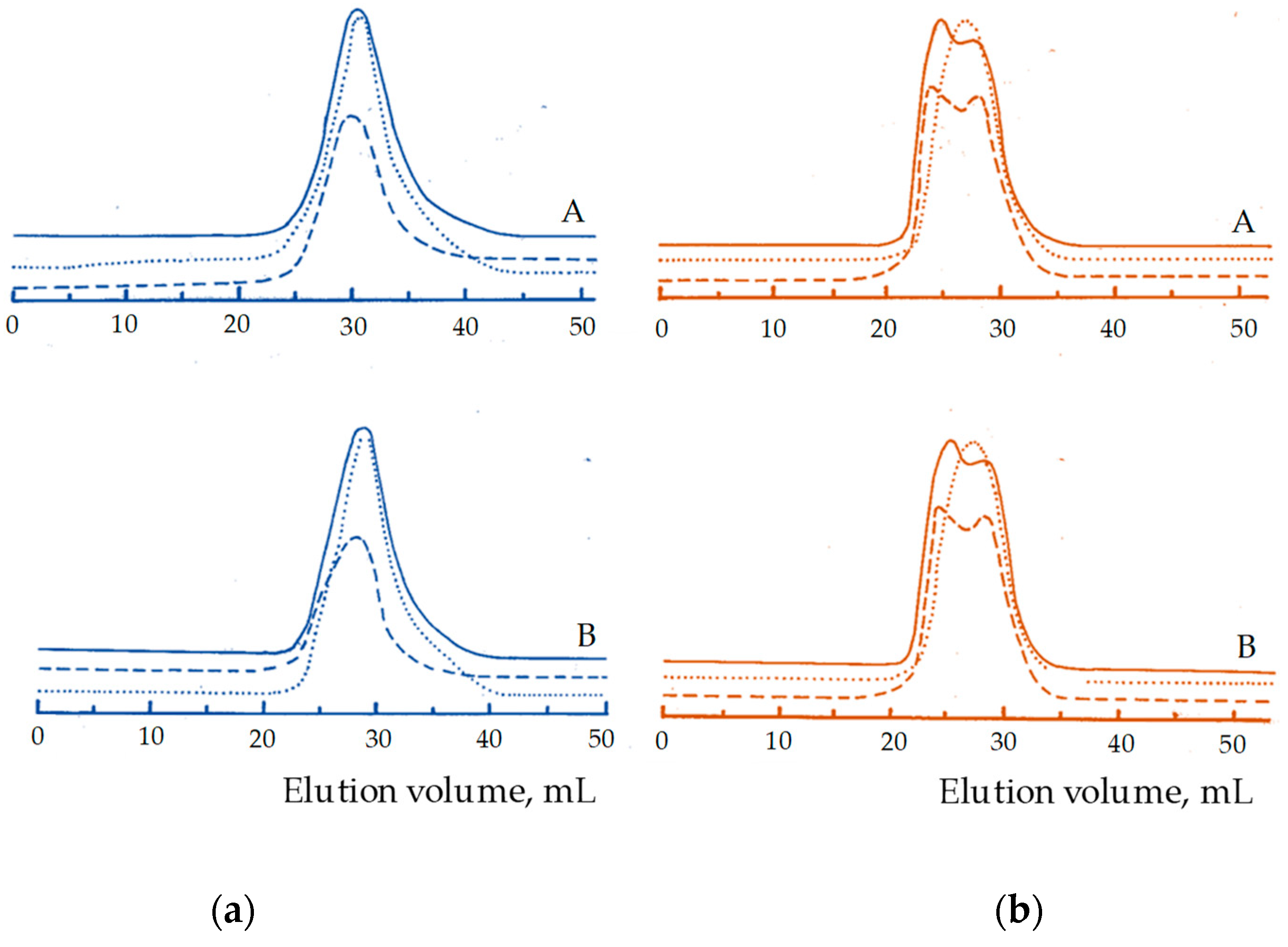
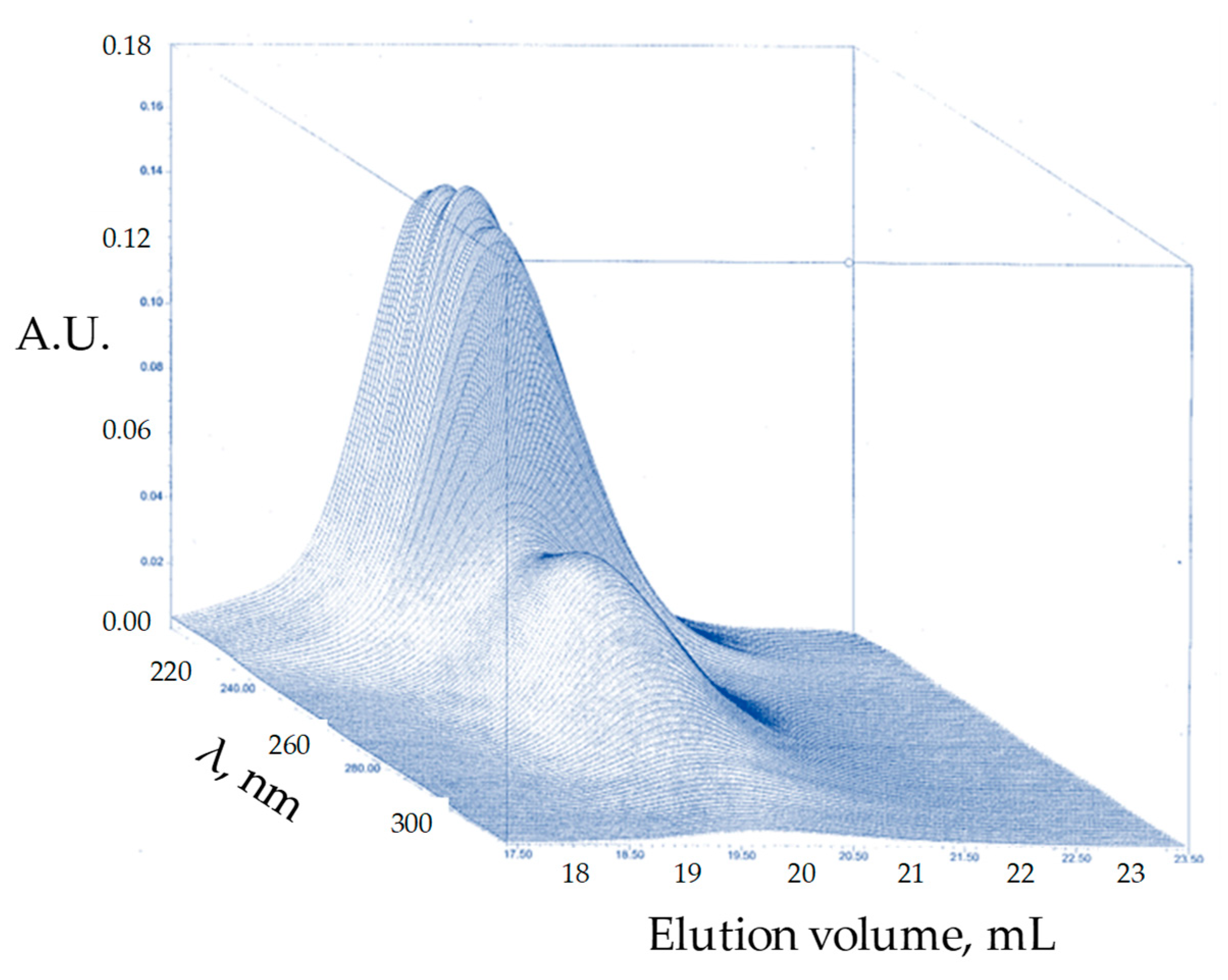
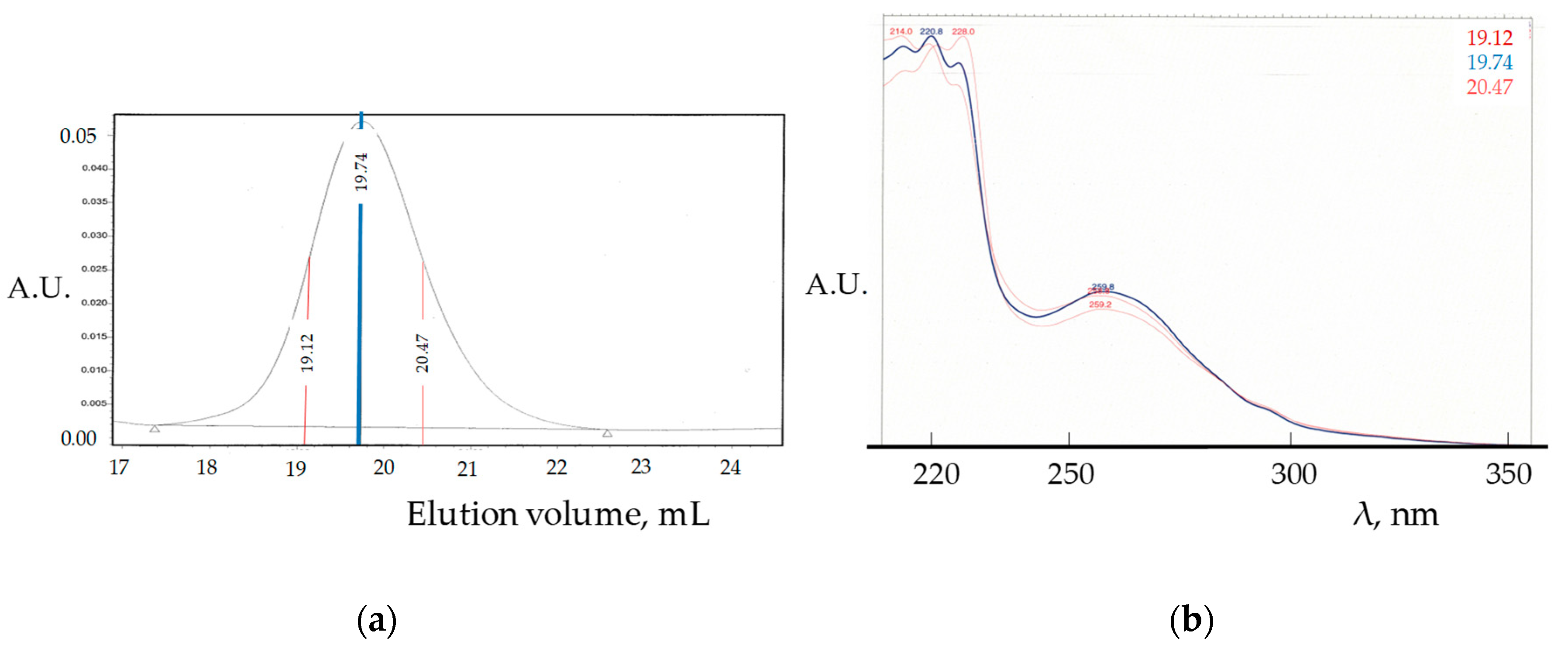
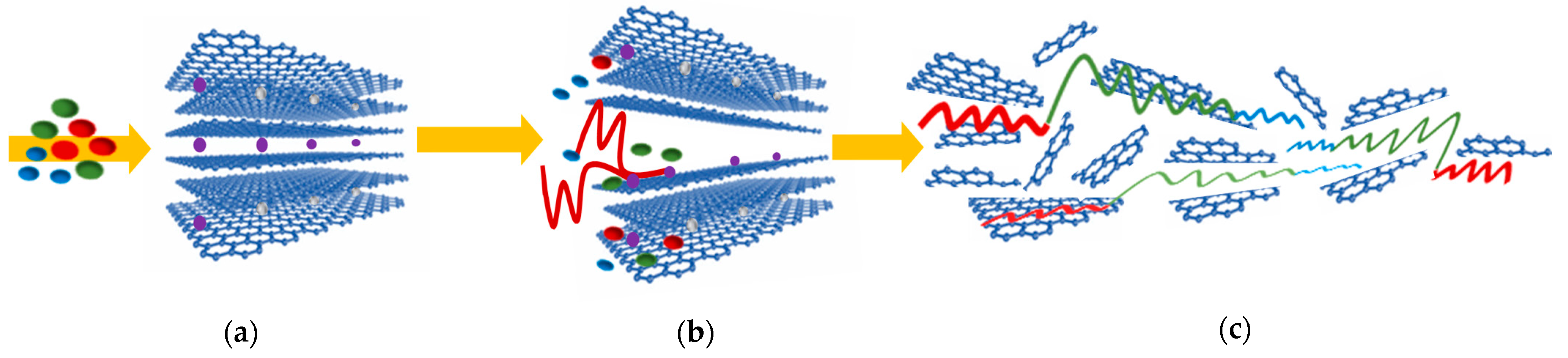
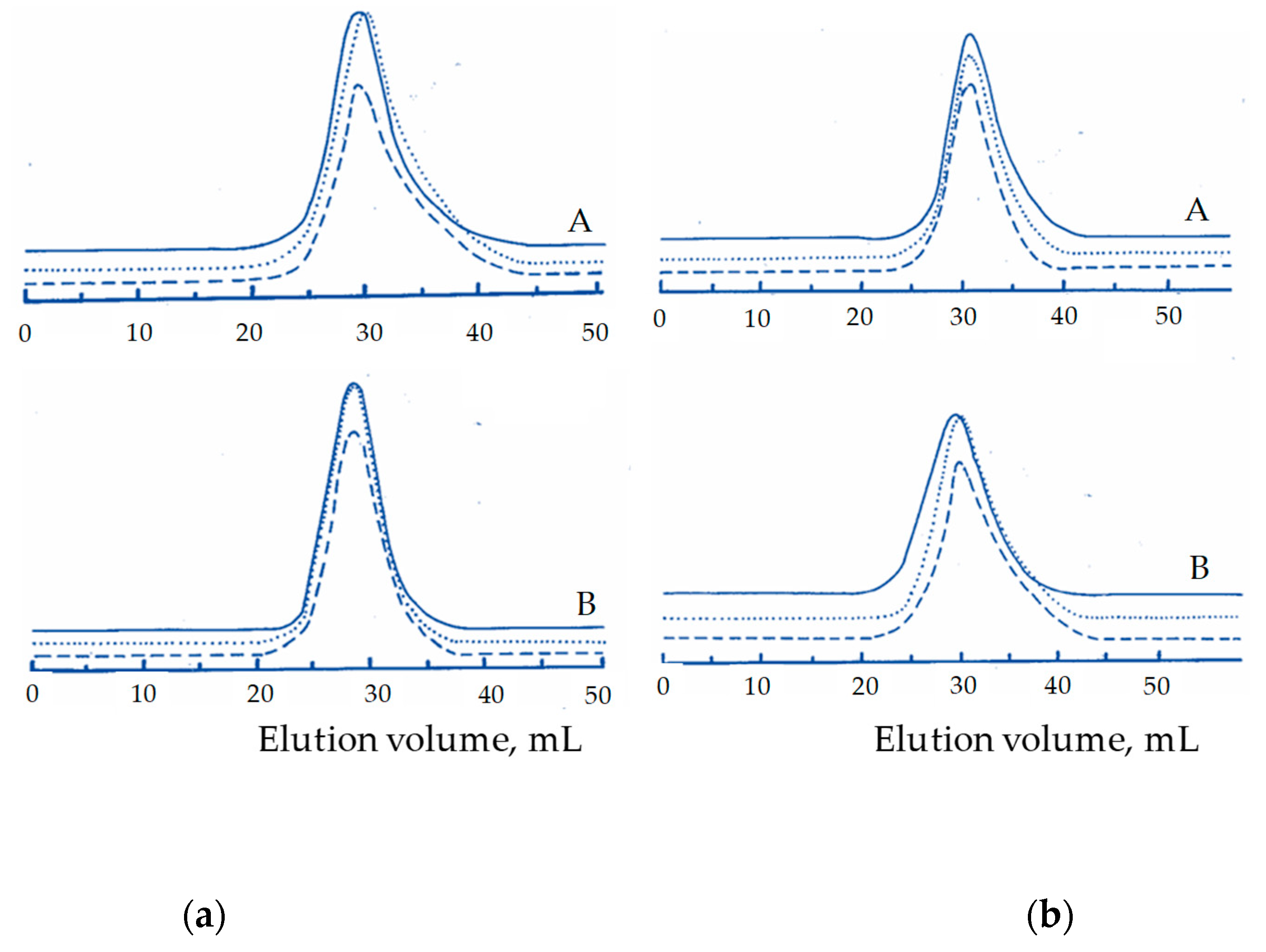
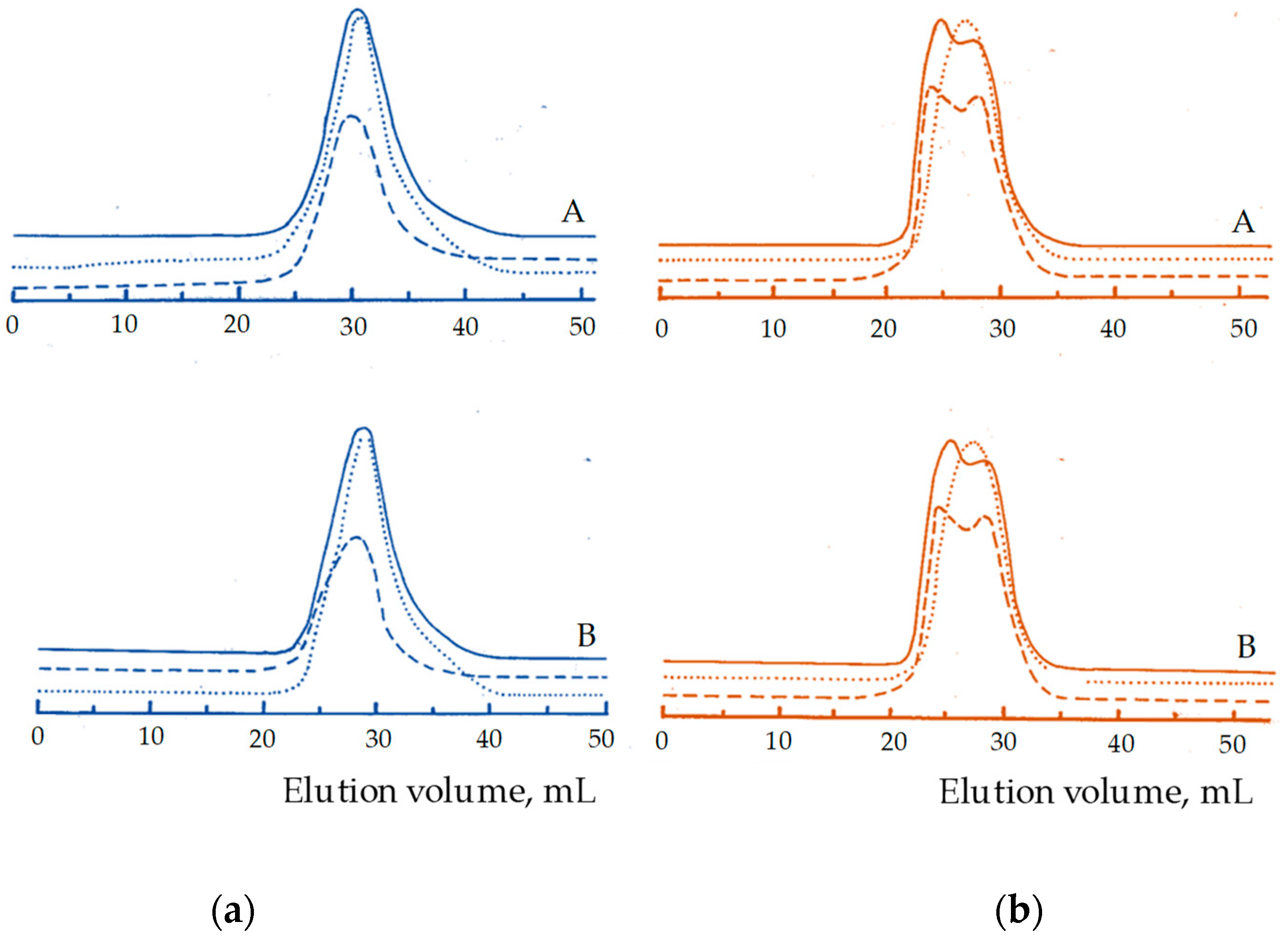
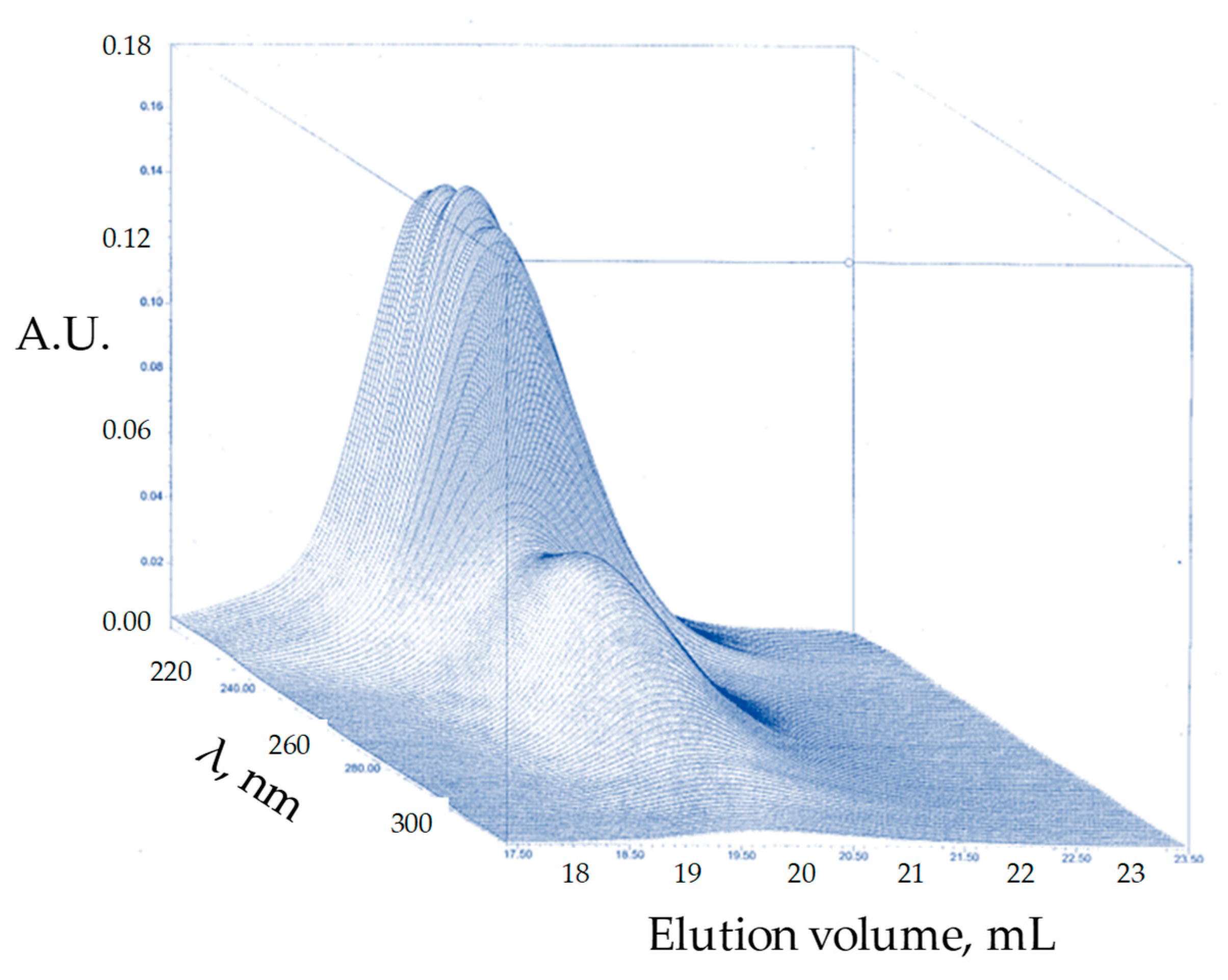
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sarkar, A.; Sasmal, R.; Das, A.; Venugopal, A.; Agasti, S.S.; George, S.J. Tricomponent Supramolecular Multiblock Copolymers with Tunable Composition via Sequential Seeded Growth. Angew. Chem. 2021, 133, 18357–18364. [Google Scholar] [CrossRef]
- Baeza, G.P. Recent advances on the structure–properties relationship of multiblock copolymers. J. Appl. Polym. Sci. 2021, 59, 2405–2433. [Google Scholar] [CrossRef]
