
Enzymatic Polymerization as a Green Approach to Synthesizing Bio-Based Polyesters



Abstract
:1. Introduction
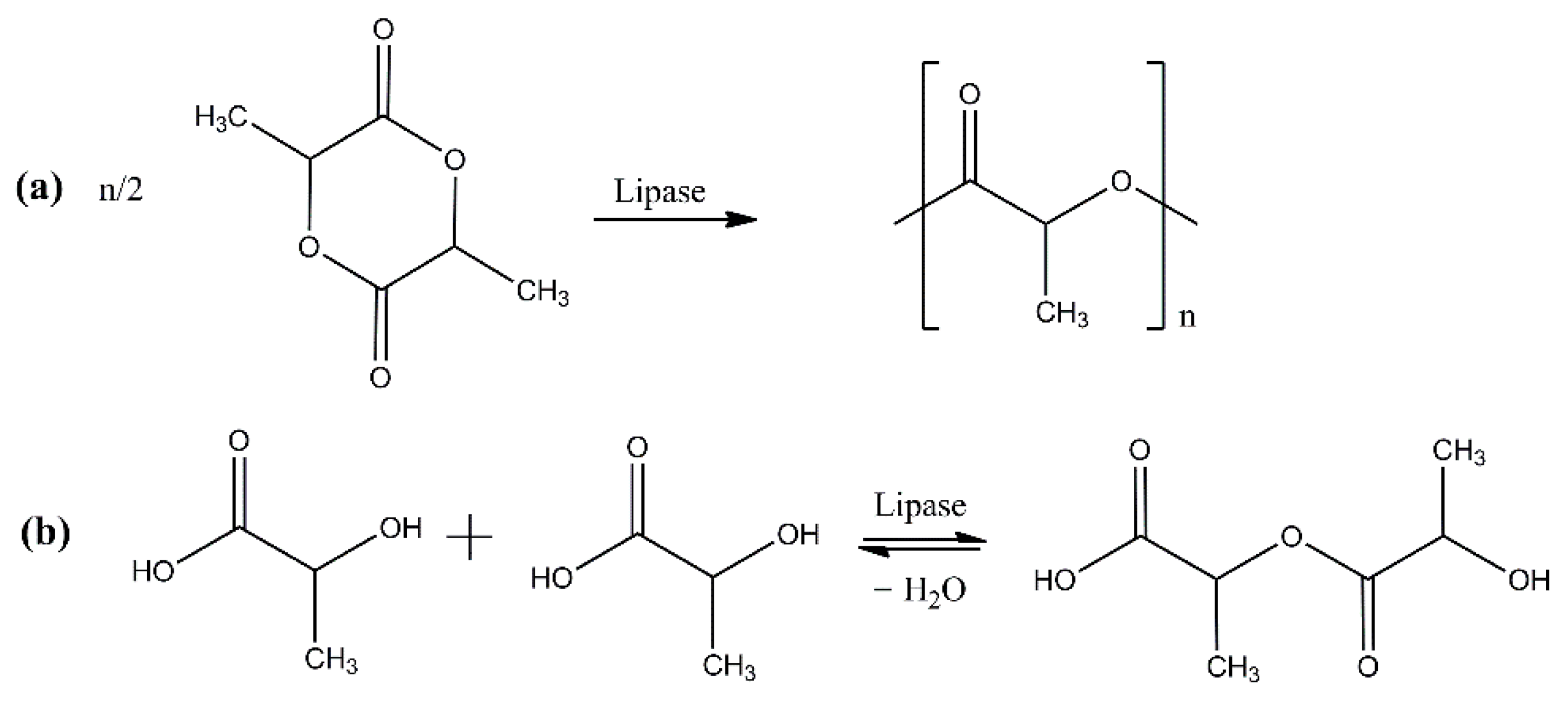
2. Enzymatic Polymerization of Aliphatic Polyesters
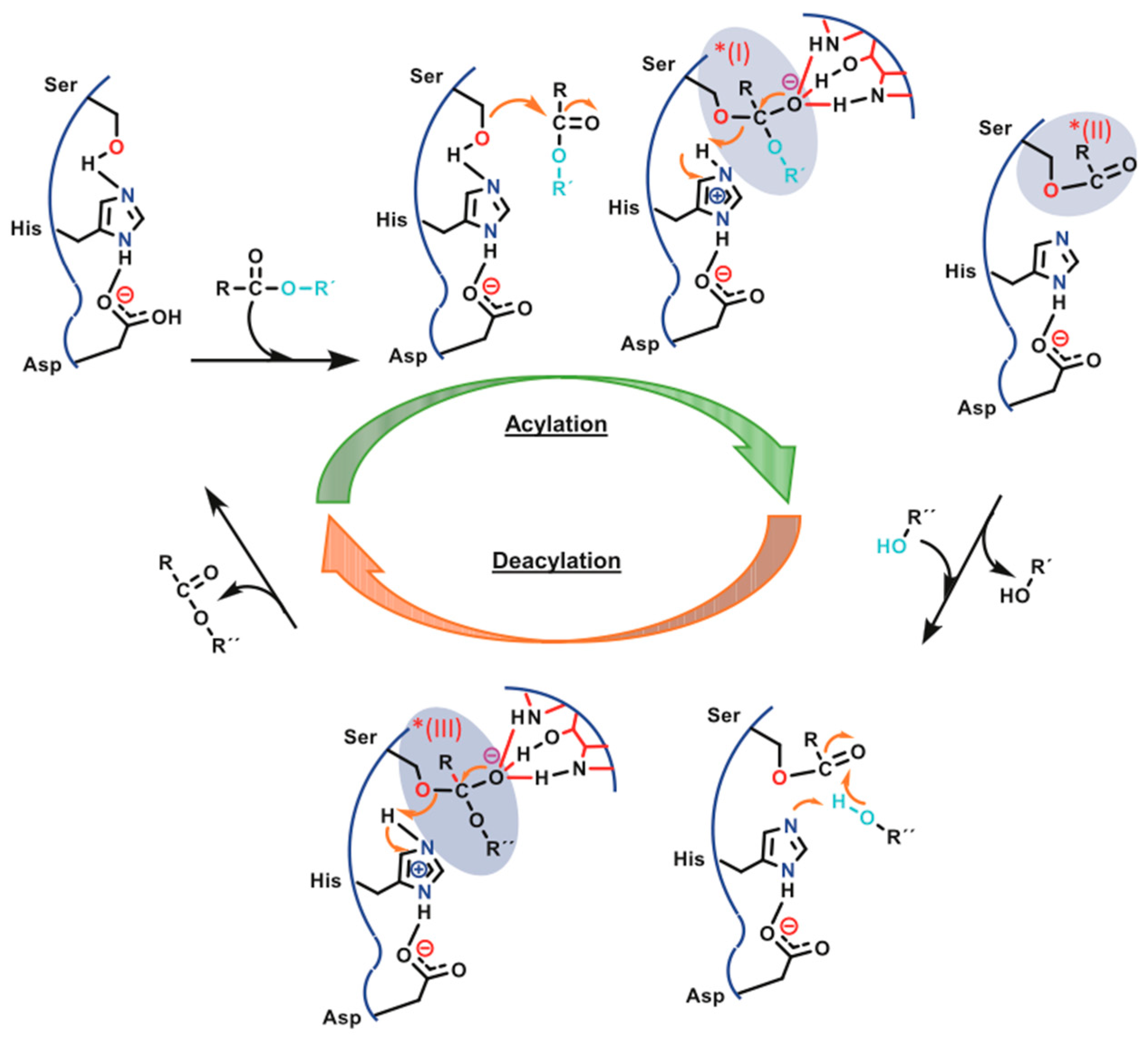
2.1. Type and Characteristics of the Used Enzyme for PLA Enzymatic Polymerization
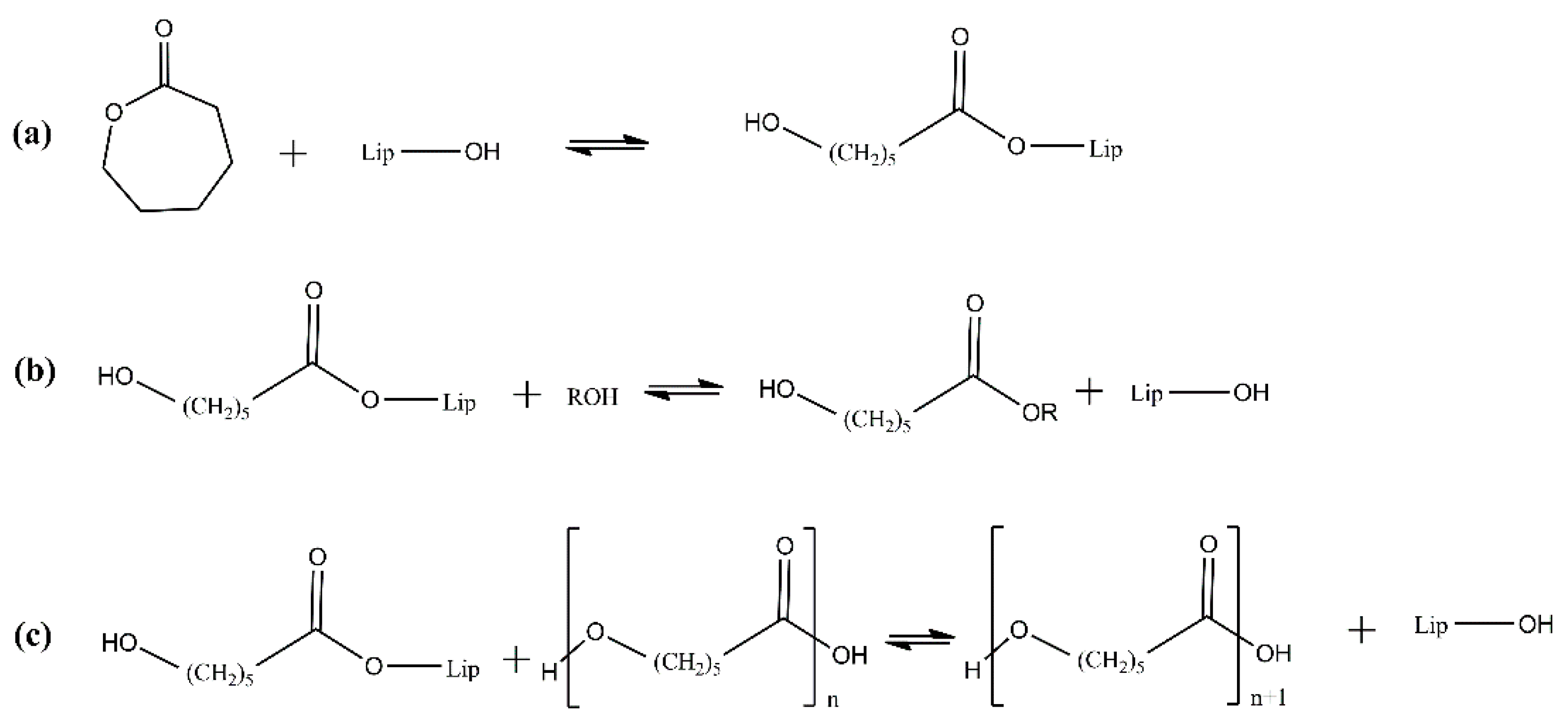
2.2. PLA Enzymatic Polymerization Key Process Parameters
2.3. PLA Enzymatic Polymerization Monitoring Variables
2.4. Type and Characteristics of the Used Enzyme for PBS Enzymatic Polymerization
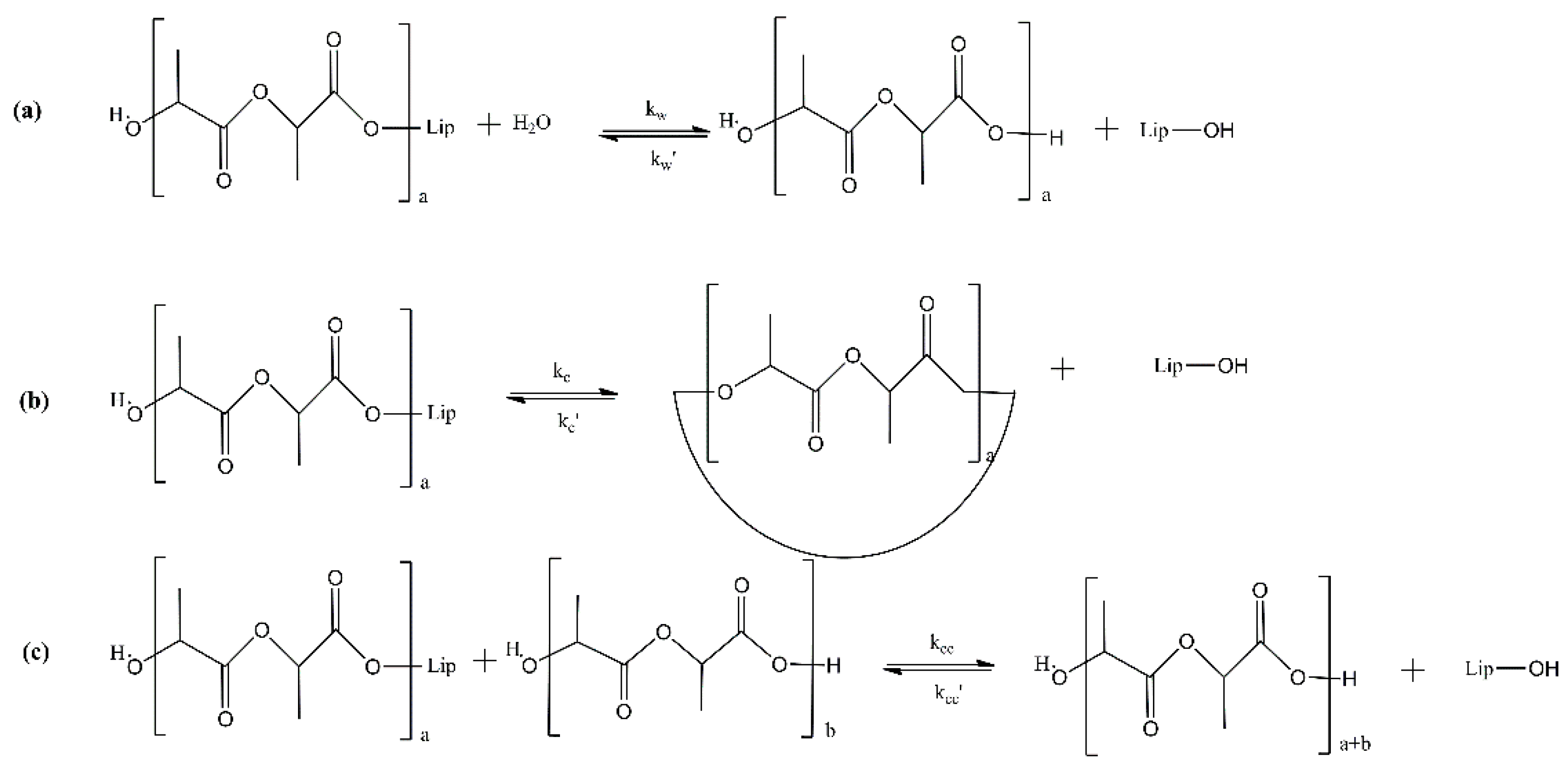
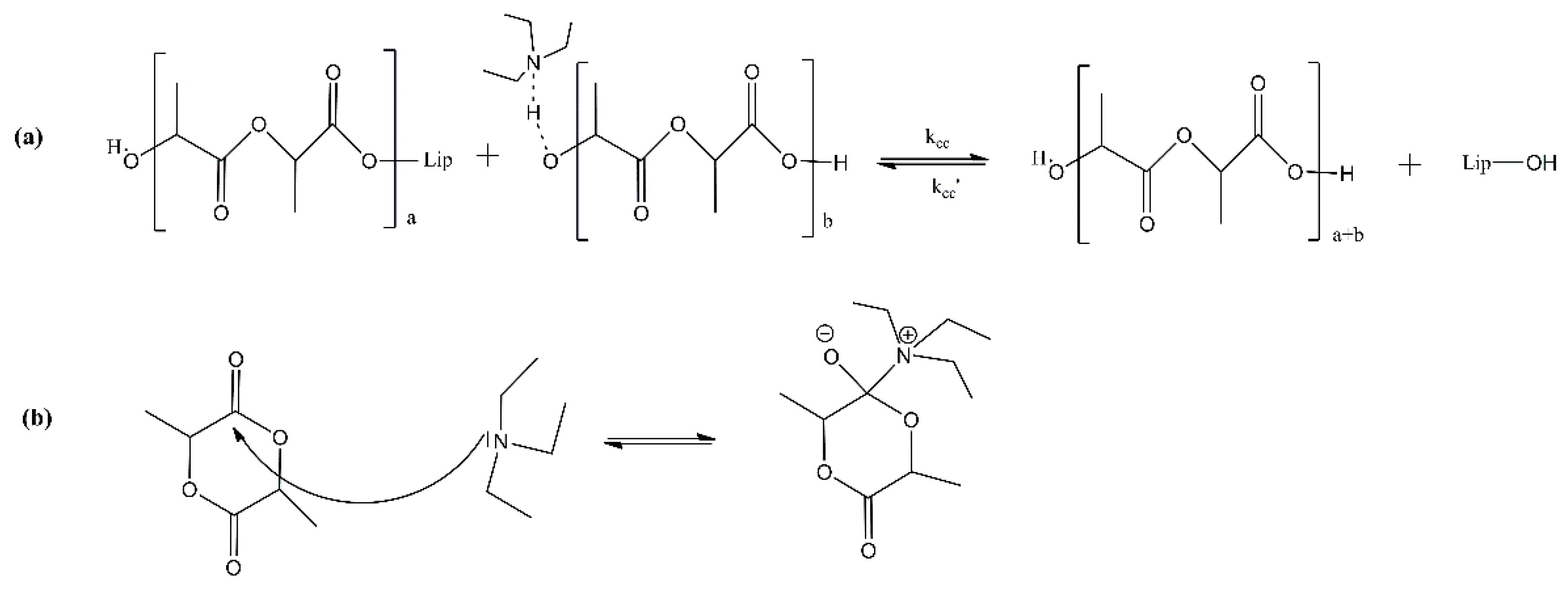
2.5. PBS Enzymatic Polymerization Key Process Parameters
2.6. PBS Enzymatic Polymerization Monitoring Variables
3. Enzymatic Polymerization of Alipharomatic Polyesters
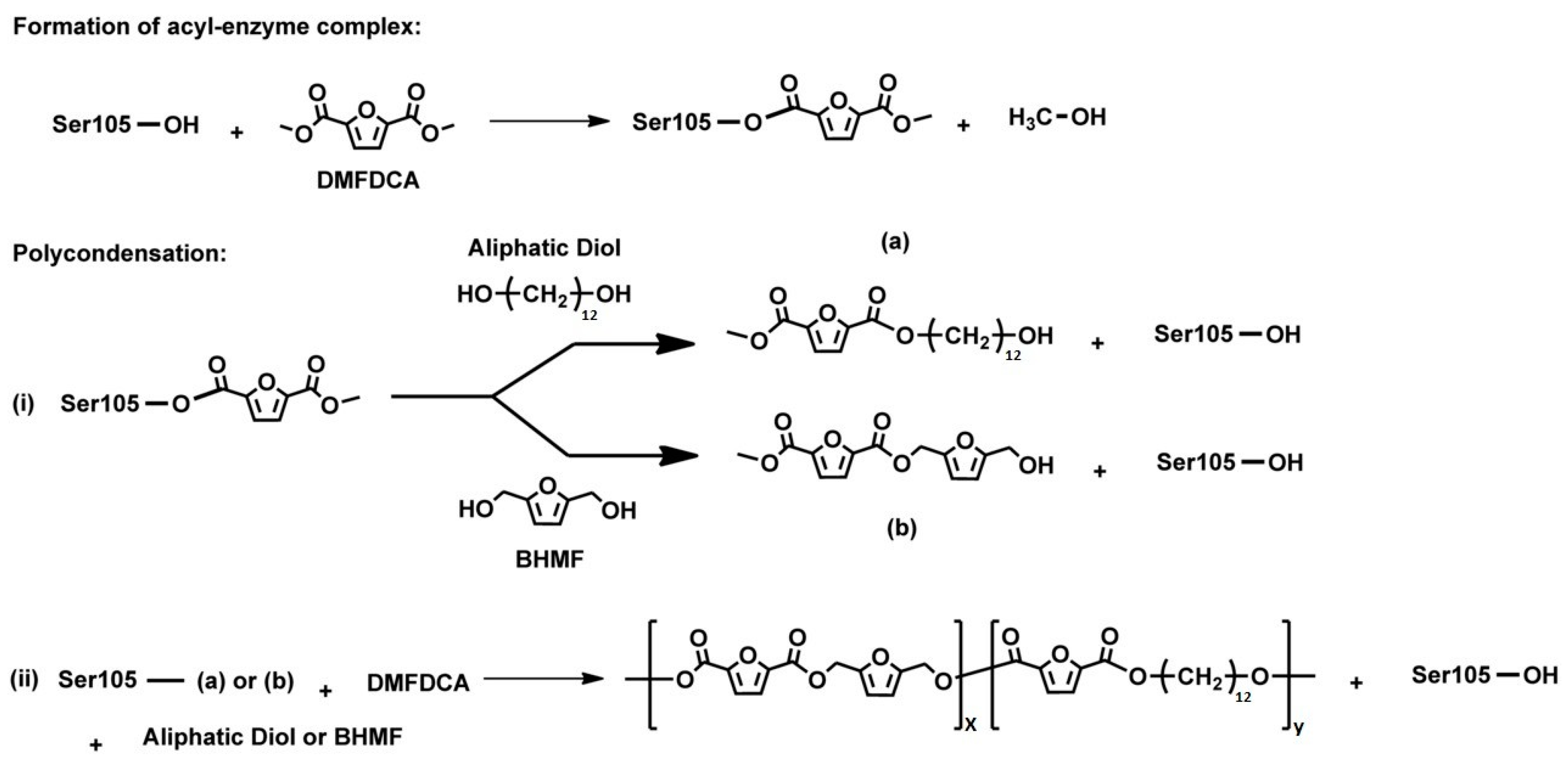
3.1. Type and Characteristics of the Used Enzyme for Furan-Based Polyesters Enzymatic Polymerization
3.2. Furan-Based Polyesters Enzymatic Polymerization Key Process Parameters
3.3. Furan-Based Polyesters Enzymatic Polymerization Monitoring Variables
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Glossary
| BDO | 1,4-butanediol |
| BHMF | 2,5- bis(hydroxymethyl)furan |
| [BMIM][PF6] | 1-butyl-3-methylimidazoliumhexa-fluorophosphate |
| CALB | Candida antarctica lipase B |
| [C4MIM][NTf2] | 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)amide |
| [C4MIM][PF6] | 1-butyl-3-methylimidazolium hexafluorophosphate |
| CNS | central nervous system |
| DES | diethyl succinate |
| DLAOH | dimer linoleic diol |
| DMA | N,N-Dimethylacetamide |
| DMFDCA | dimethyl 2,5-furandicarboxylate |
| DMS | dimethyl succinate |
| number-average degree of polymerization | |
| weight-average degree of polymerization | |
| EG | ethylene glycol |
| [EMIM][BF4] | 1-ethyl-3-methylimidazolium tetrafluoroborate |
| eROP | enzymatic ring opening polymerization |
| FDCA | 2,5-furandicarboxylic acid |
| GI | gastrointestinal |
| 1,6-HDO | 1,6 hexanediol |
| [HMIM][PF6] | 1-hexyl-3-methylimidazolium hexafluorophosphate |
| ILs | ionic liquids |
| LBC | lipase from Burkholderia cepacia |
| LCR | lipase from Candida rugosa |
| number-average molecular weight | |
| weight-average molecular weight | |
| MW | molecular weight |
| N435 | novozym 435 |
| 1,8-ODA | 1,8-octanediamine |
| PBF | poly(butylene 2,5-furandicarboxylate) |
| PBS | poly(butylene succinate) |
| PBT | poly(butylene terephthalate) |
| PCL | poly(ε-caprolactone) |
| PDLA | Poly(D-lactic acid) |
| PEAF12 | poly(dodecamethylene furanoate-co-dodecamethylene furanamide) |
| PEF | Poly(ethylene furanoate |
| PET | poly(ethylene terephthalate) |
| P(FMF-co-OF) | poly(2,5-furandimethylene furanoate-co-octamethylene furanoate) |
| PLA | poly(lactic acid) |
| PLLA | poly(L-lactic acid) |
| PPL | lipase from porcine pancreas |
| PS | polystyrene |
| PVL | poly(δ-valerolactone) |
| R134a | 1,1,1,2-tetrafluoroethane |
| ROP | ring-opening polymerization |
| scCO2 | supercritical carbon dioxide |
| SCFs | supercritical fluids |
| Tb | boiling temperature |
| TEA | triethyl amine |
| Tg | glass transition temperature |
| THF | tetrahydrofuran |
| Tm | melting temperature |
References
- Nakajima, H.; Dijkstra, P.; Loos, K. The Recent Developments in Biobased Polymers toward General and Engineering Applications: Polymers That Are Upgraded from Biodegradable Polymers, Analogous to Petroleum-Derived Polymers, and Newly Developed. Polymers 2017, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.P.; O’Connor, K.; Seeram, R. Current Progress on Bio-Based Polymers and Their Future Trends. Prog. Biomater. 2013, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilyas, R.A.; Sapuan, S.M.; Kadier, A.; Kalil, M.S.; Ibrahim, R.; Atikah, M.S.N.; Nurazzi, N.M.; Nazrin, A.; Lee, C.H.; Faiz Norrrahim, M.N.; et al. Properties and Characterization of PLA, PHA, and Other Types of Biopolymer Composites; Elsevier Inc.: Amsterdam, The Netherlands, 2020; ISBN 9780128196618. [Google Scholar]
