
Influence of Ethylene-1-Alkene Copolymers Microstructure on Thermo-Rheological Behavior of Model Blends for Enhanced Recycling


Abstract
:
1. Introduction
2. Materials and Experimental Methods
2.1. Materials
2.2. Experimental Methods
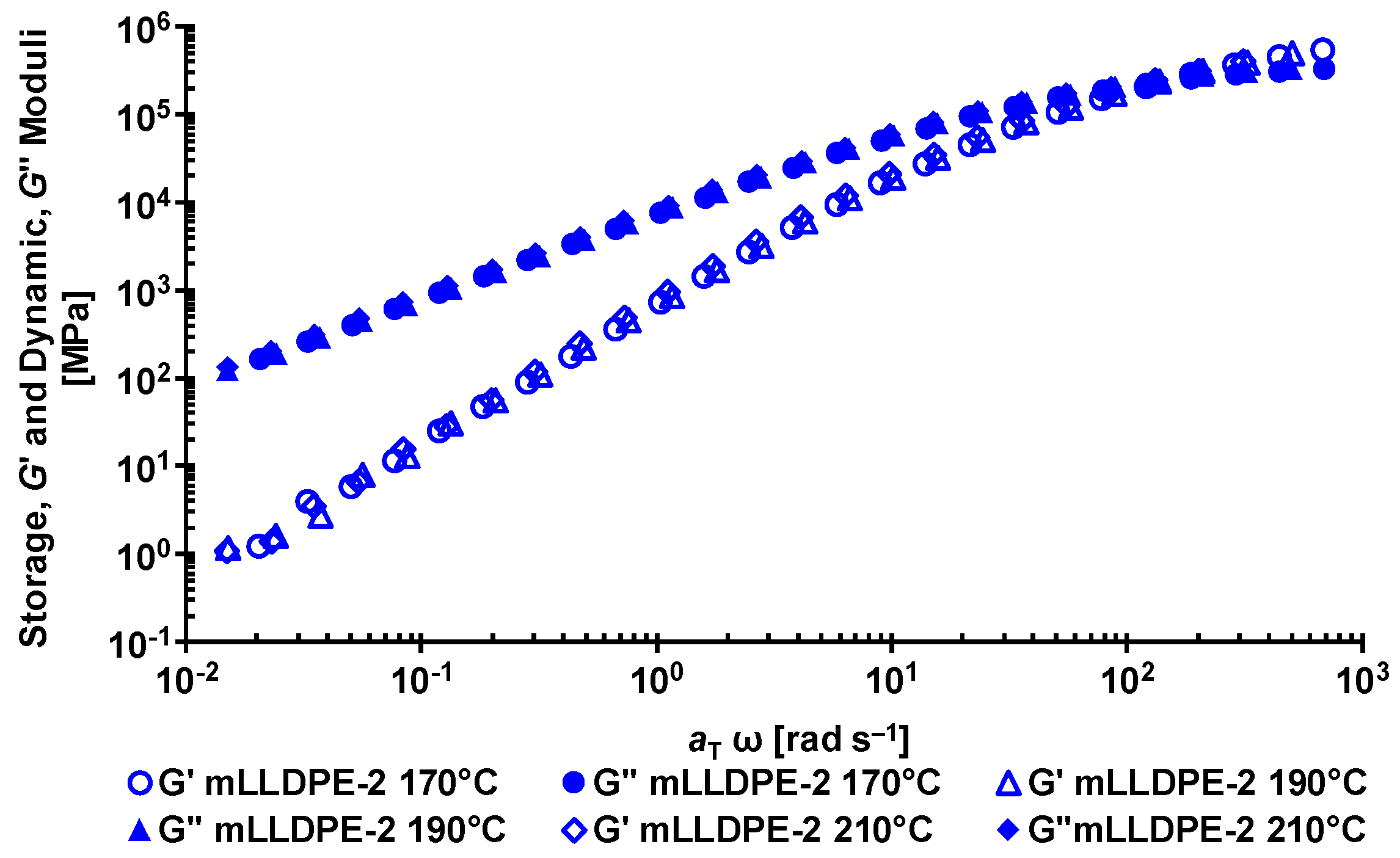
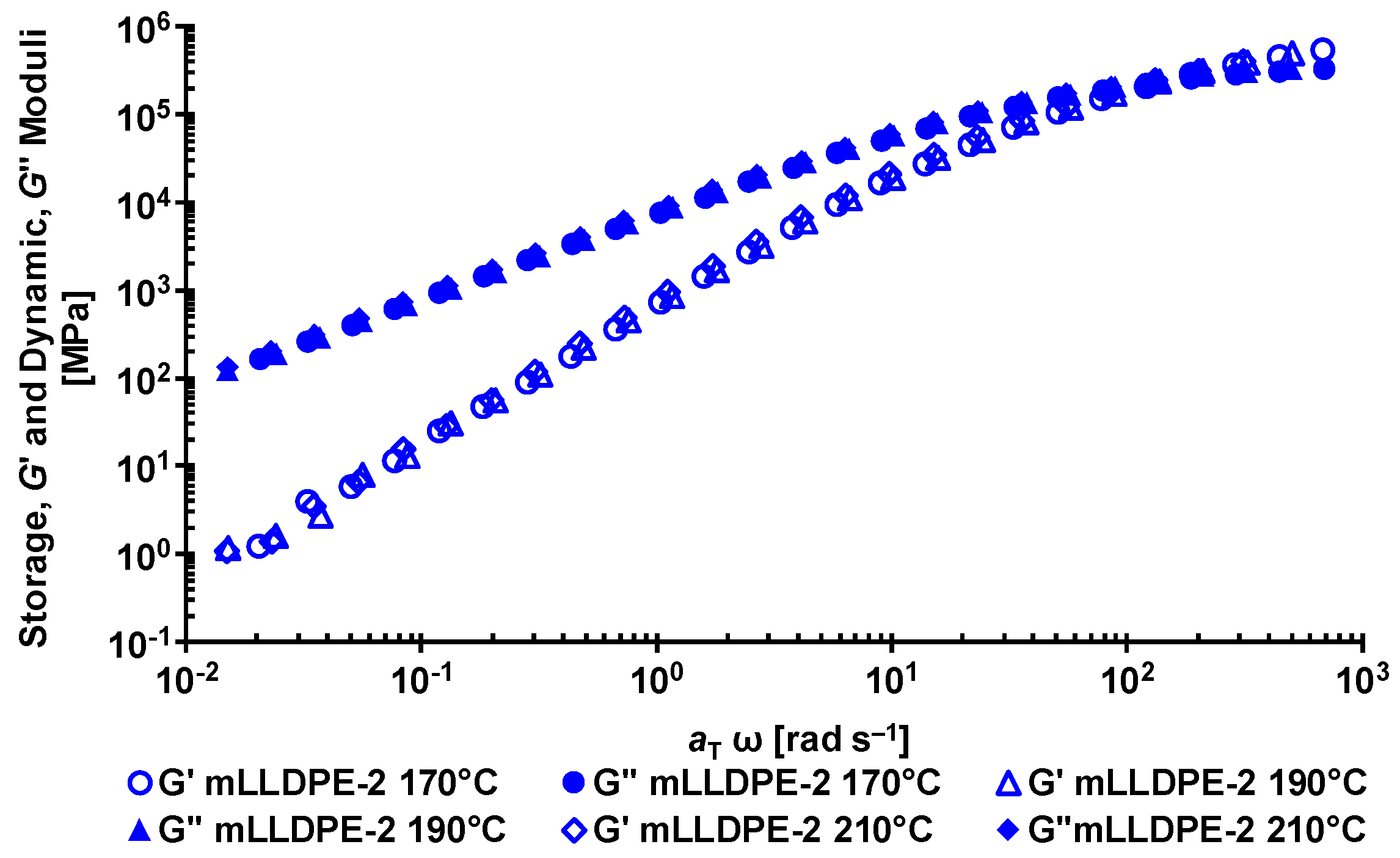