- Koo, C.M.; Hillmyer, M.A.; Bates, F.S. Structure and Properties of Semicrystalline−Rubbery Multiblock Copolymers. Macromolecules 2006, 39, 667–677. [Google Scholar] [CrossRef]
- Roy, A.; Lee, H.-S.; McGrath, J.E. Hydrophilic–hydrophobic multiblock copolymers based on poly(arylene ether sulfone)s as novel proton exchange membranes—Part B. Polymer 2008, 49, 5037–5044. [Google Scholar] [CrossRef]
- Li, Y.; Yang, H.Y.; Lee, D.S. Biodegradable and Injectable Hydrogels in Biomedical Applications. Biomacromolecules 2022, 23, 609–618. [Google Scholar] [CrossRef]
- Beyer, V.P.; Kim, J.; Becer, C.R. Synthetic approaches for multiblock copolymers. Polym. Chem. 2020, 11, 1271–1291. [Google Scholar] [CrossRef]
- Zhao, T.; Drain, B.A.; Yilmaz, G.; Becer, C.R. One-pot synthesis of amphiphilic multiblock poly(2-oxazoline)s via para-fluoro-thiol click reactions. Polym. Chem. 2021, 12, 6392–6403. [Google Scholar] [CrossRef]
- Heck, M.; Botha, C.; Wilhelm, M.; Hirschberg, V. One-Pot Synthesis of Alternating (Ultra-High Molecular Weight) Multiblock Copolymers via a Combination of Anionic Polymerization and Polycondensation. Macromol. Rapid Commun. 2021, 42, 2100448. [Google Scholar] [CrossRef]
- Pafiti, K.S.; Patrickios, C.S.; Abetz, C.; Abetz, V. High-molecular-weight symmetrical multiblock copolymers: Synthesis by RAFT polymerization and characterization. J. Polym. Sci. A Polym. Chem. 2013, 51, 4957–4965. [Google Scholar] [CrossRef]
- Scheibel, D.M.; Guo, D.; Luo, J.; Gitsov, I. A Single Enzyme Mediates the “Quasi-Living” Formation of Multiblock Copolymers with a Broad Biomedical Potential. Biomacromolecules 2020, 21, 2132–2146. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 2002, 51, 1–186. [Google Scholar] [CrossRef]
- Rashkov, I. Polymerization in Graphite Intercalation Compounds; Tcherniradev, V., Ed.; Kliment Ohridski University Press: Sofia, Bulgaria, 1989. [Google Scholar]
- Watanabe, K.; Kondow, T.; Soma, M.; Onishi, T.; Tamaru, K. Molecular-sieve type sorption on alkali graphite intercalation compounds. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1973, 333, 51–67. [Google Scholar] [CrossRef]
- Rashkov, I.; Kakuliya, T.; Vladimirov, N.; Gitsov, I. Copolymerization of styrene with some oxiranes initiated by KC24. Eur. Polym. J. 1986, 22, 407–412. [Google Scholar] [CrossRef]
- Gitsov, I.; Rashkov, I.B.; Panayotov, I.M.; Golub′, A. Anionic polymerization of lactones initiated by alkali graphitides. IV. Copolymerization of ε-caprolactone initiated by KC24. J. Polym. Sci. Part A Polym. Chem. 1989, 27, 639–646. [Google Scholar] [CrossRef]
- Franta, E.; Hogen-Esch, T.; van Beylen, M.; Smid, J. Fifty years of living polymers. J. Polym. Sci. A Polym. Chem. 2007, 45, 2576–2579. [Google Scholar] [CrossRef]
- Gitsov, I.; Frechet, J.M.J. Novel Nanoscopic Architectures. Linear-Globular ABA Copolymers with Polyether Dendrimers as A Blocks and Polystyrene as B Block. Macromolecules 1994, 27, 7309–7315. [Google Scholar] [CrossRef]
- Hérold, A. Research on the Graphite Intercalation Compounds. Bull. Soc. Chim. Fr. 1955, 187, 999–1012. [Google Scholar]
- Graham, R.K.; Dunkelberger, D.L.; Goode, W.E. Anionic Copolymerization: The Inability of the Poly-(Methyl Methacrylate) Anion to Initiate the Polymerization of Styrene1. J. Am. Chem. Soc. 1960, 82, 400–403. [Google Scholar] [CrossRef]
- Laita, Z.; Szwarc, M. Polymerization of styrene–methyl methacrylate mixtures initiated by butyllithium. J. Polym. Sci. B Polym. Lett. 1968, 6, 197–200. [Google Scholar] [CrossRef]
- Li, T.; Zhou, C.; Jiang, M. UV absorption spectra of polystyrene. Polym. Bull. 1991, 25, 211–216. [Google Scholar] [CrossRef]
- Ahmed, R.M. Optical Study on Poly(methyl methacrylate)/Poly(vinyl acetate) Blends. Int. J. Photoenergy 2009, 2009, 10389. [Google Scholar] [CrossRef]
- For PMMA UV Spectrum See Also Allresist GmbH (Strausberg, Germany) UV-Patterning of PMMA Resists. Available online: allresist.com (accessed on 8 March 2022).