- Savioli Lopes, M.; Jardini, A.L.; Maciel Filho, R. Poly(Lactic Acid) Production for Tissue Engineering Applications. Procedia Eng. 2012, 42, 1402–1413. [Google Scholar] [CrossRef] [Green Version]
- Rehman, T.U.; Shah, L.A.; Khan, M.; Irfan, M.; Khattak, N.S. Zwitterionic Superabsorbent Polymer Hydrogels for Efficient and Selective Removal of Organic Dyes. RSC Adv. 2019, 9, 18565–18577. [Google Scholar] [CrossRef] [Green Version]
- Aziz, T.; Fan, H.; Khan, F.U.; Ullah, R.; Haq, F.; Iqbal, M.; Ullah, A. Synthesis of Carboxymethyl Starch-Bio-Based Epoxy Resin and Their Impact on Mechanical Properties. Z. Phys. Chem. 2020, 234, 1759–1769. [Google Scholar] [CrossRef]
- Li, C.; Fan, H.; Aziz, T.; Bittencourt, C.; Wu, L.; Wang, D.Y.; Dubois, P. Biobased Epoxy Resin with Low Electrical Permissivity and Flame Retardancy: From Environmental Friendly High-Throughput Synthesis to Properties. ACS Sustain. Chem. Eng. 2018, 6, 8856–8867. [Google Scholar] [CrossRef]
- Song, J.H.; Murphy, R.J.; Narayan, R.; Davies, G.B.H. Biodegradable and Compostable Alternatives to Conventional Plastics. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2127–2139. [Google Scholar] [CrossRef]
- Luyt, A.S.; Malik, S.S. Can Biodegradable Plastics Solve Plastic Solid Waste Accumulation? In Plastics to Energy; Al-Salem, S.M., Ed.; Plastics Design Library; William Andrew Publishing: Norwich, NY, USA, 2019; pp. 403–423. ISBN 978-0-12-813140-4. [Google Scholar]
- Rudnik, E.; Briassoulis, D. Comparative Biodegradation in Soil Behaviour of Two Biodegradable Polymers Based on Renewable Resources. J. Polym. Environ. 2011, 19, 18–39. [Google Scholar] [CrossRef]
- Gallet, G.; Lempiäinen, R.; Karlsson, S. Characterization by Solid Phase Microextraction-Gas Chromatography-Mass Spectrometry of Matrix Changes of Poly(L-Lactide) Exposed to Outdoor Soil Environment. Polym. Degrad. Stab. 2000, 71, 147–151. [Google Scholar] [CrossRef]
- Narancic, T.; Verstichel, S.; Reddy Chaganti, S.; Morales-Gamez, L.; Kenny, S.T.; de Wilde, B.; Babu Padamati, R.; O’Connor, K.E. Biodegradable Plastic Blends Create New Possibilities for End-of-Life Management of Plastics but They Are Not a Panacea for Plastic Pollution. Environ. Sci. Technol. 2018, 52, 10441–10452. [Google Scholar] [CrossRef]
- Hans, M.; Keul, H.; Moeller, M. Ring-Opening Polymerization of DD-Lactide Catalyzed by Novozyme 435. Macromol. Biosci. 2009, 9, 239–247. [Google Scholar] [CrossRef]
- García-Arrazola, R.; López-Guerrero, D.A.; Gimeno, M.; Bárzana, E. Lipase-Catalyzed Synthesis of Poly-l-Lactide Using Supercritical Carbon Dioxide. Polym. Adv. Technol. 2008, 19, 1396–1400. [Google Scholar] [CrossRef]
- Auras, R.; Harte, B.; Selke, S. An Overview of Polylactides as Packaging Materials. Macromol. Biosci. 2004, 4, 835–864. [Google Scholar] [CrossRef]
- Li, G.; Zhao, M.; Xu, F.; Yang, B.; Li, X.; Meng, X.; Teng, L.; Sun, F.; Li, Y. Synthesis and Biological Application of Polylactic Acid. Molecules 2020, 25, 5023. [Google Scholar] [CrossRef]
- Montané, X.; Montornes, J.M.; Nogalska, A.; Olkiewicz, M.; Giamberini, M.; Garcia-Valls, R.; Badia-Fabregat, M.; Jubany, I.; Tylkowski, B. Synthesis and Synthetic Mechanism of Polylactic Acid. Phys. Sci. Rev. 2020, 5, 20190102. [Google Scholar] [CrossRef]
- Bahramian, B.; Ma, Y.; Rohanizadeh, R.; Chrzanowski, W.; Dehghani, F. A New Solution for Removing Metal-Based Catalyst Residues from a Biodegradable Polymer. Green Chem. 2016, 18, 3740–3748. [Google Scholar] [CrossRef]
- Masutani, K.; Kimura, Y. PLA Synthesis and Polymerization. In Poly(Lactic Acid) Science and Technology: Processing, Properties, Additives and Applications; Jiménez, A., Peltzer, M., Ruseckaite, R., Eds.; Royal Society of Chemistry: Cambridge, UK, 2014; pp. 3–36. ISBN 9781782624806. [Google Scholar]