2.2.1. Dynamic Rheology
2.2.2. Thermal Analysis
3. Results and Discussions
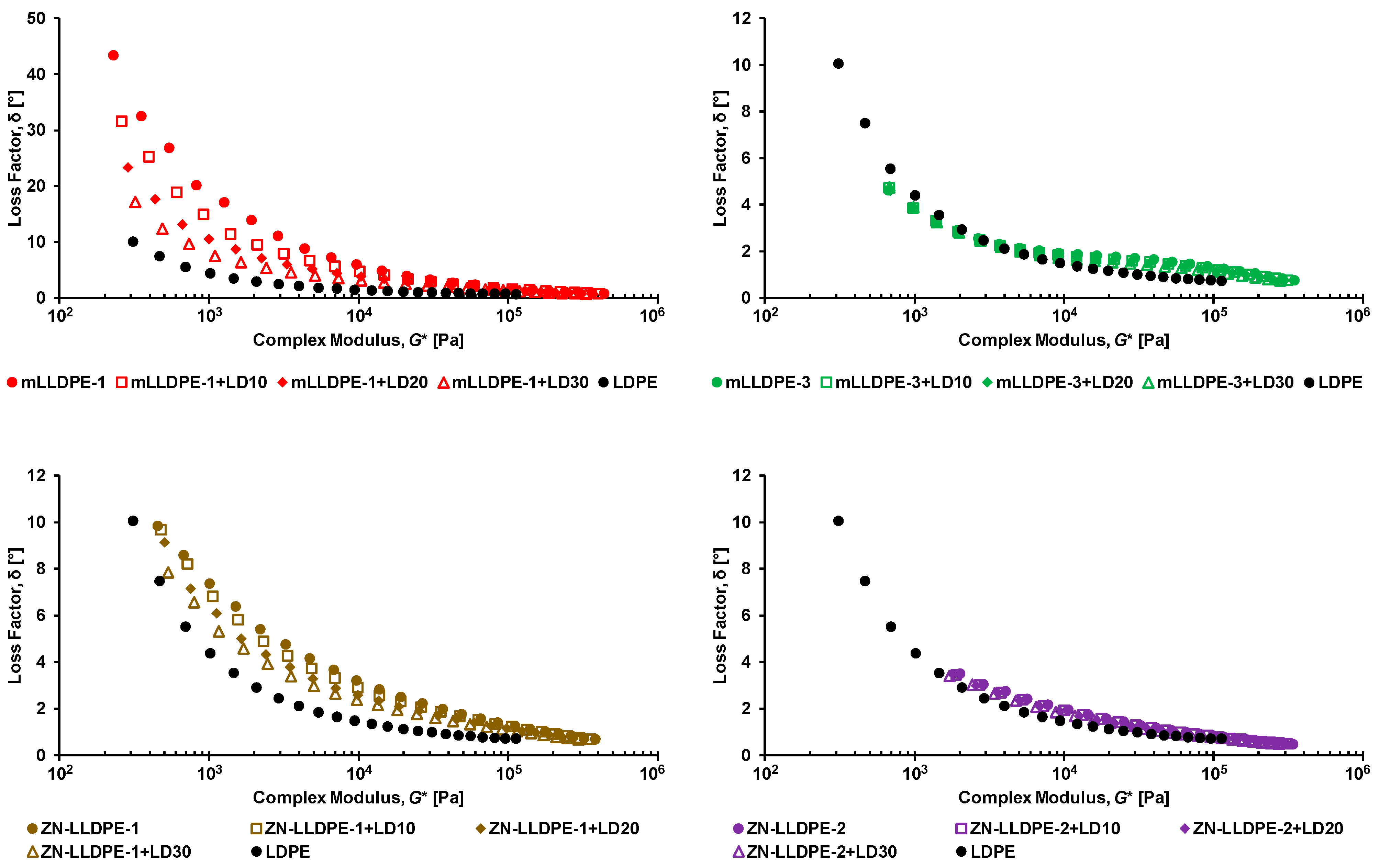
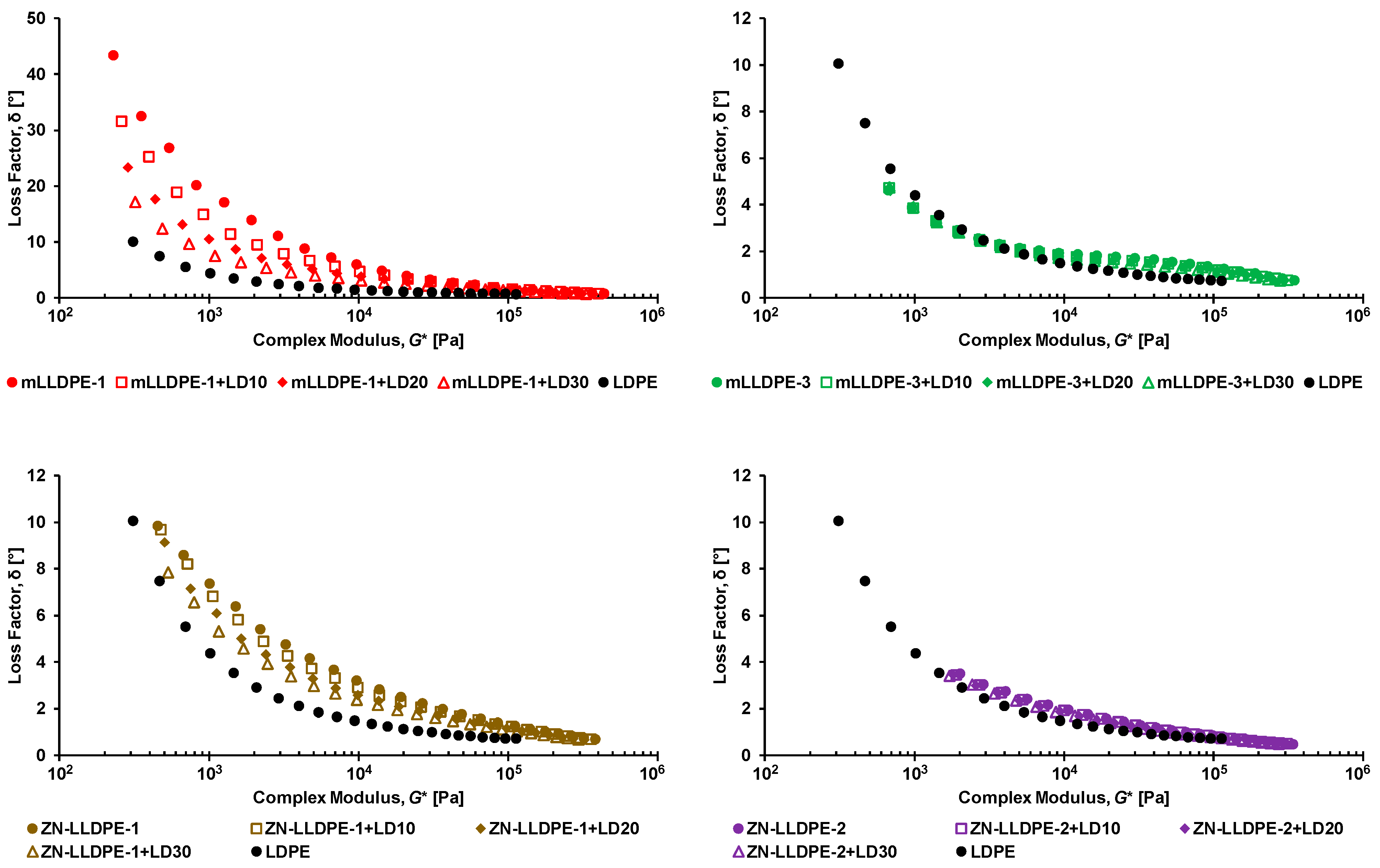
3.1. Rheological Behavior of LLDPE/LDPE Blends
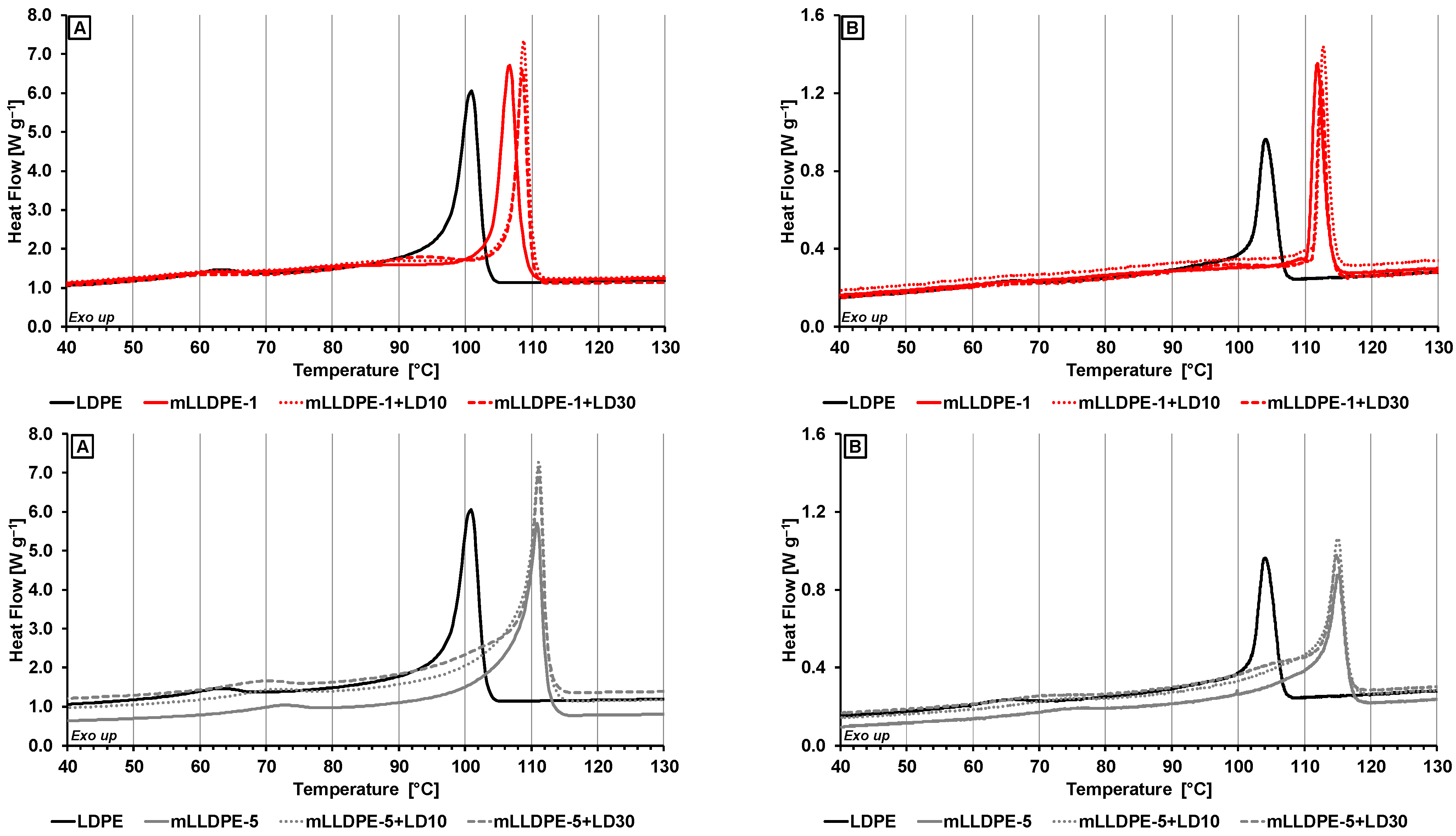
3.2. Thermal Characterization of LLDPE/LDPE Blends
