
Dynamic Patterns of Sex Chromosome Evolution in Neognath Birds: Many Independent Barriers to Recombination at the ATP5F1A Locus



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction, PCR Amplification, Cloning and Sequencing
2.2. Extraction of Data from Genome Assemblies
2.3. Identification of Transposable Element Insertions
2.4. Sequence Alignment and Phylogenetic Analyses
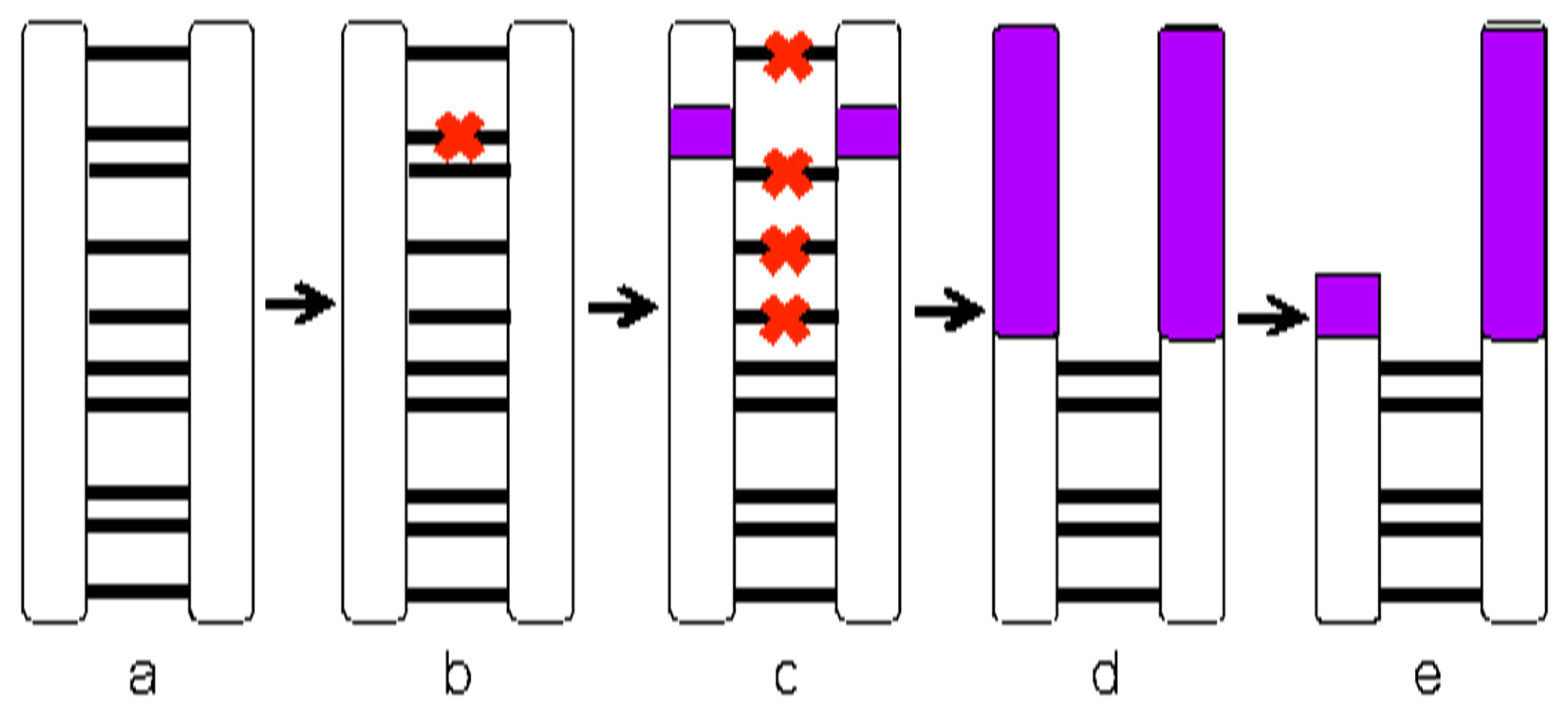
2.5. Expectations
3. Results
3.1. Success in Identifying Both Gametologs
3.2. Transposable Elements
3.3. Barriers to Recombination
3.4. Purifying Selection on ATP5F1A Proteins
4. Discussion
4.1. Avian Sex Chromosome Evolution Is Highly Dynamic
4.2. Cessation of Recombination and Relaxed Purifying Selection
4.3. Challenges in Studying Sex Chromosome Evolution
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bachtrog, D.; Kirkpatrick, M.; Mank, J.E.; McDaniel, S.F.; Pires, J.C.; Rice, W.; Valenzuela, N. Are all sex chromosomes created equal? Trends Genet. 2011, 27, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Rice, W.R. The accumulation of sexually antagonistic genes as a selective agent promoting the evolution of reduced recombination between primitive sex chromosomes. Evolution 1987, 41, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D.; Charlesworth, B.; Marais, G. Steps in the evolution of heteromorphic sex chromosomes. Heredity 2005, 95, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Ponnikas, S.; Sigeman, H.; Abbott, J.K.; Hansson, B. Why do sex chromosomes stop recombining? Trends Genet. 2018, 34, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L.S.; Metzger, D.C.H.; Darolti, I.; Wright, A.E.; Sandkam, B.A.; Almeida, P.; Shu, J.J.; Mank, J.E. Sex chromosome evolution: So many exceptions to the rules. Genome Biol. Evol. 2020, 12, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, D.L.; Gerchen, J.F.; Scharmann, M.; Pannell, J.R. A neutral model for the loss of recombination on sex chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021, 376, 20200096. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Charlesworth, D. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Mank, J.E.; Ellegren, H. Parallel divergence and degradation of the avian W sex chromosome. Trends Ecol. Evol. 2007, 22, 389–391. [Google Scholar] [CrossRef]
- Bellott, D.W.; Skaletsky, H.; Pyntikova, T.; Mardis, E.R.; Graves, T.; Kremitzki, C.; Brown, L.G.; Rozen, S.; Warren, W.C.; Wilson, R.K.; et al. Convergent evolution of chicken Z and human X chromosomes by expansion and gene acquisition. Nature 2010, 466, 612–616. [Google Scholar] [CrossRef]
- Bachtrog, D. A dynamic view of sex chromosome evolution. Curr. Opin. Genet. Dev. 2006, 16, 578–585. [Google Scholar] [CrossRef]
- Otto, S.P.; Pannell, J.R.; Peichel, C.L.; Ashman, T.-L.; Charlesworth, D.; Chippindale, A.K.; Delph, L.F.; Guerrero, R.F.; Scarpino, S.V.; McAllister, B.F. About PAR: The distinct evolutionary dynamics of the pseudoautosomal region. Trends Genet. 2011, 27, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Fridolfsson, A.K.; Cheng, H.; Copeland, N.G.; Jenkins, N.A.; Liu, H.C.; Raudsepp, T.; Woodage, T.; Chowdhary, B.; Halverson, J.; Ellegren, H. Evolution of the avian sex chromosomes from an ancestral pair of autosomes. Proc. Natl. Acad. Sci. USA 1998, 95, 8147–8152. [Google Scholar] [CrossRef] [PubMed]