- Hege, C.S.; Schiller, S.M. Non-Toxic Catalysts for Ring-Opening Polymerizations of Biodegradable Polymers at Room Temperature for Biohybrid Materials. Green Chem. 2014, 16, 1410–1416. [Google Scholar] [CrossRef]
- Ojansivu, M.; Johansson, L.; Vanhatupa, S.; Tamminen, I.; Hannula, M.; Hyttinen, J.; Kellomäki, M.; Miettinen, S. Knitted 3D Scaffolds of Polybutylene Succinate Support Human Mesenchymal Stem Cell Growth and Osteogenesis. Stem Cells Int. 2018, 2018, 5928935. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chang, J.; Cao, A.; Wang, J. In Vitro Evaluation of Biodegradable Poly(Butylene Succinate) as a Novel Biomaterial. Macromol. Biosci. 2005, 5, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Rafiqah, S.A.; Khalina, A.; Harmaen, A.S.; Tawakkal, I.A.; Zaman, K.; Asim, M.; Nurrazi, M.N.; Lee, C.H. A Review on Properties and Application of Bio-based Poly(Butylene Succinate). Polymers 2021, 13, 1436. [Google Scholar] [CrossRef] [PubMed]
- Łopusiewicz, Ł.; Zdanowicz, M.; Macieja, S.; Kowalczyk, K.; Bartkowiak, A. Development and Characterization of Bioactive Poly(Butylene-Succinate) Films Modified with Quercetin for Food Packaging Applications. Polymers 2021, 13, 1798. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, N.; Mazlan, M.M.; Tawakkal, I.S.M.A.; Talib, R.A.; Kian, L.K.; Jawaid, M. Characterization of Active Polybutylene Succinate Films Filled Essential Oils for Food Packaging Application. J. Polym. Environ. 2021, 1–12. [Google Scholar] [CrossRef]
- Jacquel, N.; Freyermouth, F.; Fenouillot, F.; Rousseau, A.; Pascault, J.P.; Fuertes, P.; Saint-Loup, R. Synthesis and Properties of Poly(Butylene Succinate): Efficiency of Different Transesterification Catalysts. J. Polym. Sci. Part A Polym. Chem. 2011, 48, 5301–5312. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Achilias, D.S. Synthesis of Poly(Alkylene Succinate) Biodegradable Polyesters, Part II: Mathematical Modelling of the Polycondensation Reaction. Polymer 2008, 49, 3677–3685. [Google Scholar] [CrossRef]
- Papaspyrides, C.D.; Vouyiouka, S.; Georgousopoulou, I.N.; Marinkovic, S.; Estrine, B.; Joly, C.; Dole, P. Feasibility of Solid-State Postpolymerization on Fossil- and Bio-Based Poly(Butylene Succinate) Including Polymer Upcycling Routes. Ind. Eng. Chem. Res. 2016, 55, 5832–5842. [Google Scholar] [CrossRef]
- Pellis, A.; Malinconico, M.; Guarneri, A.; Gardossi, L. Renewable Polymers and Plastics: Performance beyond the Green. New Biotechnol. 2021, 60, 146–158. [Google Scholar] [CrossRef]
- Jiang, Y.; Loos, K. Enzymatic Synthesis of Biobased Polyesters and Polyamides. Polymers 2016, 8, 243. [Google Scholar] [CrossRef] [Green Version]
- Loos, K.; Zhang, R.; Pereira, I.; Agostinho, B.; Hu, H.; Maniar, D. A Perspective on PEF Synthesis, Properties, and End-Life. Front. Chem. 2020, 8, 585. [Google Scholar] [CrossRef]
- Douka, A.; Vouyiouka, S.; Papaspyridi, L.M.; Papaspyrides, C.D. A Review on Enzymatic Polymerization to Produce Polycondensation Polymers: The Case of Aliphatic Polyesters, Polyamides and Polyesteramides. Prog. Polym. Sci. 2018, 79, 1–25. [Google Scholar] [CrossRef]
- Hu, Y.; Daoud, W.A.; Cheuk, K.K.L.; Lin, C.S.K. Newly Developed Techniques on Polycondensation, Ring-Opening Polymerization and Polymer Modification: Focus on Poly(Lactic Acid). Materials 2016, 9, 133. [Google Scholar] [CrossRef] [Green Version]
- Rahmayetty; Whulanza, Y.; Sukirno; Rahman, S.F.; Suyono, E.A.; Yohda, M.; Gozan, M. Use of Candida Rugosa Lipase as a Biocatalyst for L-Lactide Ring-Opening Polymerization and Polylactic Acid Production. Biocatal. Agric. Biotechnol. 2018, 16, 683–691. [Google Scholar] [CrossRef]
- Takwa, M.; Larsen, M.W.; Hult, K.; Martinelle, M. Rational Redesign of Candida Antarctica Lipase B for the Ring Opening Polymerization of d,d-Lactide. Chem. Commun. 2011, 47, 7392–7394. [Google Scholar] [CrossRef] [PubMed]