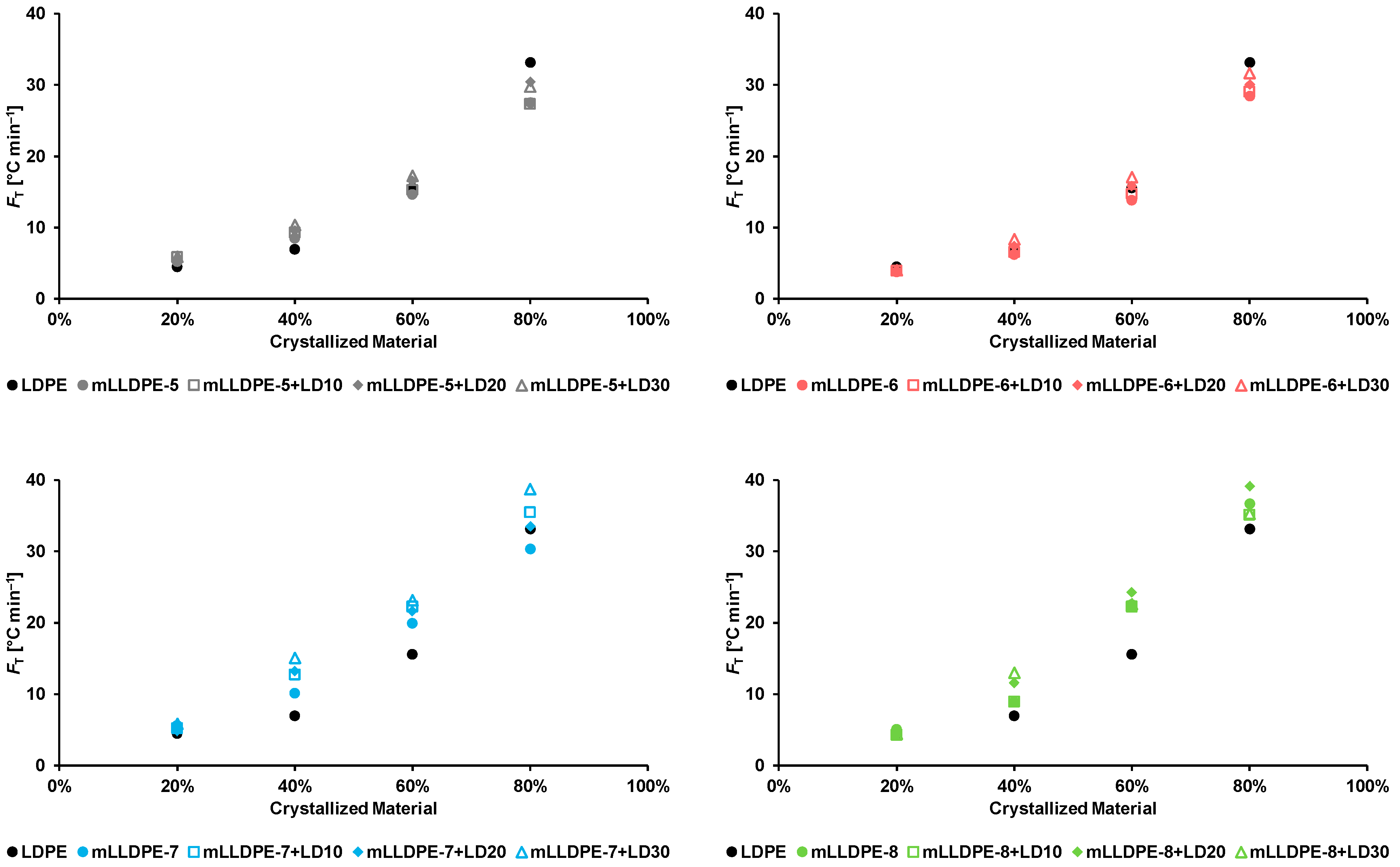
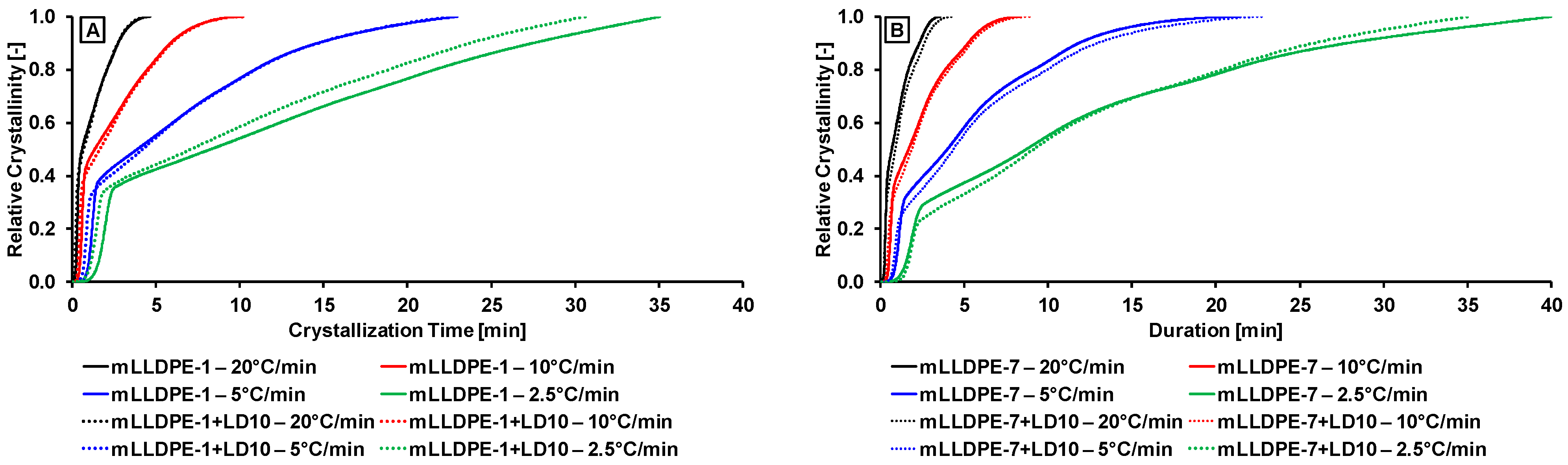
3.2.1. Effect of Cooling Rate
3.2.2. Effect of Blending LDPE to LLDPE
- High-temperature crystallization region (100–120 °C) corresponding to crystallization behavior of pristine LLDPE and of LLDPE in the presence of LDPE;