- Kotani, Y.; Kamigaito, M.; Sawamoto, M. Living Random Copolymerization of Styrene and Methyl Methacrylate with a Ru(II) Complex and Synthesis of ABC-Type “Block-Random” Copolymers. Macromolecules 1998, 31, 5582–5587. [Google Scholar] [CrossRef]
- Panayotov, I.M.; Rashkov, I.B. Anionic polymerization and copolymerization of vinyl monomers initiated by potassium–graphite inclusion compounds. J. Polym. Sci. A-1 Polym. Chem. 1972, 10, 1267–1270. [Google Scholar] [CrossRef]
- Rashkov, I.; Panayotov, I.; Gitsov, I. Mechanism of the anionic polymerization of lactones, initiated by intercalation graphite compounds. Polym. Bull. 1981, 4, 97–103. [Google Scholar] [CrossRef]
- Rashkov, I.B.; Gitsov, I.; Panayotov, I.M. Anionic polymerization of lactones initiated by alkali graphitides. II. Changes in the KC24 structure during polymerization of lactones. J. Polym. Sci. Polym. Chem. Ed. 1983, 21, 937–941. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
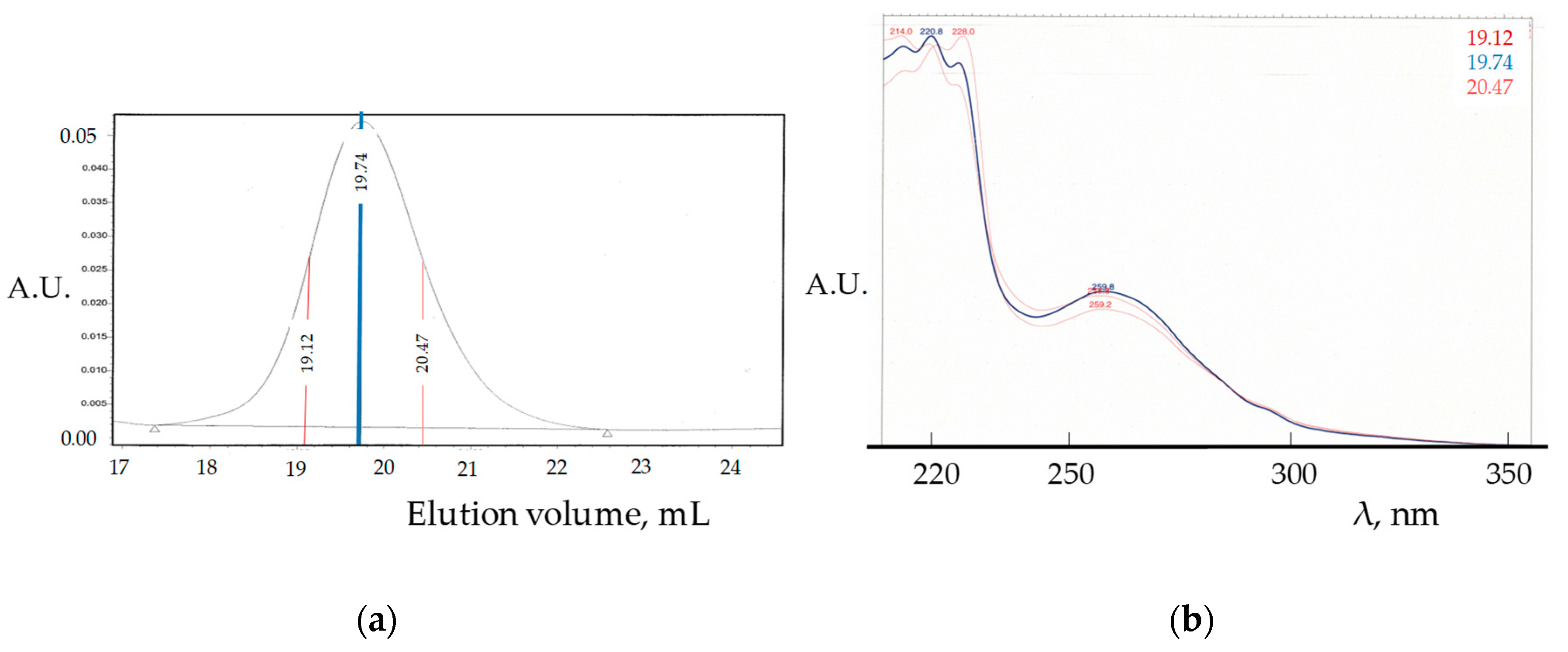{kind=link}
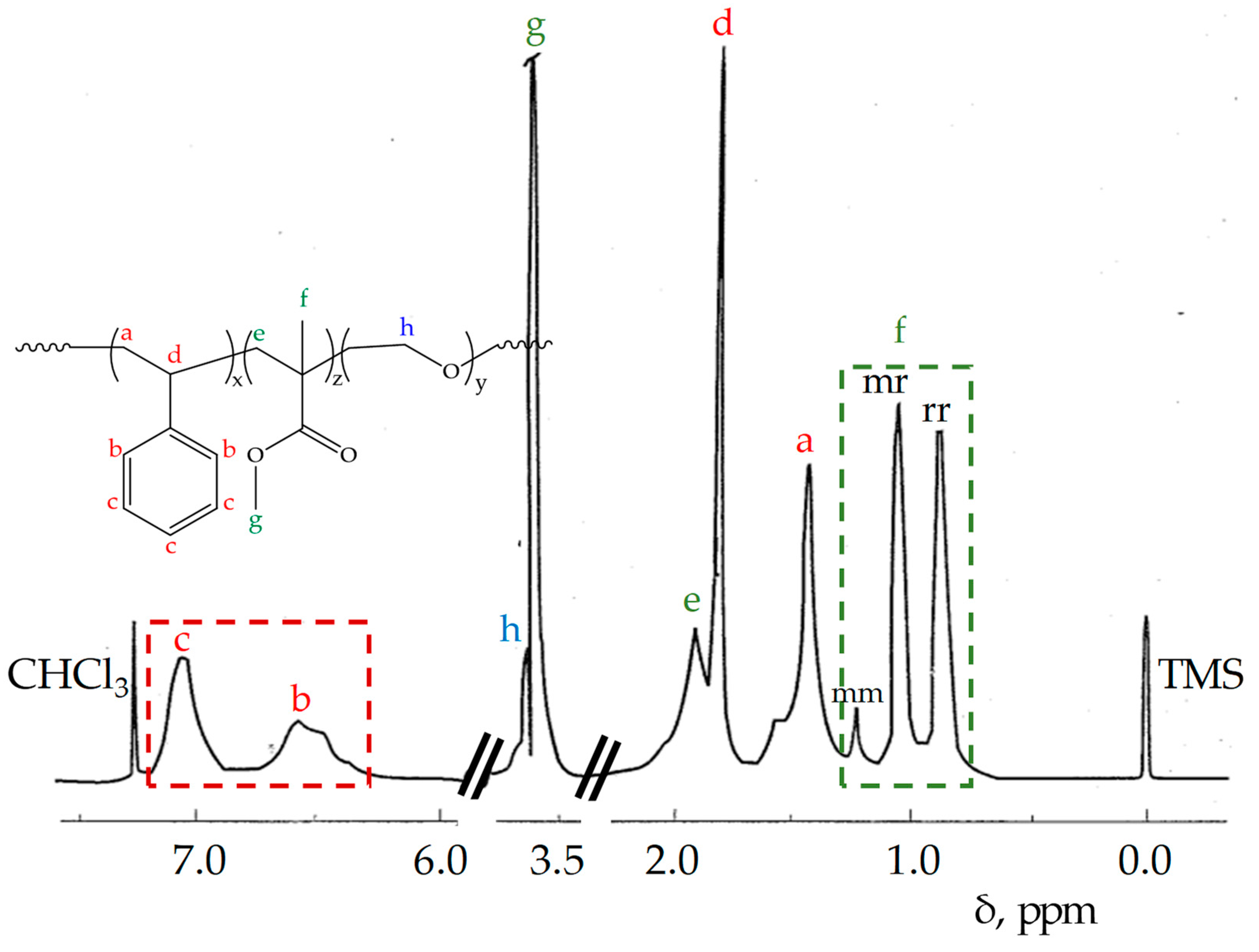{kind=link}
| Product | M 2 Addition Sequence | Temperature (°C) | Time (h) | Yield 3 (%) |
|---|---|---|---|---|
| STEM 1 4 | All | 25 | 27 | 80 |
| STEM 2 4 | St/MMA/EO | 25 | 27 | 68 |
| STEM 3 5 | St/EO/MMA | 25 | 42 | 78 |