- Målberg, S.; Finne-Wistrand, A.; Albertsson, A.C. The Environmental Influence in Enzymatic Polymerization of Aliphatic Polyesters in Bulk and Aqueous Mini-Emulsion. Polymer 2010, 51, 5318–5322. [Google Scholar] [CrossRef]
- Düşkünkorur, H.Ö.; Bégué, A.; Pollet, E.; Phalip, V.; Güvenilir, Y.; Avérous, L. Enzymatic Ring-Opening (Co)Polymerization of Lactide Stereoisomers Catalyzed by Lipases. Toward the in Situ Synthesis of Organic/Inorganic Nanohybrids. J. Mol. Catal. B Enzym. 2015, 115, 20–28. [Google Scholar] [CrossRef]
- Duchiron, S.W.; Pollet, E.; Givry, S.; Avérous, L. Mixed Systems to Assist Enzymatic Ring Opening Polymerization of Lactide Stereoisomers. RSC Adv. 2015, 5, 84627–84635. [Google Scholar] [CrossRef]
- Guzmán-Lagunes, F.; López-Luna, A.; Gimeno, M.; Bárzana, E. Enzymatic Synthesis of Poly-l-Lactide in Supercritical R134a. J. Supercrit. Fluids 2012, 72, 186–190. [Google Scholar] [CrossRef]
- Chanfreau, S.; Mena, M.; Porras-Domínguez, J.R.; Ramírez-Gilly, M.; Gimeno, M.; Roquero, P.; Tecante, A.; Bárzana, E. Enzymatic Synthesis of Poly-L-Lactide and Poly-L-Lactide-Co-Glycolide in an Ionic Liquid. Bioprocess Biosyst. Eng. 2010, 33, 629–638. [Google Scholar] [CrossRef]
- Zhao, H.; Nathaniel, G.A.; Merenini, P.C. Enzymatic Ring-Opening Polymerization (ROP) of Lactides and Lactone in Ionic Liquids and Organic Solvents: Digging the Controlling Factors. RSC Adv. 2017, 7, 48639–48648. [Google Scholar] [CrossRef] [Green Version]
- Omay, D.; Guvenilir, Y. Synthesis and Characterization of Poly(d,l-Lactic Acid) via Enzymatic Ring Opening Polymerization by Using Free and Immobilized Lipase. Biocatal. Biotransform. 2013, 31, 132–140. [Google Scholar] [CrossRef]
- Mena, M.; Shirai, K.; Tecante, A.; Bárzana, E.; Gimeno, M. Enzymatic Syntheses of Linear and Hyperbranched Poly-L-Lactide Using Compressed R134a–Ionic Liquid Media. J. Supercrit. Fluids 2015, 103, 77–82. [Google Scholar] [CrossRef]
- Gkountela, C.; Rigopoulou, M.; Barampouti, E.M.; Vouyiouka, S. Enzymatic Prepolymerization Combined with Bulk Post-Polymerization towards the Production of Bio-Based Polyesters: The Case of Poly(Butylene Succinate). Eur. Polym. J. 2021, 143, 110197. [Google Scholar] [CrossRef]
- Lassalle, V.; Ferreira, M. Lipase-Catalyzed Synthesis of Polylactic Acid: An Overview of the Experimental Aspects. J. Chem. Technol. Biotechnol. 2008, 83, 1493–1502. [Google Scholar] [CrossRef]
- Rahmayetty; Barleany, D.R.; Suhendi, E.; Prasetya, B.; Andiyani, T. Polylactic Acid Synthesis via Direct Polycondensation Method Using Candida Rugosa Lipase Catalyst. World Chem. Eng. J. 2017, 1, 70–74. [Google Scholar]
- Panyachanakul, T.; Lomthong, T.; Lorliam, W.; Prajanbarn, J.; Tokuyama, S.; Kitpreechavanich, V.; Krajangsang, S. New Insight into Thermo-Solvent Tolerant Lipase Produced by Streptomyces Sp. A3301 for Re-Polymerization of Poly(DL-Lactic Acid). Polymer 2020, 204, 122812. [Google Scholar] [CrossRef]
- Yeniad, B.; Naik, H.; Heise, A. Lipases in Polymer Chemistry. Adv. Biochem. Eng. Biotechnol. 2011, 125, 69–95. [Google Scholar] [CrossRef]
- Nikulin, M.; Švedas, V. Prospects of Using Biocatalysis for the Synthesis and Modification of Polymers. Molecules 2021, 26, 2750. [Google Scholar] [CrossRef]
- Pellis, A.; Herrero Acero, E.; Ferrario, V.; Ribitsch, D.; Guebitz, G.M.; Gardossi, L. The Closure of the Cycle: Enzymatic Synthesis and Functionalization of Bio-Based Polyesters. Trends Biotechnol. 2016, 34, 316–328. [Google Scholar] [CrossRef]
- Filho, D.G.; Silva, A.G.; Guidini, C.Z. Lipases: Sources, Immobilization Methods, and Industrial Applications. Appl. Microbiol. Biotechnol. 2019, 103, 7399–7423. [Google Scholar] [CrossRef]
- Mokhtar, N.F.; Noor, R.; Raja, Z.; Rahman, A.; Dina, N.; Noor, M. The Immobilization of Lipases on Porous Support by Adsorption and Hydrophobic Interaction Method. Catalysts 2020, 10, 744. [Google Scholar] [CrossRef]