- Medium-temperature crystallization region (90–110 °C) corresponding to crystallization behavior of LDPE in the presence of LLDPE;
- Low-temperature region (60–80 °C) corresponding to crystallization behavior of relatively highly branched polymer fraction.
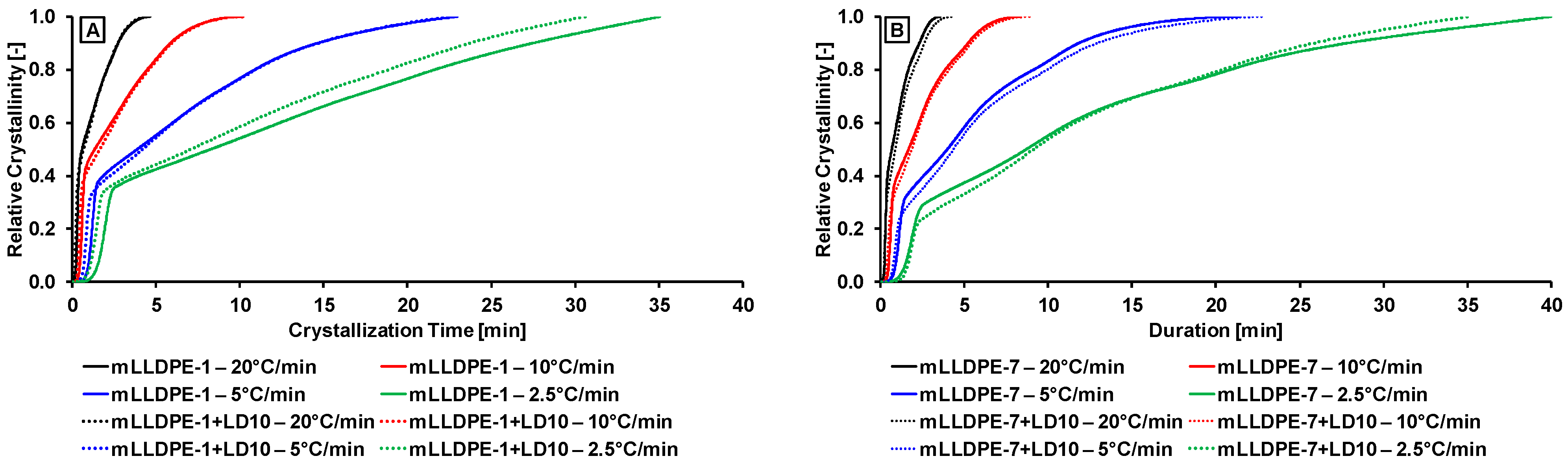
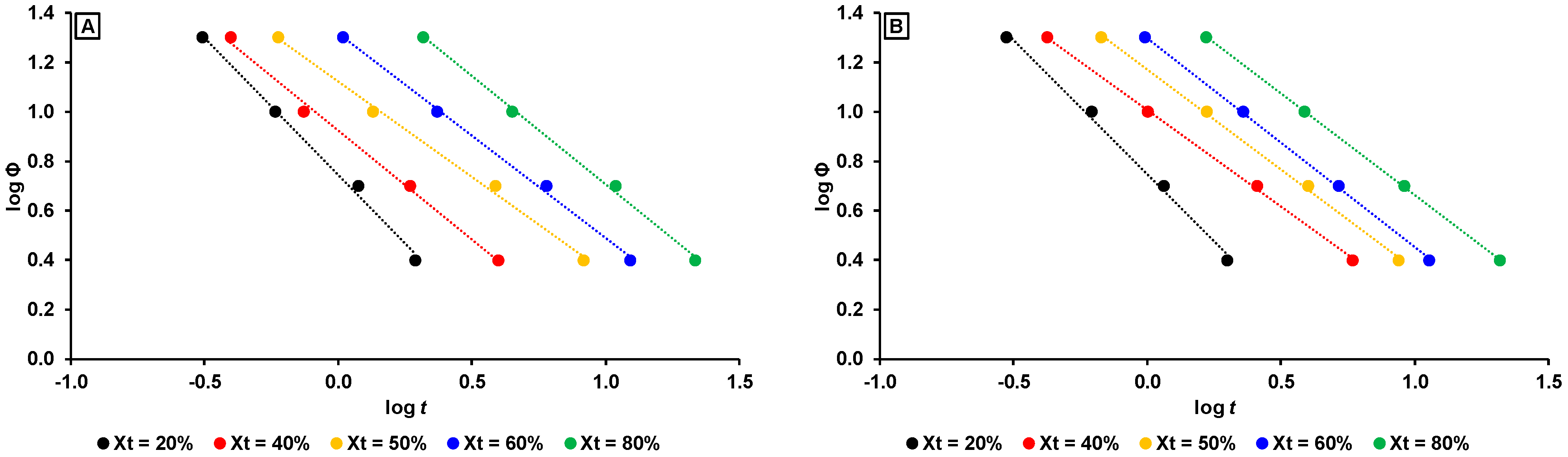