- Kratochvíl, L.; Gamble, T.; Rovatsos, M. Sex chromosome evolution among amniotes: Is the origin of sex chromosomes non-random? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021, 376, 20200108. [Google Scholar] [CrossRef] [PubMed]
- Johnson Pokorná, M.; Kratochvíl, L. What was the ancestral sex-determining mechanism in amniote vertebrates? Biol. Rev. Camb. Philos. Soc. 2016, 91, 1–12. [Google Scholar] [CrossRef]
- Nam, K.; Ellegren, H. The chicken (Gallus gallus) Z chromosome contains at least three nonlinear evolutionary strata. Genetics 2008, 180, 1131–1136. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, J.; Bachtrog, D.; An, N.; Huang, Q.; Jarvis, E.D.; Gilbert, M.T.P.; Zhang, G. Complex evolutionary trajectories of sex chromosomes across bird taxa. Science 2014, 346, 1246338. [Google Scholar] [CrossRef]
- Xu, L.; Auer, G.; Peona, V.; Suh, A.; Deng, Y.; Feng, S.; Zhang, G.; Blom, M.P.K.; Christidis, L.; Prost, S.; et al. Dynamic evolutionary history and gene content of sex chromosomes across diverse songbirds. Nat. Ecol. Evol. 2019, 3, 834–844. [Google Scholar] [CrossRef]
- Fridolfsson, A.-K.; Ellegren, H. A Simple and Universal Method for Molecular Sexing of Non-Ratite Birds. J. Avian Biol. 1999, 30, 116. [Google Scholar] [CrossRef]
- García-Moreno, J.; Mindell, D.P. Rooting a phylogeny with homologous genes on opposite sex chromosomes (gametologs): A case study using avian CHD. Mol. Biol. Evol. 2000, 17, 1826–1832. [Google Scholar] [CrossRef]
- Ellegren, H.; Carmichael, A. Multiple and independent cessation of recombination between avian sex chromosomes. Genetics 2001, 158, 325–331. [Google Scholar] [CrossRef]
- Handley, L.-J.L.; Ceplitis, H.; Ellegren, H. Evolutionary strata on the chicken Z chromosome: Implications for sex chromosome evolution. Genetics 2004, 167, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Suh, A.; Kriegs, J.O.; Brosius, J.; Schmitz, J. Retroposon insertions and the chronology of avian sex chromosome evolution. Mol. Biol. Evol. 2011, 28, 2993–2997. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, J.; Lagisz, M.; Nakagawa, S. The long and the short of avian W chromosomes: No evidence for gradual W shortening. Biol. Lett. 2012, 8, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Pala, I.; Hasselquist, D.; Bensch, S.; Hansson, B. Patterns of molecular evolution of an avian neo-sex chromosome. Mol. Biol. Evol. 2012, 29, 3741–3754. [Google Scholar] [CrossRef]
- Sigeman, H.; Strandh, M.; Proux-Wéra, E.; Kutschera, V.E.; Ponnikas, S.; Zhang, H.; Lundberg, M.; Soler, L.; Bunikis, I.; Tarka, M.; et al. Avian neo-sex chromosomes reveal dynamics of recombination suppression and W degeneration. Mol. Biol. Evol. 2021, 38, 5275–5291. [Google Scholar] [CrossRef]
- Kretschmer, R.; Gunski, R.J.; Garnero, A.D.V.; de Freitas, T.R.O.; Toma, G.A.; de Cioffi, M.B.; de Oliveira, E.H.C.; O’Connor, R.E.; Griffin, D.K. Chromosomal Analysis in Crotophaga ani (Aves, Cuculiformes) Reveals Extensive Genomic Reorganization and an Unusual Z-Autosome Robertsonian Translocation. Cells 2020, 10, 4. [Google Scholar] [CrossRef]
- Huang, Z.; Furo, I.; Peona, V.; Liu, J.; Gomes, A.J.B.; Cen, W.; Huang, H.; Zhang, Y.; Chen, D.; Ting, X.; et al. Recurrent chromosome reshuffling and the evolution of neo-sex chromosomes in parrots. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, W.H.; Yi, S.; Makova, K. Male-driven evolution. Curr. Opin. Genet. Dev. 2002, 12, 650–656. [Google Scholar] [CrossRef]