- Noel, M.; Combes, D. Effects of Temperature and Pressure on Rhizomucor Miehei Lipase Stability. J. Biotechnol. 2003, 102, 23–32. [Google Scholar] [CrossRef]
- Kundys, A.; Białecka-Florjańczyk, E.; Fabiszewska, A.; Małajowicz, J. Candida Antarctica Lipase B as Catalyst for Cyclic Esters Synthesis, Their Polymerization and Degradation of Aliphatic Polyesters. J. Polym. Environ. 2018, 26, 396–407. [Google Scholar] [CrossRef]
- Loeker, F.C.; Duxbury, C.J.; Kumar, R.; Gao, W.; Gross, R.A.; Howdle, S.M. Enzyme-Catalyzed Ring-Opening Polymerization of ε-Caprolactone in Supercritical Carbon Dioxide. Macromolecules 2004, 37, 2450–2453. [Google Scholar] [CrossRef]
- Poojari, Y.; Beemat, J.S.; Clarson, S.J. Enzymatic Synthesis of Poly(ε-Caprolactone): Thermal Properties, Recovery, and Reuse of Lipase B from Candida Antarctica Immobilized on Macroporous Acrylic Resin Particles. Polym. Bull. 2013, 70, 1543–1552. [Google Scholar] [CrossRef]
- Adhami, W.; Bakkour, Y.; Rolando, C. Polylactones Synthesis by Enzymatic Ring Opening Polymerization in Flow. Polymer 2021, 230, 124040. [Google Scholar] [CrossRef]
- Luna, C.; Luna, D.; Bautista, F.M.; Estevez, R.; Calero, J.; Posadillo, A.; Romero, A.A.; Sancho, E.D. Application of Enzymatic Extracts from a CALB Standard Strain as Biocatalyst within the Context of Conventional Biodiesel Production Optimization. Molecules 2017, 22, 2025. [Google Scholar] [CrossRef] [Green Version]
- Pedro, K.C.N.R.; Parreira, J.M.; Correia, I.N.; Henriques, C.A.; Langone, M.A.P. Enzymatic Biodiesel Synthesis from Acid Oil Using a Lipase Mixture. Quim. Nova 2018, 41, 284–291. [Google Scholar] [CrossRef]
- Yilmaz, E.; Soylak, M. Type of Green Solvents Used in Separation and Preconcentration Methods; Elsevier Inc.: Amsterdam, The Netherlands, 2020; ISBN 9780128185698. [Google Scholar]
- Pellis, A.; Byrne, F.P.; Sherwood, J.; Vastano, M.; Comerford, J.W.; Farmer, T.J. Safer Bio-Based Solvents to Replace Toluene and Tetrahydrofuran for the Biocatalyzed Synthesis of Polyesters. Green Chem. 2019, 21, 1686–1694. [Google Scholar] [CrossRef] [Green Version]
- Joshi, D.R.; Adhikari, N. An Overview on Common Organic Solvents and Their Toxicity. J. Pharm. Res. Int. 2019, 28, 1–18. [Google Scholar] [CrossRef]
- Gu, Y.; Jérôme, F. Bio-Based Solvents: An Emerging Generation of Fluids for the Design of Eco-Efficient Processes in Catalysis and Organic Chemistry. Chem. Soc. Rev. 2013, 42, 9550–9570. [Google Scholar] [CrossRef]
- Häckl, K.; Kunz, W. Some Aspects of Green Solvents. Comptes Rendus Chim. 2018, 21, 572–580. [Google Scholar] [CrossRef]
- Gardella, L.; Furaro, D.; Galimberti, M.; Monticelli, O. On the Development of a Facile Approach Based on the Use of Ionic Liquids: Preparation of PLLA (Sc-PLA)/High Surface Area Nano-Graphite Systems. Green Chem. 2015, 17, 4082–4088. [Google Scholar] [CrossRef]
- Knez; Markočič, E.; Leitgeb, M.; Primožič, M.; Knez Hrnčič, M.; Škerget, M. Industrial Applications of Supercritical Fluids: A Review. Energy 2014, 77, 235–243. [Google Scholar] [CrossRef]
- Yoshizawa-Fujita, M.; Saito, C.; Takeoka, Y.; Rikukawa, M. Lipase-Catalyzed Polymerization of L-Lactide in Ionic Liquids. Polym. Adv. Technol. 2008, 19, 1396–1400. [Google Scholar] [CrossRef]
- Kaar, J.L.; Jesionowski, A.M.; Berberich, J.A.; Moulton, R.; Russell, A.J. Impact of Ionic Liquid Physical Properties on Lipase Activity and Stability. J. Am. Chem. Soc. 2003, 125, 4125–4131. [Google Scholar] [CrossRef]
- Horváth, T.; Marossy, K.; Szabó, T.J. Ring-Opening Polymerization and Plasticization of Poly(L-Lactic)Acid by Adding of Glycerol-Dioleate. J. Therm. Anal. Calorim. 2021, 147, 2221–2227. [Google Scholar] [CrossRef]
- Vouyiouka, S.N.; Papaspyrides, C.D. Mechanistic Aspects of Solid-State Polycondensation. Polym. Sci. A Compr. Ref. 2012, 4, 857–874. [Google Scholar] [CrossRef]