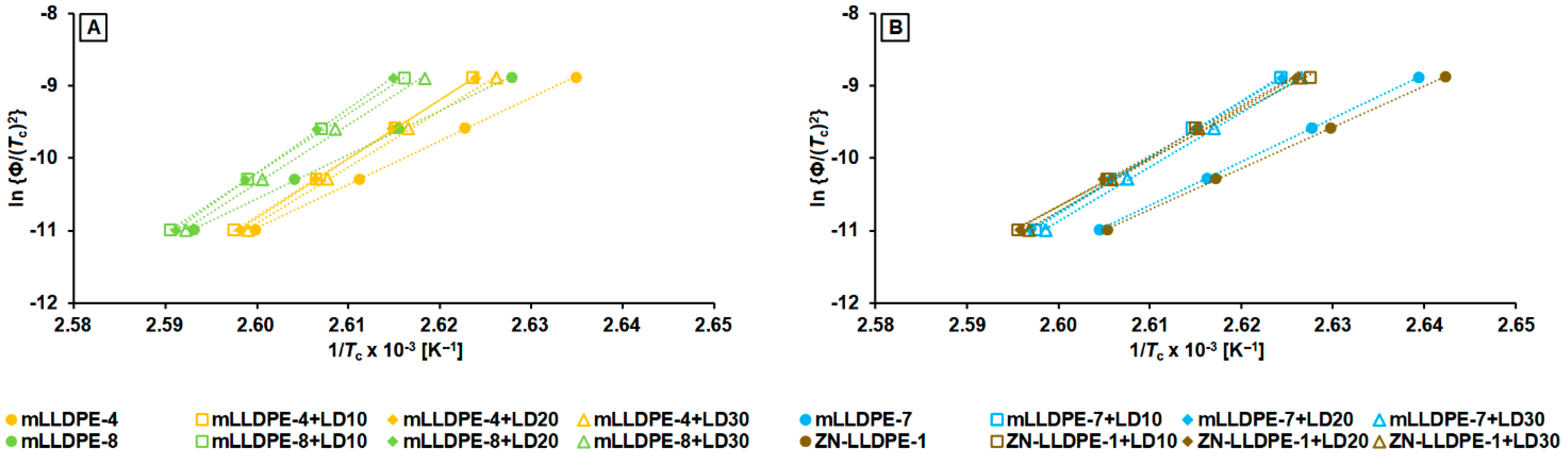
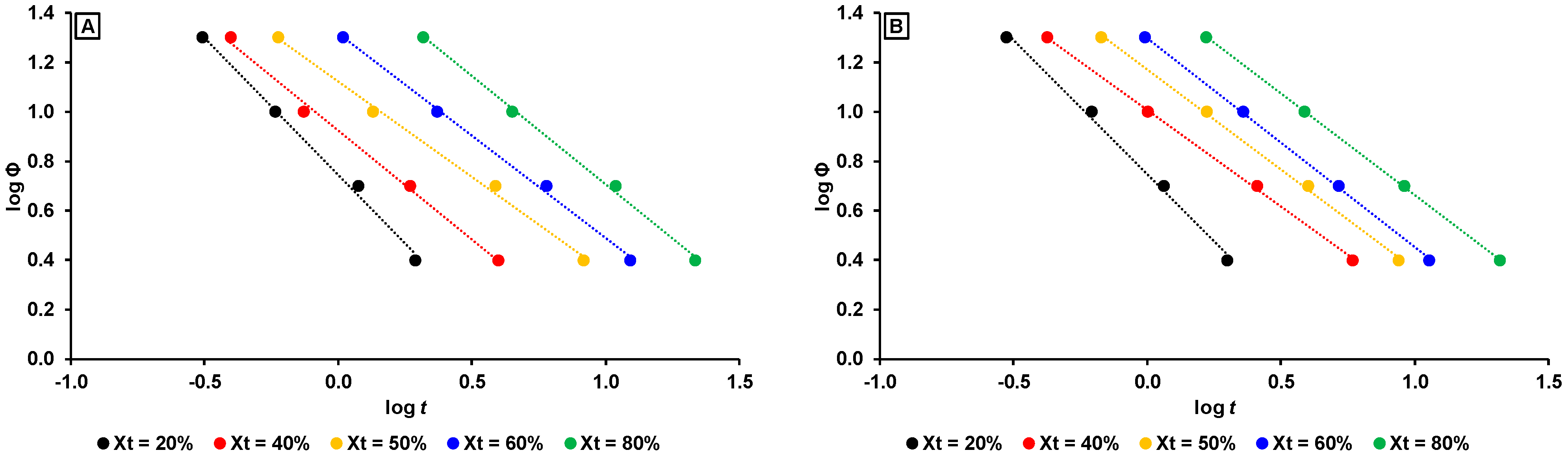
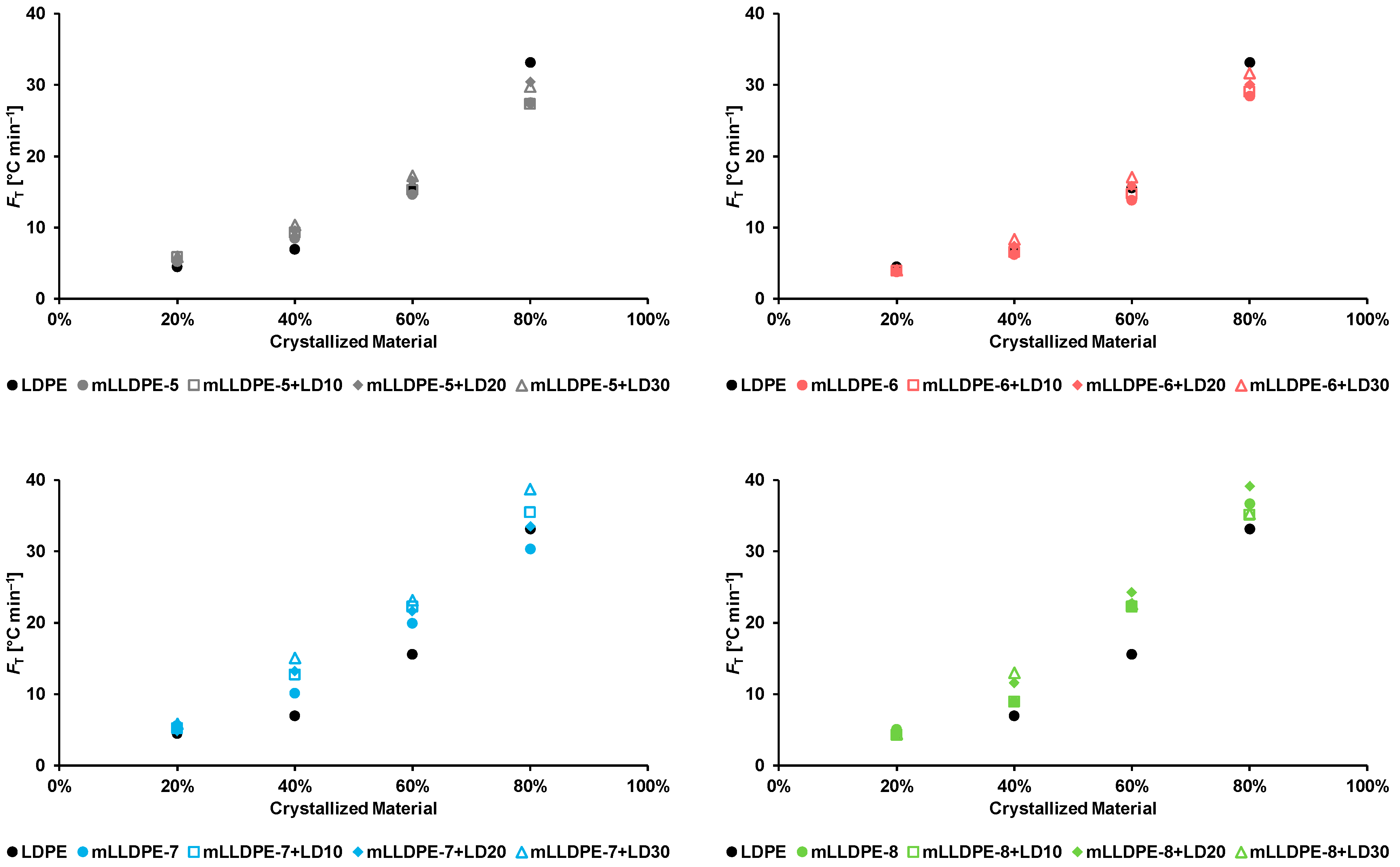
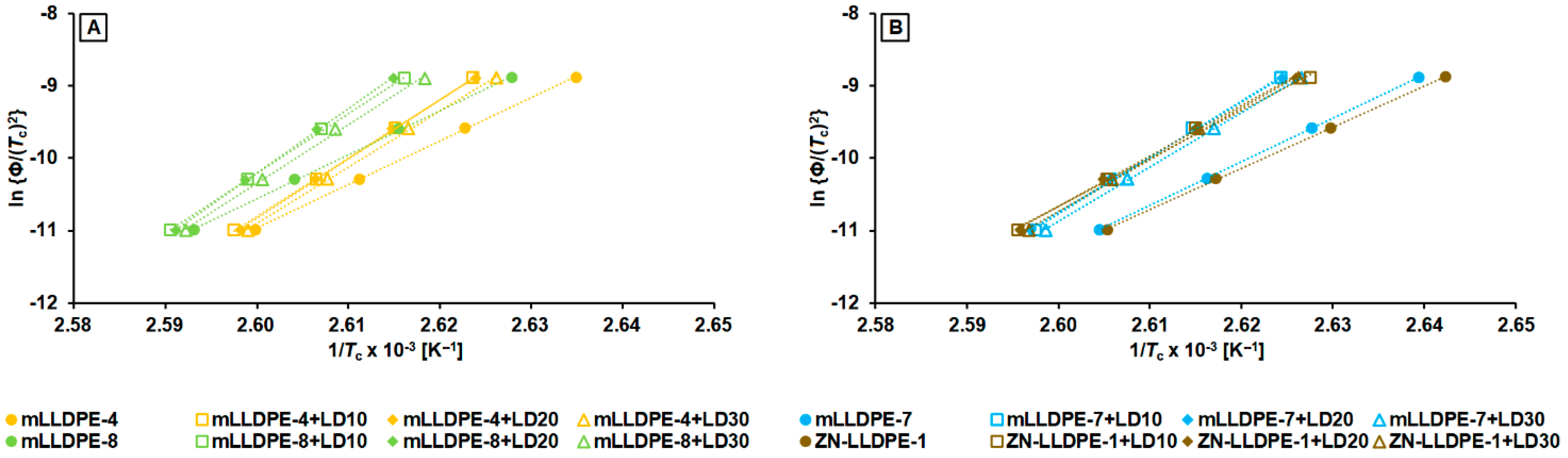
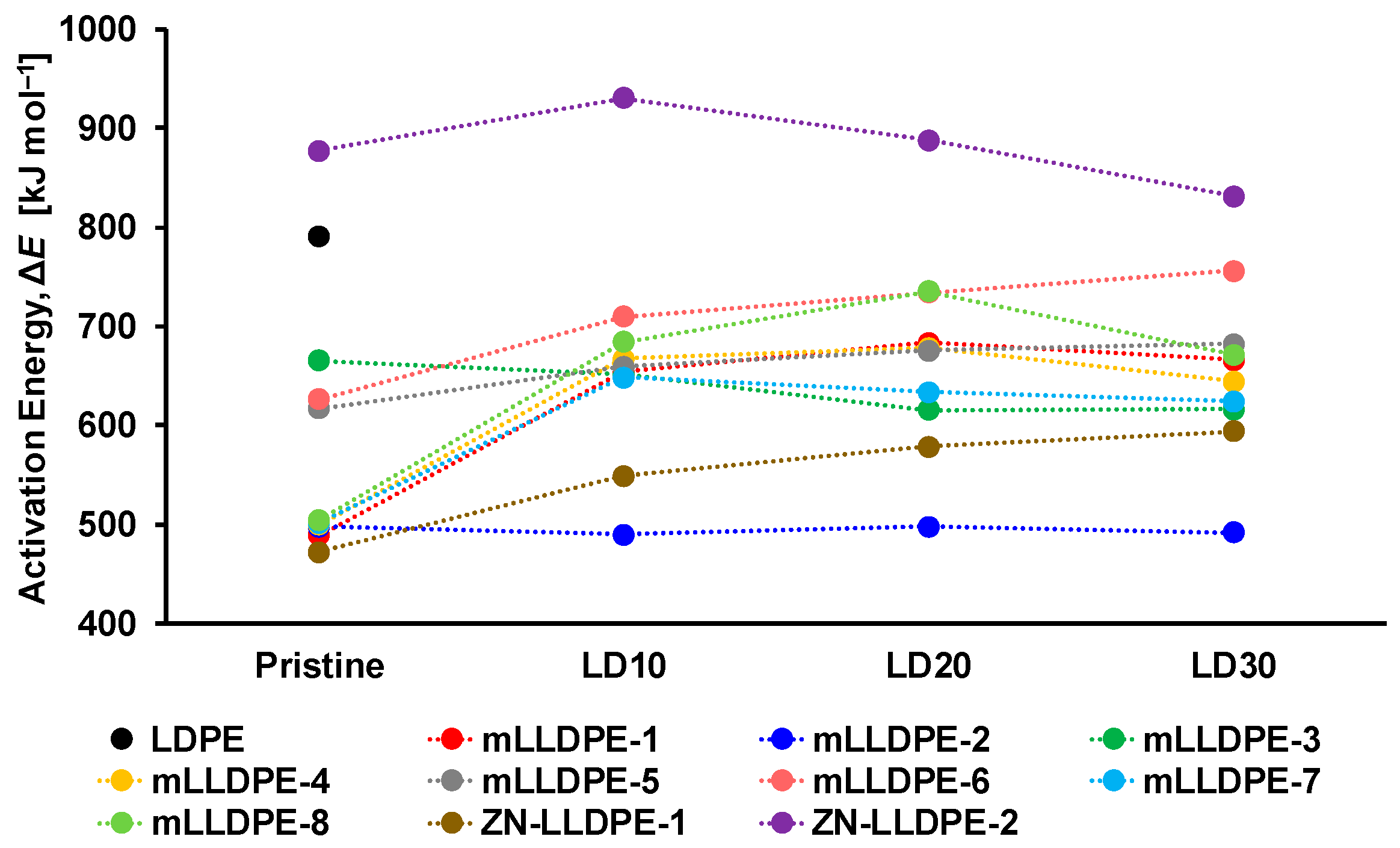
3.2.3. Non-Isothermal Crystallization Kinetics
- mLLDPE with SCB/1000C < 12, (viz., mLLDPE-2, mLLDPE-3, mLLDPE-5 and mLLDPE-6): For these mLLDPE matrices, blending with LDPE did not significantly influence Ti,c and Tc of mLLDPE. Their LPDE blends tend to crystallize faster than LDPE. In these blends, concurrent and/or separate crystallization was found to be the more preferred mode than co-crystallization of mLLDPE and LDPE.