- Charlesworth, B.; Coyne, J.A.; Barton, N.H. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 1987, 130, 113–146. [Google Scholar] [CrossRef]
- Mank, J.E.; Axelsson, E.; Ellegren, H. Fast-X on the Z: Rapid evolution of sex-linked genes in birds. Genome Res. 2007, 17, 618–624. [Google Scholar] [CrossRef]
- Veeramah, K.R.; Gutenkunst, R.N.; Woerner, A.E.; Watkins, J.C.; Hammer, M.F. Evidence for increased levels of positive and negative selection on the X chromosome versus autosomes in humans. Mol. Biol. Evol. 2014, 31, 2267–2282. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J.; Yang, W.; An, N.; Zhang, P.; Zhang, G.; Zhou, Q. Temporal genomic evolution of bird sex chromosomes. BMC Evol. Biol. 2014, 14, 250. [Google Scholar] [CrossRef] [PubMed]
- Berlin, S.; Tomaras, D.; Charlesworth, B. Low mitochondrial variability in birds may indicate Hill-Robertson effects on the W chromosome. Heredity 2007, 99, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Hickey, A.J.R. Avian mtDNA diversity? An alternate explanation for low mtDNA diversity in birds: An age-old solution? Heredity 2008, 100, 443. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Mitochondria and the W chromosome: Low variability on the W chromosome in birds is more likely to indicate selection on mitochondrial genes. Heredity 2008, 100, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Smeds, L.; Warmuth, V.; Bolivar, P.; Uebbing, S.; Burri, R.; Suh, A.; Nater, A.; Bureš, S.; Garamszegi, L.Z.; Hogner, S.; et al. Evolutionary analysis of the female-specific avian W chromosome. Nat. Commun. 2015, 6, 7330. [Google Scholar] [CrossRef]
- Berlin, S.; Ellegren, H. Fast accumulation of nonsynonymous mutations on the female-specific W chromosome in birds. J. Mol. Evol. 2006, 62, 66–72. [Google Scholar] [CrossRef]
- de Souza, M.S.; Kretschmer, R.; Barcellos, S.A.; Costa, A.L.; de Cioffi, M.B.; de Oliveira, E.H.C.; Garnero, A.D.V.; Gunski, R.J. Repeat sequence mapping shows different W chromosome evolutionary pathways in two caprimulgiformes families. Birds 2020, 1, 4. [Google Scholar] [CrossRef]
- Peona, V.; Palacios-Gimenez, O.M.; Blommaert, J.; Liu, J.; Haryoko, T.; Jønsson, K.A.; Irestedt, M.; Zhou, Q.; Jern, P.; Suh, A. The avian W chromosome is a refugium for endogenous retroviruses with likely effects on female-biased mutational load and genetic incompatibilities. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021, 376, 20200186. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Chitra, B. Nucleotide sequence of a cDNA for the α subunit of human mitochondrial ATP synthase. Biochim. Biophys. Acta (BBA)—Gene Struct. Expr. 1991, 1089, 393–395. [Google Scholar] [CrossRef]
- Carmichael, A.N.; Fridolfsson, A.K.; Halverson, J.; Ellegren, H. Male-biased mutation rates revealed from Z and W chromosome-linked ATP synthase alpha-subunit (ATP5A1) sequences in birds. J. Mol. Evol. 2000, 50, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Takagi, N.; Sasaki, M. A phylogenetic study of bird karyotypes. Chromosoma 1974, 46, 91–120. [Google Scholar] [CrossRef] [PubMed]
- Belterman, R.H.R.; De Boer, L.E.M. A karyological study of 55 species of birds, including karyotypes of 39 species new to cytology. Genetica 1984, 65, 39–82. [Google Scholar] [CrossRef]