- Wcislek, A.; Olalla, A.S.; McClain, A.; Piegat, A.; Sobolewski, P.; Puskas, J.; Fray, M. el Enzymatic Degradation of Poly(Butylene Succinate) Copolyesters Synthesized with the Use of Candida Antarctica Lipase, B. Polymers 2018, 10, 688. [Google Scholar] [CrossRef] [Green Version]
- Guinault, A.; Sollogoub, C.; Domenek, S.; Grandmontagne, A.; Ducruet, V. Influence of Crystallinity on Gas Barrier and Mechanical Properties of Pla Food Packaging Films. Int. J. Mater. Form. 2010, 3, 603–606. [Google Scholar] [CrossRef]
- Linko, Y.Y.; Lämsä, M.; Wu, X.; Uosukainen, E.; Seppälä, J.; Linko, P. Biodegradable Products by Lipase Biocatalysis. J. Biotechnol. 1998, 66, 41–50. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Fernandez-Lafuente, R. Lipase from Rhizomucor Miehei as an Industrial Biocatalyst in Chemical Process. J. Mol. Catal. B Enzym. 2010, 64, 1–22. [Google Scholar] [CrossRef]
- Pellis, A.; Comerford, J.W.; Maneffa, A.J.; Sipponen, M.H.; Clark, J.H.; Farmer, T.J. Elucidating Enzymatic Polymerisations: Chain-Length Selectivity of Candida Antarctica Lipase B towards Various Aliphatic Diols and Dicarboxylic Acid Diesters. Eur. Polym. J. 2018, 106, 79–84. [Google Scholar] [CrossRef]
- Uyama, H.; Inada, K.; Kobayashi, S. Lipase-Catalyzed Synthesis of Aliphatic Polyesters by Polycondensation of Dicarboxylic Acids and Glycols in Solvent-Free System. Polym. J. 2000, 32, 440–443. [Google Scholar] [CrossRef] [Green Version]
- El Fray, M.; Gradzik, B. Synteza Enzymatyczna Poli(Bursztynianu Butylenu) (Pbs) Katalizowana Lipazą B Ze Szczepu Candida Obiecujący Materiał Dla Enzymatic Synthesis of Poly(Butylene Succinate) (Pbs) Catalyzed By Lipase B From Candida Antarctica: A New Promising Material. Eng. Biomater. 2012, 115, 26–31. [Google Scholar]
- Kanelli, M.; Douka, A.; Vouyiouka, S.; Papaspyrides, C.D.; Topakas, E.; Papaspyridi, L.M.; Christakopoulos, P. Production of Biodegradable Polyesters via Enzymatic Polymerization and Solid State Finishing. J. Appl. Polym. Sci. 2014, 131, 2–9. [Google Scholar] [CrossRef]
- Jiang, Y.; Woortman, A.J.J.; Alberda van Ekenstein, G.O.R.; Loos, K. Enzyme-Catalyzed Synthesis of Unsaturated Aliphatic Polyesters Based on Green Monomers from Renewable Resources. Biomolecules 2013, 3, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Azim, H.; Dekhterman, A.; Jiang, Z.; Gross, R.A. Candida Antarctica Lipase B Catalyzed Synthesis of Poly(Butylene Succinate): Shorter Chain Building Blocks Also Work. Biomacromolecules 2006, 7, 3093–3097. [Google Scholar] [CrossRef]
- Pospiech, D.; Choińska, R.; Flugrat, D.; Sahre, K.; Jehnichen, D.; Korwitz, A.; Friedel, P.; Werner, A.; Voit, B. Enzymatic Synthesis of Poly(Alkylene Succinate)s: Influence of Reaction Conditions. Processes 2021, 9, 411. [Google Scholar] [CrossRef]
- An, S.; Zhu, J.; Lu, D.; Liu, Z. Lipase-Catalyzed Synthesis and Characterization of High-Molecular-Weight PBS. CIESC J. 2013, 64, 1855–1861. [Google Scholar]
- Ren, L.; Wang, Y.; Ge, J.; Lu, D.; Liu, Z. Enzymatic Synthesis of High-Molecular-Weight Poly(Butylene Succinate) and Its Copolymers. Macromol. Chem. Phys. 2015, 216, 636–640. [Google Scholar] [CrossRef]
- Sugihara, S.; Toshima, K.; Matsumura, S. New Strategy for Enzymatic Synthesis of High-Molecular-Weight Poly(Butylene Succinate) via Cyclic Oligomers. Macromol. Rapid Commun. 2006, 27, 203–207. [Google Scholar] [CrossRef]
- Hevilla, V.; Sonseca, A.; Echeverría, C.; Muñoz-Bonilla, A.; Fernández-García, M. Enzymatic Synthesis of Polyesters and Their Bioapplications: Recent Advances and Perspectives. Macromol. Biosci. 2021, 21, 2100156. [Google Scholar] [CrossRef] [PubMed]
- Sonseca, A.; McClain, A.; Puskas, J.E.; el Fray, M. Kinetic Studies of Biocatalyzed Copolyesters of Poly(Butylene Succinate)(PBS)Containing Fully Bio-Based Dilinoleic Diol. Eur. Polym. J. 2019, 116, 515–525. [Google Scholar] [CrossRef]