- mLLDPE with SCB/1000C > 12, (viz., mLLDPE-1, mLLDPE-4, mLLDPE-7 and mLLDPE-8): For these mLLDPE matrices, blending with LDPE did significantly influence Ti,c and Tc of mLLDPE. Their LPDE blends tend to crystallize slower than LDPE. In these blends, co-crystallization was found to be the more preferred mode than concurrent and/or separate crystallization of mLLDPE and LDPE.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steffl, T. Rheological and Film Blowing Properties of Various Low Density Polyethylenes and Their Blends. Ph.D. Thesis, Universität Erlangen-Nürnberg, Erlangen, Germany, June 2004. [Google Scholar]
- Simanke, A.G.; Galland, G.B.; Baumhardt, R.; Quijada, R.; Mauler, R.S. Influence of the type and the comonomer contents on the mechanical behavior of ethylene/α-olefin copolymers. J. Appl. Polym. Sci. 1999, 74, 1194–1200. [Google Scholar] [CrossRef]
- Usami, T.; Gotoh, Y.; Takayama, S. Generation mechanism of short-chain branching distribution in linear low-density polyethylene. Macromolecules 1986, 19, 2722–2726. [Google Scholar] [CrossRef]
- Munstedt, H.; Kurzbeck, S.; Egersdorfer, L. Influence of molecular structure on rheological properties of polyethylenes. Rheol. Acta 1998, 37, 21–29. [Google Scholar] [CrossRef]
- Wignal, G.D.; Alamo, R.G.; Ritchson, E.J.; Mandelkern, L.; Schwahn, D. SANS studies of liquid− liquid phase separation in heterogeneous and metallocene-based linear low-density polyethylenes. Macromolecules 2001, 34, 8160–8165. [Google Scholar] [CrossRef]
- Hussein, I.A.; Williams, M.C. Rheological Study of the Influence of Branch Content on the Miscibility of Octene m-LLDPE and ZNLLDPEin LDPE. Polym. Eng. Sci. 2004, 44, 660–672. [Google Scholar] [CrossRef]
- Knuuttila, H.; Lehtinen, A.; Nummila-Pakarinen, A. Advanced polyethylene technologies—Controlled material properties. In Long Term Properties of Polyolefins; Springer: Berlin/Heidelberg, Germany, 2004; pp. 13–28. [Google Scholar]
- Malpass, D.B. An overview of industrial polyethylene processes. In Introduction to Industrial Polyethylene: Properties, Catalysts, Processes; Scrivener Publishing LLC: Beverly, MA, USA, 2010; pp. 71–82. [Google Scholar]
- Hussein, I.A. Influence of composition distribution and branch content on the miscibility of m-LLDPE and HDPE blends: Rheological investigation. Macromolecules 2003, 36, 2024–2031. [Google Scholar] [CrossRef]
- Hussein, I.A.; Hameed, T.; Abu Sharkh, B.F.; Mezghani, K. Miscibility of hexene-LLDPE and LDPE blends: Influence of branch content and composition distribution. Polymer 2003, 44, 4665–4672. [Google Scholar] [CrossRef] [Green Version]
- Pérez, R.; Rojo, E.; Fernández, M.; Leal, V.; Lafuente, P.; Antamaría, A.S. Basic and applied rheology of m-LLDPE/LDPE blends: Miscibility and processing features. Polymer 2005, 46, 8045–8053. [Google Scholar] [CrossRef]
- Delgadillo-Velázquez, O.; Hatzikiriakos, S.G.; Sentmanat, M. Thermorheological properties of LLDPE/LDPE blends. Rheol. Acta 2008, 47, 19–31. [Google Scholar] [CrossRef]
- Mamun, A.; Chen, X.; Alamo, R.G. Interplay between a strong memory effect of crystallization and liquid–liquid phase separation in melts of broadly distributed ethylene–1-alkene copolymers. Macromolecules 2014, 47, 7958–7970. [Google Scholar] [CrossRef]
- Groeninckx, G.; Vanneste, M.; Everaert, V. Crystallization, morphological structure and melting of polymer blends. In Polymer Blends Handbook; Utracki, L.A., Ed.; Kluwer Academic Publishers: Alphen aan den Rijn, The Netherlands, 2003; pp. 215–240. [Google Scholar]
- Galgali, G.; Kaliappan, S.K. Microstructure–sealing performance linkage of metallocene linear low density polyethylene and low density polyethylene blends. Polym. Cryst. 2021, 4, e10206. [Google Scholar] [CrossRef]
- ISO 6721-10:2015; Plastics—Determination of Dynamic Mechanical Properties—Part 10: Complex Shear Viscosity Using a Parallel-Plate Oscillatory Rheometer. International Organization for Standardization: Geneva, Switzerland, 2015.
- ISO 11357-3:2018; Plastics—Differential Scanning Calorimetry (DSC)—Part 3: Determination of Temperature and Enthalpy of melting and Crystallization. International Organization for Standardization: Geneva, Switzerland, 2018.
- Fan, Z.J.; Williams, M.C.; Choi, P.A. molecular dynamics study of the effects of branching characteristics of LDPE on its miscibility with HDPE. Polymer 2002, 43, 1497–1502. [Google Scholar] [CrossRef]
- Abu-Sharkh, B.F.; Giri, A.M.; Hussein, I.A. Influence of branch content on the microstructure of blends of linear and octene-branched polyethylene: A MD simulation study. Eur. Polym. J. 2004, 40, 1177–1182. [Google Scholar] [CrossRef]
- Van Gurp, M.; Palmen, J. Time-temperature superposition for polymeric blends. J. Rheol. Bull. 1998, 67, 5–8. [Google Scholar]
- Hatzikiriakos, S.G. Long chain branching and polydispersity effects on the rheological properties of polyethylenes. Polym. Eng. Sci. 2000, 40, 2279–2287. [Google Scholar] [CrossRef]
- Kanso, M.A.; Giacomin, A. Van Gurp–Palmen relations for long-chain branching from general rigid bead-rod theory. J. Phys. Fluids 2020, 32, 033101-15. [Google Scholar] [CrossRef]
- Hassager, O. Kinetic theory and rheology of bead—Rod models for macromolecular solutions. II. Linear unsteady flow properties. J. Chem. Phys. 1974, 60, 4001–4008. [Google Scholar] [CrossRef]
- Olabishi, O.; Robeson, L.M.; Shaw, M.Y. Polymer-Polymer Miscibility; Academic Press: New York, NY, USA, 1979; pp. 105–133. [Google Scholar]
- Mandelkern, L. Crystallisation of Polymers: Volume 2. Kinetics and Mechanisms; Cambridge University Press: Cambridge, UK, 2004; pp. 67–96. [Google Scholar]
- Young, R.J.; Lovell, P.A. Introduction to Polymers; CRC Press: New York, NY, USA, 2011; pp. 168–186. [Google Scholar]