- Yazdi, H.P.; Silva, W.T.A.F.; Suh, A. Why do some sex chromosomes degenerate more slowly than others? the odd case of ratite sex chromosomes. Genes 2020, 11, 1153. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Xu, X.; Witt, C.; Deng, Y.; Chenc, G.; Meng, G.; Feng, S.; Xu, L.; Szekely, T.; et al. Phylogeny and sex chromosome evolution of palaeognathae. J. Genet. Genom. 2021; in press. [Google Scholar] [CrossRef]
- Cummings, S.M.; McMullan, M.; Joyce, D.A.; van Oosterhout, C. Solutions for PCR, cloning and sequencing errors in population genetic analysis. Conserv. Genet. 2010, 11, 1095–1097. [Google Scholar] [CrossRef]
- Peona, V.; Blom, M.P.K.; Xu, L.; Burri, R.; Sullivan, S.; Bunikis, I.; Liachko, I.; Haryoko, T.; Jønsson, K.A.; Zhou, Q.; et al. Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird-of-paradise. Mol. Ecol. Resour. 2021, 21, 263–286. [Google Scholar] [CrossRef]
- Yazdi, H.P.; Ellegren, H. A genetic map of ostrich Z chromosome and the role of inversions in avian sex chromosome evolution. Genome Biol. Evol. 2018, 10, 2049–2060. [Google Scholar] [CrossRef]
- Gill, F.; Donsker, D.; Rasmussen, P. IOC World Bird List (v11.2). Available online: https://www.worldbirdnames.org/new/ (accessed on 26 November 2021).
- Kimball, R.T.; Crawford, D.J.; Smith, E.B. Evolutionary processes in the genus Coreocarpus: Insights from molecular phylogenetics. Evolution 2003, 57, 52–61. [Google Scholar] [CrossRef]
- De Kloet, S.R. Loss of the gene for the alpha subunit of ATP synthase (ATP5A1) from the W chromosome in the African grey parrot (Psittacus erithacus). J. Mol. Evol. 2001, 53, 135–143. [Google Scholar] [CrossRef]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232-5. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. Ufboot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Braun, E.L. Protein evolution is structure dependent and non-homogeneous across the tree of life. In Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Virtual Event, 21–24 September 2020; ACM: New York, NY, USA, 2020; pp. 1–11. [Google Scholar]
- Minh, B.Q.; Dang, C.C.; Vinh, L.S.; Lanfear, R. Qmaker: Fast and accurate method to estimate empirical models of protein evolution. Syst. Biol. 2021, 70, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods); Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Swofford, D.L.; Maddison, W.P. Reconstructing ancestral character states under Wagner parsimony. Math. Biosci. 1987, 87, 199–229. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Kimball, R.T.; Braun, E.L. Introns outperform exons in analyses of basal avian phylogeny using clathrin heavy chain genes. Gene 2008, 410, 89–96. [Google Scholar] [CrossRef]
- Hill, W.G.; Robertson, A. The effect of linkage on limits to artificial selection. Genet. Res. 1966, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Sassi, S.O.; Braun, E.L.; Benner, S.A. The evolution of seminal ribonuclease: Pseudogene reactivation or multiple gene inactivation events? Mol. Biol. Evol. 2007, 24, 1012–1024. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Oliveira Furo, I.; Kretschmer, R.; Dos Santos, M.S.; de Lima Carvalho, C.A.; Gunski, R.J.; O’Brien, P.C.M.; Ferguson-Smith, M.A.; Cioffi, M.B.; de Oliveira, E.H.C. Chromosomal Mapping of Repetitive DNAs in Myiopsitta monachus and Amazona aestiva (Psittaciformes, Psittacidae) with Emphasis on the Sex Chromosomes. Cytogenet. Genome Res. 2017, 151, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Braun, E.L.; Kimball, R.T. Data types and the phylogeny of neoaves. Birds 2021, 2, 1. [Google Scholar] [CrossRef]