- Misset, O.; van Dijk, A. Diagnosing the Inactivating Process of Enzymes. In Stability and Stabilization of Biocatalysts; Ballesteros, A., Plou, F.J., Iborra, J.L., Halling, P.J., Eds.; Progress in Biotechnology; Elsevier: Amsterdam, The Netherlands, 1998; Volume 15, pp. 3–18. [Google Scholar]
- Nasr, K.; Meimoun, J.; Favrelle-Huret, A.; de Winter, J.; Raquez, J.M.; Zinck, P. Enzymatic Polycondensation of 1,6-Hexanediol and Diethyl Adipate: A Statistical Approach Predicting the Key-Parameters in Solution and in Bulk. Polymers 2020, 12, 1907. [Google Scholar] [CrossRef] [PubMed]
- Burgard, A.; Burk, M.J.; Osterhout, R.; van Dien, S.; Yim, H. Development of a Commercial Scale Process for Production of 1,4-Butanediol from Sugar. Curr. Opin. Biotechnol. 2016, 42, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Sokołowska, M.; Fray, M.E. “Green” Poly(Butylene Succinate-Co-Dilinoleic Succinate) Copolymers Synthesized Using Candida Antarctica Lipase B (CAL-B) as Biocatalyst. Proceedings 2020, 69, 33. [Google Scholar] [CrossRef]
- Bhatia, S. Introduction to Enzymes and Their Applications. In Introduction to Pharmaceutical Biotechnology, Volume 2: Enzymes, Proteins and Bioinformatics; IOP SCIENCE: Bristol, UK, 2018; pp. 1–29. ISBN 9780750313025. [Google Scholar]
- Comerford, J.W.; Byrne, F.P.; Weinberger, S.; Farmer, T.J.; Guebitz, G.M.; Gardossi, L.; Pellis, A. Thermal Upgrade of Enzymatically Synthesized Aliphatic and Aromatic Oligoesters. Materials 2020, 13, 368. [Google Scholar] [CrossRef] [Green Version]
- Georgousopoulou, I.N.; Vouyiouka, S.; Dole, P.; Papaspyrides, C.D. Thermo-Mechanical Degradation and Stabilization of Poly(Butylene Succinate). Polym. Degrad. Stab. 2016, 128, 182–192. [Google Scholar] [CrossRef]
- Carlos Morales-Huerta, J.; Martínez De Ilarduya, A.; Muñoz-Guerra, S. Poly(Alkylene 2,5-Furandicarboxylate)s (PEF and PBF) by Ring Opening Polymerization. Polymer 2016, 87, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Gruter, G.-J.M.; van Aken, T.B. Making an Impact: Sustainable Success Stories. In How to Commercialize Chemical Technologies for a Sustainable Future; Clark, T.J., Pasternak, A.S., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2021; pp. 263–270. [Google Scholar]
- Papadopoulos, L.; Zamboulis, A.; Kasmi, N.; Wahbi, M.; Nannou, C.; Lambropoulou, D.A.; Kostoglou, M.; Papageorgiou, G.Z.; Bikiaris, D.N. Investigation of the Catalytic Activity and Reaction Kinetic Modeling of Two Antimony Catalysts in the Synthesis of Poly(Ethylene Furanoate). Green Chem. 2021, 23, 2507–2524. [Google Scholar] [CrossRef]
- Zhu, J.; Cai, J.; Xie, W.; Chen, P.H.; Gazzano, M.; Scandola, M.; Gross, R.A. Poly(Butylene 2,5-Furan Dicarboxylate), a Biobased Alternative to PBT: Synthesis, Physical Properties, and Crystal Structure. Macromolecules 2013, 46, 796–804. [Google Scholar] [CrossRef]
- Poulopoulou, N.; Guigo, N.; Sbirrazzuoli, N.; Papageorgiou, D.G.; Bikiaris, D.N.; Nikolaidis, G.N.; Papageorgiou, G.Z. Towards Increased Sustainability for Aromatic Polyesters: Poly(Butylene 2,5-Furandicarboxylate) and Its Blends with Poly(Butylene Terephthalate). Polymer 2021, 212, 123157. [Google Scholar] [CrossRef]
- Jiang, Y.; Woortman, A.J.J.; Alberda Van Ekenstein, G.O.R.; Loos, K. A Biocatalytic Approach towards Sustainable Furanic-Aliphatic Polyesters. Polym. Chem. 2015, 6, 5198–5211. [Google Scholar] [CrossRef] [Green Version]
- Maniar, D.; Jiang, Y.; Woortman, A.J.J.; van Dijken, J.; Loos, K. Furan-Based Copolyesters from Renewable Resources: Enzymatic Synthesis and Properties. ChemSusChem 2019, 12, 990–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniar, D.; Silvianti, F.; Ospina, V.M.; Woortman, A.J.J.; van Dijken, J.; Loos, K. On the Way to Greener Furanic-Aliphatic Poly(Ester Amide)s: Enzymatic Polymerization in Ionic Liquid. Polymer 2020, 205, 122662. [Google Scholar] [CrossRef]
