1. Introduction
Radiation emergency management in a nuclear radiation incident over a large geographic region or affects many individuals involves an extraordinary degree of coordination between first responders, testing laboratories, and clinical personnel. The scope of testing in population-based scenarios has been recognized to exceed the capacity most biodosimetry laboratories. Not only would the volumes of samples overwhelm these laboratories, but the impact of large volume testing on accuracy has not been established.
The dicentric chromosome assay (DCA [
1]) is the gold standard test within the clinically relevant and treatable radiation exposure range. DCA has been the preferred radiation biodosimeter for estimation of individual exposures after large scale accidents at Chernobyl [
2,
3,
4], Goiania [
5,
6,
7], and Fukushima-Daiichi [
8,
9,
10]. While rapid tests are under development for triage purposes (micronucleus, H2AX, etc [
11,
12,
13,
14,
15,
16]), the calibration curves for these assays tend to exhibit high variance, which impacts the confidence of the estimated dose. This can possibly lead to overtesting of worried well or inadequate testing of at risk, exposed populations.
Several international exercises and proposals have envisioned cooperative biodosimetry testing could be distributed over multiple laboratories to overcome the bottleneck in generating dose estimates for exposed populations (RENEB, Can Bio, US Bio network [
17,
18,
19,
20,
21]). There are a number of significant challenges inherent in implementing such a strategy. The degree of interlaboratory coordination required—including sample transport—may not be adequate to handle the workloads in an actual population-scale event. Furthermore, reliable internet resources connecting laboratories may not be available to collate results across international borders and interpretation would have to be standardized between laboratories.
Performance of the conventional DCA on all or most suspected cases of Acute Radiation Syndrome in a mass casualty would cause bottlenecks in sample processing, specifically cell culture, capture of metaphase cell images, and interpretation of those images. Without automation, the throughput of this test will probably not be adequate to triage patients for life saving cytokine therapies [
8]. Semi-automated testing is efficient for small sample volumes, but processing and analysis of thousands of samples will surpass recommended therapeutic windows in a mass casualty [
22]. Full automation of sample preparation, metaphase cell imaging, and interpretations of the DCA could substantially contribute to meeting testing capacity requirements to ensure timely administration of therapies. Equipment is commercially available to automate sample preparation (from Hanabi) and metaphase cell imaging (from Leica Biosystems and MetaSystems). Interpretation of images and dose estimation is performed manually or at best, semi-automatically with editorial supervision.
The Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) is a medical image processing system that leverages machine learning to analyze metaphase cell samples from different individuals, in the form of images, to identify dicentric chromosomes as an indicator of the patient’s level of radiation exposure that would then be used to determine the treatment needed, if any [
23,
24,
25]. The current Windows-based implementation of the system substantially reduces the time required for laboratories to estimate radiation exposures without compromising accuracy. However, in a large-scale radiation accident or mass casualty, data from many different individuals would need to be processed quickly. A bank of personal computers running ADCI may not be able to meet the throughput required to triage entire populations on the scale of a moderate sized city.
The present study investigates large-scale automation of analysis of cytogenetic data required for the DCA and radiation exposure assessment. To accelerate ADCI, image processing components of ADCI were parallelized on a multiprocessor supercomputer to determine if performance is sufficient to handle the demands of population-scale exposures. This simultaneously processed metaphase images from multiple samples, with the goal of expediting the analysis of the full set of samples. The advantage of parallelization is that it would provide dose of exposure information for clinical decision making for many exposed individuals at the same time. It is assumed that testing laboratories have the capacity to automate culturing of multiple blood samples, harvest and prepare slides of metaphase cells, and possess microscope systems for capture of metaphase cell images. We previously demonstrated a proof of concept implementation with a subset of the components of the ADCI system [
26], indicating that the software could be adapted for large scale processing of many samples by master-slave rank-based scheduling of available computing resources across all samples and images. This was the starting point for the current implementation of the fully functional ADCI application on the IBM BG/Q supercomputer (BG/Q). This high-throughput ADCI (ADCI-HT) software was designed to rapidly analyze thousands of images in parallel, obtaining near identical dose predictions quickly compared to the time required via a commercial Windows desktop. Nuclear incident results were simulated for 15 US cities by calibrating radiation plumes predicted from physical dosimetry with population data. ADCI-HT was then used to analyze individuals exposed to >1 Gy and >2 Gy of radiation. Finally, analysis of actual international exercise data was performed, confirming ADCI-HT software predictions meets IAEA triage analysis requirements.
2. Methods
ADCI-HT automates the selection of metaphase images, interprets dicentric chromosomes in these images, generates calibration curves, and estimates exposures from the dicentric chromosome frequencies for partial or whole-body dose estimates. It does not carry out laboratory procedures, including cell culture, fixation, slide preparation, Giemsa staining, or image capture. These are performed in different biodosimetry laboratories, either manually or with semi-automated systems.