- Kimball, R.T.; Oliveros, C.H.; Wang, N.; White, N.D.; Barker, F.K.; Field, D.J.; Ksepka, D.T.; Chesser, R.T.; Moyle, R.G.; Braun, M.J.; et al. A phylogenomic supertree of birds. Diversity 2019, 11, 109. [Google Scholar] [CrossRef]
- Backström, N.; Brandström, M.; Gustafsson, L.; Qvarnström, A.; Cheng, H.; Ellegren, H. Genetic mapping in a natural population of collared flycatchers (Ficedula albicollis): Conserved synteny but gene order rearrangements on the avian Z chromosome. Genetics 2006, 174, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Kampf, K.; Arnold, A.P. Comparison of the chicken and zebra finch Z chromosomes shows evolutionary rearrangements. Chromosome Res. 2006, 14, 805–815. [Google Scholar] [CrossRef]
- Nanda, I.; Schmid, M. Conservation of avian Z chromosomes as revealed by comparative mapping of the Z-linked aldolase B gene. Cytogenet. Genome Res. 2002, 96, 176–178. [Google Scholar] [CrossRef]
- Xu, L.; Zhou, Q. The Female-Specific W Chromosomes of Birds Have Conserved Gene Contents but Are Not Feminized. Genes 2020, 11, 1126. [Google Scholar] [CrossRef]
- Bachtrog, D. The temporal dynamics of processes underlying Y chromosome degeneration. Genetics 2008, 179, 1513–1525. [Google Scholar] [CrossRef]
- Suh, A. The specific requirements for CR1 retrotransposition explain the scarcity of retrogenes in birds. J. Mol. Evol. 2015, 81, 18–20. [Google Scholar] [CrossRef]
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 1994, 370, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Uebbing, S.; Künstner, A.; Mäkinen, H.; Ellegren, H. Transcriptome sequencing reveals the character of incomplete dosage compensation across multiple tissues in flycatchers. Genome Biol. Evol. 2013, 5, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Braun, E.L.; Kimball, R.T. Testing hypotheses about the sister group of the passeriformes using an independent 30-locus data set. Mol. Biol. Evol. 2012, 29, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Berv, J.S.; Field, D.J. Genomic Signature of an Avian Lilliput Effect across the K-Pg Extinction. Syst. Biol. 2018, 67, 1–13. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total | Included | Excluded | ||||
|---|---|---|---|---|---|---|
| Method | # Orders | # Species | # Orders | # Species | # Orders | # Species |
| PCR/cloning | 20 | 30 | 14 | 17 | 10 | 13 |
| Genomes | 13 | 32 | 10 | 25 | 5 | 7 |
| Published | 6 | 8 | 6 | 8 | 0 | 0 |
| Combined | 25 | 69 1 | 22 | 49 1 | 13 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimball, R.T.; Braun, E.L. Dynamic Patterns of Sex Chromosome Evolution in Neognath Birds: Many Independent Barriers to Recombination at the ATP5F1A Locus. Birds 2022, 3, 51-70. https://doi.org/10.3390/birds3010004
Kimball RT, Braun EL. Dynamic Patterns of Sex Chromosome Evolution in Neognath Birds: Many Independent Barriers to Recombination at the ATP5F1A Locus. Birds. 2022; 3(1):51-70. https://doi.org/10.3390/birds3010004
Chicago/Turabian StyleKimball, Rebecca T., and Edward L. Braun. 2022. "Dynamic Patterns of Sex Chromosome Evolution in Neognath Birds: Many Independent Barriers to Recombination at the ATP5F1A Locus" Birds 3, no. 1: 51-70. https://doi.org/10.3390/birds3010004
APA StyleKimball, R. T., & Braun, E. L. (2022). Dynamic Patterns of Sex Chromosome Evolution in Neognath Birds: Many Independent Barriers to Recombination at the ATP5F1A Locus. Birds, 3(1), 51-70. https://doi.org/10.3390/birds3010004