- Jiang, Y.; Maniar, D.; Woortman, A.J.J.; Loos, K. Enzymatic Synthesis of 2,5-Furandicarboxylic Acid-Based Semi-Aromatic Polyamides: Enzymatic Polymerization Kinetics, Effect of Diamine Chain Length and Thermal Properties. RSC Adv. 2016, 6, 67941–67953. [Google Scholar] [CrossRef] [Green Version]
- Silvianti, F.; Maniar, D.; Boetje, L.; Loos, K. Green Pathways for the Enzymatic Synthesis of Furan-Based Polyesters and Polyamides. ACS Symp. Ser. 2020, 1373, 3–29. [Google Scholar] [CrossRef]

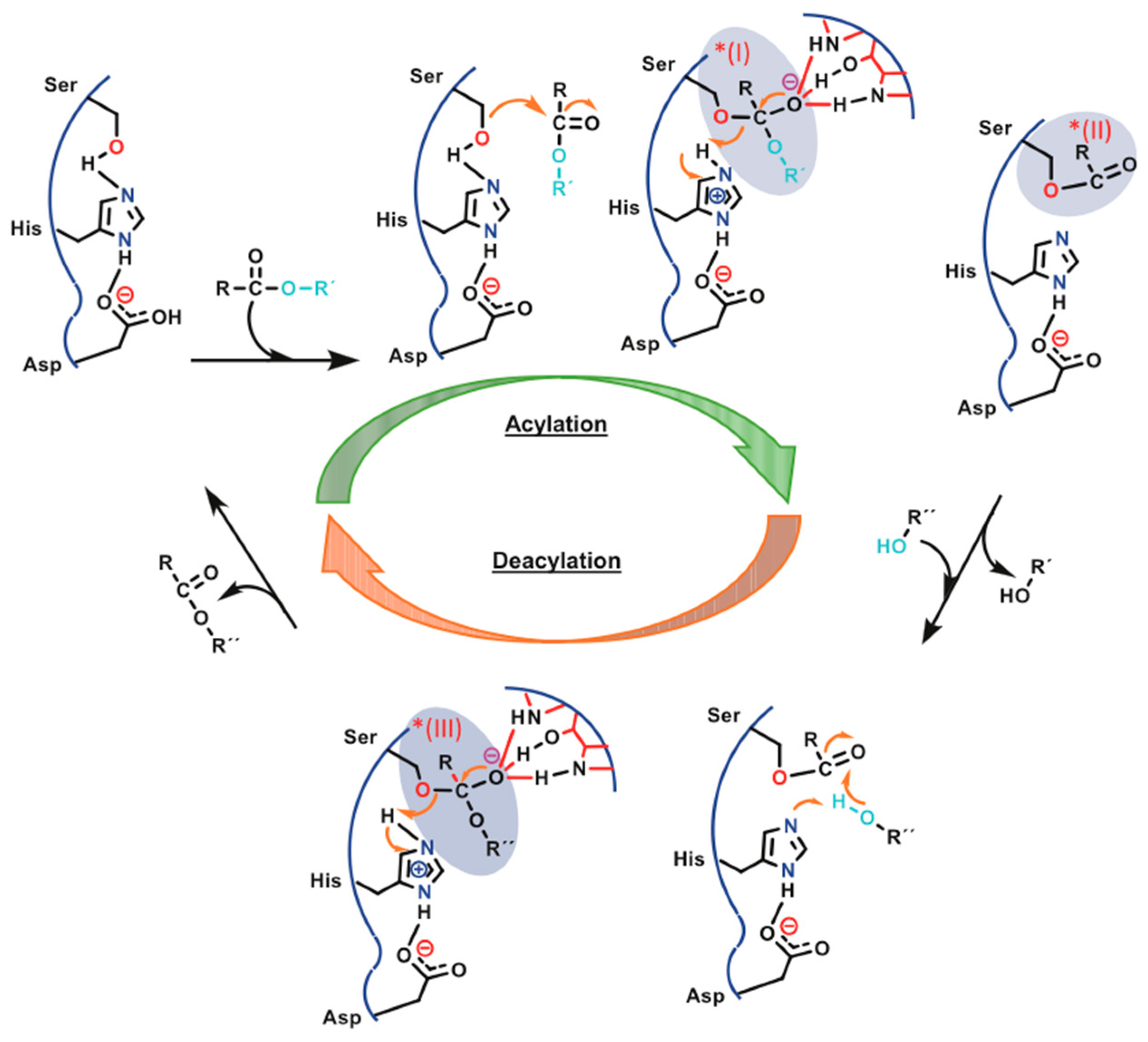

{kind=link}
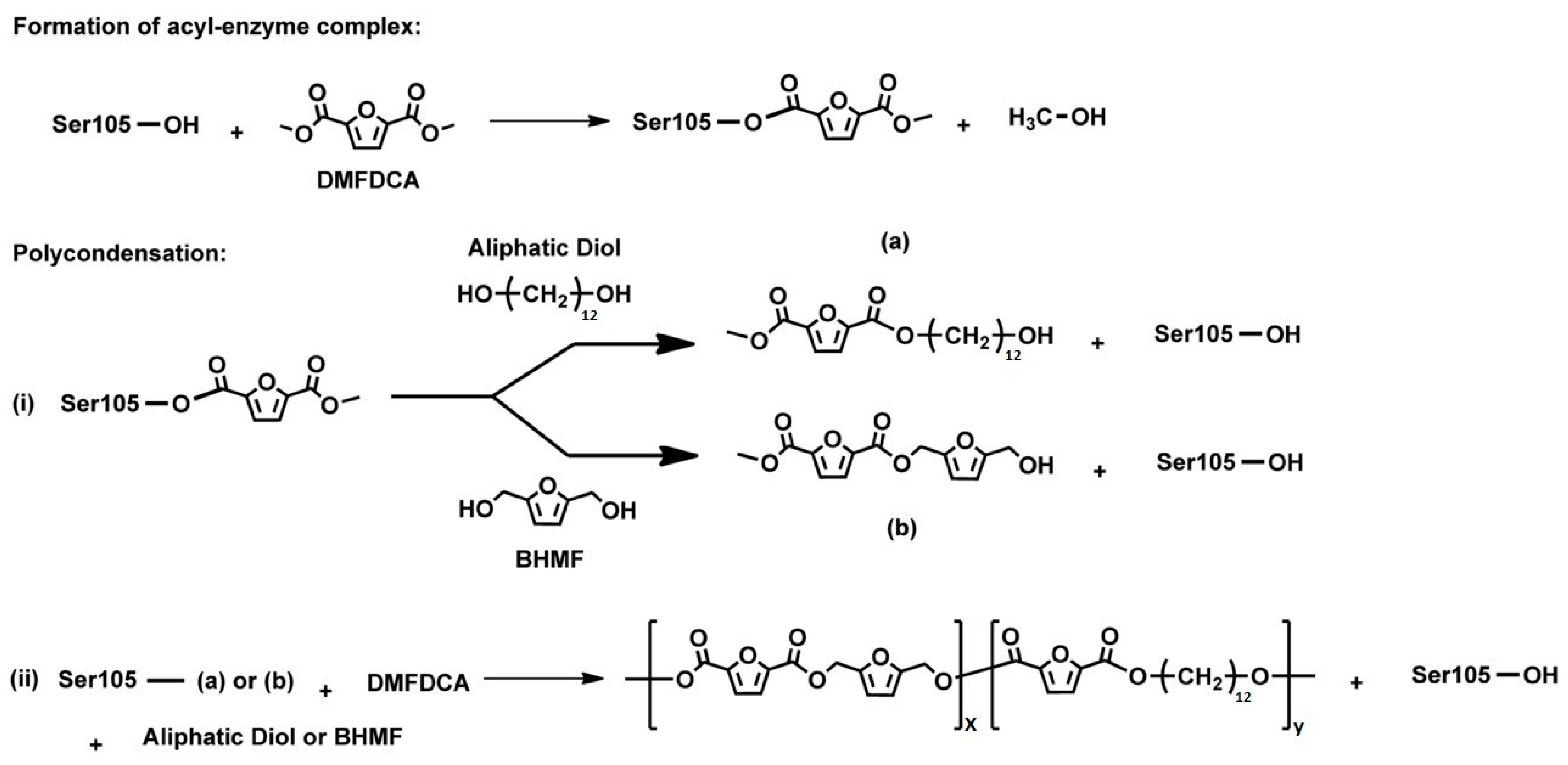
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Structure |
|---|---|
| PLA | 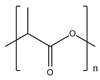 |
| PBS |  |
| PBF | 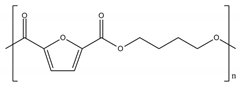 |
| Ref. | Enzyme | Polymerization Method | Solvent | T (°C) | Range of Achieved MW (g/mol) | |
|---|---|---|---|---|---|---|
| 1 | 2 | |||||
| [47] | Streptomyces lipase | polycondensation | Toluene | 60 | - | 525 |
| [46] | Candida rugosa lipase | direct polycondensation | - | 100 | 1430–1480 | - |
| [35] | CALB redesigned | eROP | Toluene | 60 | - | 780 |
| [45] | Im. CALB | polycondesation | Isopropyl ether | 60 | - | 2460 |
| [37] | Im. CALB | eROP | Toluene | 70 | - | 2600 |
| [34] | Candida rugosa lipase | eROP | - | 90 | 5400 | 2850 |
| [38] | N435 | eROP | Toluene, TEA | 70 | - | 4900 |
| [14] | N435 | eROP | scCO2 | 65 | 12,900 | - |
| [41] | N435 | eROP | N,N-Dimethylacetamide (DMA) | 130 | 18,800 | - |
| [13] | N435 | eROP | Toluene | 60 | - | 12,000 |
| [39] | B. cepacia lipase | eROP | scR134a | 105 | - | 14,000 |
| [42] | N435 | eROP | Toluene | 80 | - | 26,000 |
| [43] | Im. CALB | eROP | compressed R134a and the IL [C4MIM] [PF6] 3 | 65 | - | 28,000 |
| [40] | N435 | eROP | IL [HMIM] [PF6] 4 | 90 | - | 37,800 |
| [36] | B. cepacia lipase | eROP | - | 125 | - | 78,100 |
| Ref. | Enzyme | Solvent | T (°C) | Range of Achieved MW (g/mol) | |
|---|---|---|---|---|---|
| [75] | N435 | - | 85 | 1094 | 851 |
| [44] | N435 | Isooctane | 50 | 2000 | - |
| [76] | N435 | - | 60 | 2550 | 1700 |
| [77] | N435 | Diphenyl ether | 80 | - | 840–2550 |
| [78] | N435 | Diphenyl ether | 75 | - | 3910 |
| [79] | N435 | Diphenyl ether | 80 | 11,520 | 4463–6017 |
| [80] | N435 | Diphenyl ether | 60–90 | - | 2000–7000 |
| [81] | N435 | - | 130 | 23,600 | 11,700 |
| [80] | N435 | - | 80–95 | 38,000 | 27,340 |
| [82] | N435 | - | 95 | 44,000 | - |
| [83] | N435 | Toluene | 95 | 73,000 | - |
| [84] | N435 | Toluene | 100–120 | 130,000 | 81,250 |
| Ref. | Polymer | Enzyme | Solvent | T (°C) | Range of Achieved MW (g/mol) | |
|---|---|---|---|---|---|---|
| [92] | PBF | N435 | - | 50 | 600 | 500 |
| [99] | PBF | N435 | Diphenylether | 80–140 | 5500 | 1600 |
| [100] | Furan-based copolyesters (P(FMF-co-OF)) | N435 | Diphenylether | 80–95 | 35,000 | - |
| [101] | Furanic-aliphatic poly(ester amide)s (PEAF12) | N435 | Toluene | 90 | 21,000 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkountela, C.I.; Vouyiouka, S.N. Enzymatic Polymerization as a Green Approach to Synthesizing Bio-Based Polyesters. Macromol 2022, 2, 30-57. https://doi.org/10.3390/macromol2010003
Gkountela CI, Vouyiouka SN. Enzymatic Polymerization as a Green Approach to Synthesizing Bio-Based Polyesters. Macromol. 2022; 2(1):30-57. https://doi.org/10.3390/macromol2010003
Chicago/Turabian StyleGkountela, Christina I., and Stamatina N. Vouyiouka. 2022. "Enzymatic Polymerization as a Green Approach to Synthesizing Bio-Based Polyesters" Macromol 2, no. 1: 30-57. https://doi.org/10.3390/macromol2010003
APA StyleGkountela, C. I., & Vouyiouka, S. N. (2022). Enzymatic Polymerization as a Green Approach to Synthesizing Bio-Based Polyesters. Macromol, 2(1), 30-57. https://doi.org/10.3390/macromol2010003