- Yamaguchi, M.; Abe, S. LLDPE/LDPE blends. I. Rheological, thermal, and mechanical properties. J. Appl. Polym. Sci. 1999, 74, 3153–3159. [Google Scholar] [CrossRef]
- Sweed, M. Co-Crystallization in Polyolefin Blends Studied by Various Crystallization Analysis Techniques. Master’s Thesis, University of Stellenbosch, Stellenbosch, South Africa, April 2006. [Google Scholar]
- Liu, T.; Mo, Z.; Wang, S.; Zhang, H. Nonisothermal melt and cold crystallization kinetics of poly (aryl ether ether ketone ketone). Polym. Eng. Sci. 1997, 37, 568–575. [Google Scholar] [CrossRef]
- Kissinger, H.E. Variation of peak temperature with heating rate in differential thermal analysis. J. Res. Natl. Bur. Stand. 1956, 57, 217–221. [Google Scholar] [CrossRef]
- Vyazovkin, S. Kissinger Method in Kinetics of Materials: Things to Beware and Be Aware of. Molecules 2020, 25, 2813. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, J.J.; Kim, J.; Kim, Y.C. Effect of starch content on the non-isothermal crystallization behavior of HDPE/silicate nanocomposites. J. Ind. Eng. Chem. 2010, 16, 406–410. [Google Scholar] [CrossRef]
- Jin, J.; Chen, S.J.; Zhang, J. Non-isothermal crystallization kinetics of partially miscible ethylene-vinyl acetate copolymer/low density polyethylene blends. Express Polym. Lett. 2010, 4, 141–152. [Google Scholar] [CrossRef]
- Abareshi, M.; Zebarjad, S.M.; Goharshadi, E.K. Non-isothermal crystallization kinetics of polyethylene–clay nanocomposites prepared by high-energy ball milling. Bull. Mater. Sci. 2004, 37, 1113–1121. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | MFR2 [g 10 min−1] | Density [kg m−3] | Mn [kg mol−1] | Mw [kg mol−1] | MWD = Mw/Mn [-] | SHI(1/100) [-] |
|---|---|---|---|---|---|---|
| LDPE | 2.0 | 923 | 17 | 108 | 6.3 | 27.4 |
| mLLDPE-1 | 1.5 | 918 | 25 | 104 | 4.2 | 2.0 |
| mLLDPE-2 | 1.0 | 918 | 32 | 113 | 3.5 | 1.5 |
| mLLDPE-3 | 1.0 | 920 | 24 | 114 | 4.7 | 7.4 |
| mLLDPE-4 | 1.0 | 918 | 26 | 118 | 4.6 | 2.7 |
| mLLDPE-5 | 1.3 | 927 | 28 | 107 | 3.9 | 1.5 |
| mLLDPE-6 | 1.3 | 927 | 24 | 106 | 4.4 | 1.8 |
| mLLDPE-7 | 1.0 | 918 | 25 | 105 | 4.2 | 4.5 |
| mLLDPE-8 | 1.5 | 918 | 27 | 111 | 4.1 | 1.7 |
| ZN-LLDPE-1 | 0.9 | 919 | 23 | 119 | 5.1 | 3.7 |
| ZN-LLDPE-2 | 0.2 | 931 | 11 | 192 | 18.2 | 10.2 |
| Sample | Comonomer Type [-] | Total Comonomer Content [mol.%] | SCB/1000C [-] |
|---|---|---|---|
| mLLDPE-1 | C4 + C6 | 2.8 | 14.4 |
| mLLDPE-2 | C6 | 2.1 | 10.8 |
| mLLDPE-3 | C6 | 1.1 | 6.1 |
| mLLDPE-4 | C4 | 3.5 | 18.1 |
| mLLDPE-5 | C6 | 1.1 | 6.2 |
| mLLDPE-6 | C4 + C6 | 1.4 | 7.8 |
| mLLDPE-7 | C8 | 2.5 | 14.1 |
| mLLDPE-8 | C4 | 4.2 | 21.7 |
| ZN-LLDPE-1 | C8 | 2.1 | 10.8 |
| ZN-LLDPE-2 | C4 + C6 | 2.0 | 11.9 |
| Sample | Ea [kJ mol−1] |
|---|---|
| mLLDPE-1 | 36.4 |
| mLLDPE-1 + LD30 | 40.2 |
| mLLDPE-2 | 35.0 |
| mLLDPE-2 + LD30 | 37.3 |
| mLLDPE-3 | 39.8 |
| mLLDPE-3 + LD30 | 43.2 |
| Sample | 20 °C min−1 | 2.5 °C min−1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Ti,c [°C] | Tc [°C] | Tf,c [°C] | ΔHc [J g−1] | Ti,c [°C] | Tc [°C] | Tf,c [°C] | ΔHc [J g−1] | |
| LDPE | 105.6 | 100.9 | 27.3 | 130.6 | 108.3 | 104.0 | 10.9 | 159.5 |
| mLLDPE-1 | 112.5 | 106.6 | 19.4 | 129.8 | 116.7 | 111.9 | 29.0 | 155.4 |
| mLLDPE-1 + LD20 | 113.0 | 108.9 | 21.8 | 130.6 | 116.5 | 112.6 | 38.1 | 156.2 |
| mLLDPE-2 | 115.2 | 107.1 | 39.5 | 118.6 | 118.1 | 112.3 | 29.9 | 138.1 |
| mLLDPE-2 + LD20 | 114.9 | 107.1 | 37.7 | 121.3 | 118.7 | 112.2 | 35.9 | 151.8 |
| mLLDPE-3 | 107.7 | 103.2 | 42.9 | 115.9 | 110.5 | 107.0 | 19.7 | 137.0 |
| mLLDPE-3 + LD20 | 108.3 | 103.2 | 28.1 | 133.5 | 110.6 | 107.3 | 19.5 | 148.4 |
| mLLDPE-4 | 111.9 | 106.4 | 28.6 | 120.9 | 115.8 | 111.5 | 27.6 | 135.6 |
| mLLDPE-4 + LD20 | 112.2 | 108.0 | 24.4 | 121.0 | 115.9 | 111.7 | 11.0 | 158.2 |
| mLLDPE-5 | 116.3 | 110.9 | 39.9 | 121.7 | 119.0 | 115.1 | 40.3 | 135.9 |
| mLLDPE-5 + LD20 | 116.4 | 111.1 | 36.5 | 139.5 | 119.6 | 114.9 | 40.7 | 157.4 |
| mLLDPE-6 | 113.7 | 109.2 | 31.5 | 142.6 | 116.4 | 113.3 | 27.7 | 167.6 |
| mLLDPE-6 + LD20 | 113.4 | 109.6 | 33.6 | 143.9 | 116.5 | 113.2 | 35.3 | 163.4 |
| mLLDPE-7 | 111.4 | 105.7 | 39.9 | 110.6 | 115.4 | 110.8 | 15.4 | 129.2 |
| mLLDPE-7 + LD20 | 112.4 | 107.9 | 30.5 | 117.0 | 115.2 | 111.9 | 34.7 | 149.0 |
| mLLDPE-8 | 114.2 | 107.4 | 23.1 | 125.5 | 116.5 | 112.5 | 21.2 | 139.6 |
| mLLDPE-8 + LD20 | 113.5 | 109.3 | 24.9 | 128.8 | 116.3 | 112.8 | 24.4 | 149.6 |
| ZN-LLDPE-1 | 111.5 | 105.3 | 28.3 | 126.2 | 117.2 | 110.7 | 25.7 | 144.8 |
| ZN-LLDPE-1 + LD30 | 112.7 | 107.7 | 22.6 | 131.1 | 115.7 | 112.1 | 40.4 | 144.3 |
| ZN-LLDPE-2 | 119.1 | 114.7 | 33.3 | 158.4 | 121.7 | 117.7 | 25.4 | 184.3 |
| ZN-LLDPE-2 + LD30 | 118.4 | 114.6 | 39.7 | 142.6 | 120.9 | 117.6 | 34.1 | 173.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galgali, G.; Kaliappan, S.K.; Pandit, T. Influence of Ethylene-1-Alkene Copolymers Microstructure on Thermo-Rheological Behavior of Model Blends for Enhanced Recycling. Macromol 2022, 2, 168-183. https://doi.org/10.3390/macromol2020011
Galgali G, Kaliappan SK, Pandit T. Influence of Ethylene-1-Alkene Copolymers Microstructure on Thermo-Rheological Behavior of Model Blends for Enhanced Recycling. Macromol. 2022; 2(2):168-183. https://doi.org/10.3390/macromol2020011
Chicago/Turabian StyleGalgali, Girish, Senthil Kumar Kaliappan, and Tej Pandit. 2022. "Influence of Ethylene-1-Alkene Copolymers Microstructure on Thermo-Rheological Behavior of Model Blends for Enhanced Recycling" Macromol 2, no. 2: 168-183. https://doi.org/10.3390/macromol2020011
APA StyleGalgali, G., Kaliappan, S. K., & Pandit, T. (2022). Influence of Ethylene-1-Alkene Copolymers Microstructure on Thermo-Rheological Behavior of Model Blends for Enhanced Recycling. Macromol, 2(2), 168-183. https://doi.org/10.3390/macromol2020011