| Product | Mw × 103 (SEC) 1 | Dispersity 2 (Ð) | PMMA Microstructure mm/mr/rr | Molar Content 3 St/MMA/EO |
|---|---|---|---|---|
| STEM 1 4 | 175 | 3.16 | 0.20/0.50/0.30 | 0.19/0.66/0.15 |
| STEM 2 4 | 279 | 5.90 | 0.16/0.52/0.32 | 0.30/0.50/0.20 |
| STEM 3 5 | 175 | 3.60 | 0.08/0.43/0.49 | 0.28/0.62/0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vladimirov, N.G.; Gitsov, I. Polymerization Initiated by Graphite Intercalation Compounds Revisited: One-Pot Synthesis of Amphiphilic Pentablock Copolymers. Macromol 2022, 2, 184-193. https://doi.org/10.3390/macromol2020012
Vladimirov NG, Gitsov I. Polymerization Initiated by Graphite Intercalation Compounds Revisited: One-Pot Synthesis of Amphiphilic Pentablock Copolymers. Macromol. 2022; 2(2):184-193. https://doi.org/10.3390/macromol2020012
Chicago/Turabian StyleVladimirov, Nikolay G., and Ivan Gitsov. 2022. "Polymerization Initiated by Graphite Intercalation Compounds Revisited: One-Pot Synthesis of Amphiphilic Pentablock Copolymers" Macromol 2, no. 2: 184-193. https://doi.org/10.3390/macromol2020012
APA StyleVladimirov, N. G., & Gitsov, I. (2022). Polymerization Initiated by Graphite Intercalation Compounds Revisited: One-Pot Synthesis of Amphiphilic Pentablock Copolymers. Macromol, 2(2), 184-193. https://doi.org/10.3390/macromol2020012