2.1. Underlying Principles for Accelerating ADCI by High-Throughput Supercomputing
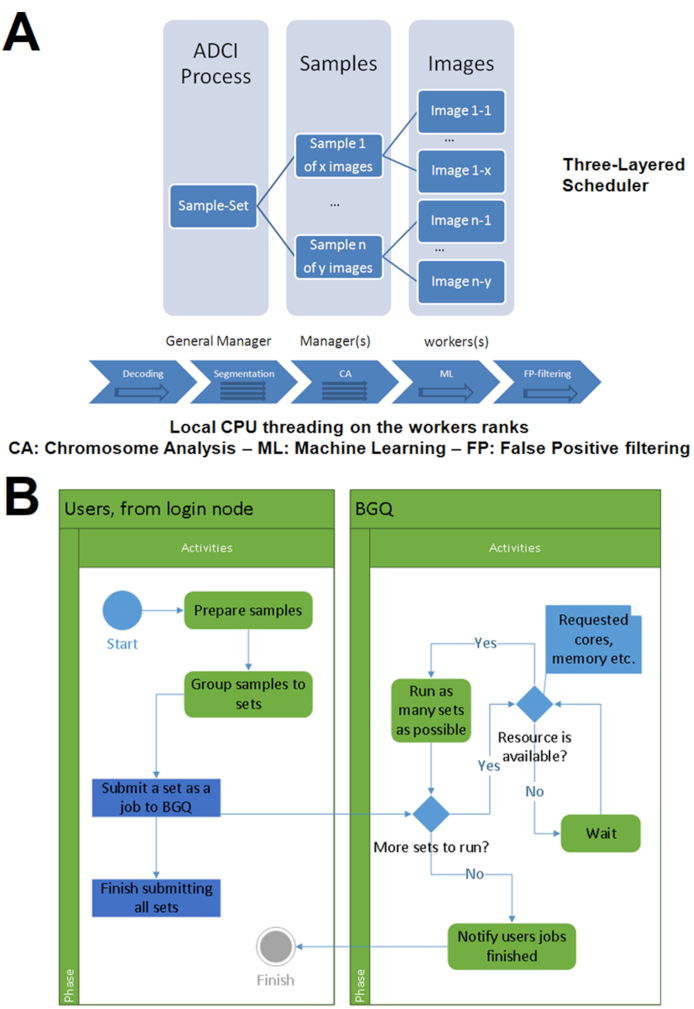
Elements of the ADCI software were redesigned to port it to the BG/Q hardware platform for high throughput radiation biodosimetry. ADCI-HT emphasizes the throughput of the software to handle many samples in a single run. The high-throughput (HT) version leverages previous analyses using the Windows Desktop version of the program that would be used to generate a calibration curve and derive optimal parameters for image model selection. The HT version lacks a graphical user interface for dynamic interaction. Thus, elements of ADCI-HT software were either replicated, modified or replaced in order to manage parallel analyses of samples with thousands of computer processors. Some compilation differences between Intel and PowerPC architectures used in the BG/Q supercomputer had to be addressed during implementation. The emphasis on large scale processing capability on BG/Q limits available input/output (I/O) resources and the system is not interactive. BG/Q sacrifices I/O performance for compute power, since there is only a single node that performs I/O operations. Images from multiple patients were therefore combined into sample sets by creating file archives before transferring these large files to BG/Q. Subsequently, simulated patient sample cohorts whose sizes were based on estimated population sizes (see below) were created by randomly sampling 500 or more images from a larger pool of images from each laboratory source. These were recombined to form new archives of sets of samples from the same laboratory source, that were subsequently processed by ADCI-HT. These archives were used to benchmark the performance of ADCI-HT. ADCI-HT reads and decodes compressed archives of multiple samples (typically 50–400), each consisting of at least 500 microscope cell images (TIF or PNG format) into memory. The elements of the system used by both versions are described in
Figure 1, which is updated from our previously described Windows Desktop Version of ADCI software [
27]. The elements of program code that perform image selection, calibration curve derivation and dose estimation are identical in ADCI-HT and Windows-based ADCI. Differences between physical and biodosimetry exposures for whole body radiation fulfill IAEA accuracy criteria of ≤0.5 Gy [
28].The HT implementation replicates the 3 layers below the “Application” level of the ADCI-Windows-Desktop version (IV). Additionally, a new function named “Sample::calculateDose()” was added to use the ported classes to read the dosimetry curve and image selection model files, and uses these files to select the images in each sample and estimate the dose of radiation exposure.
2.2. Scheduling System
Efficient automated cytogenetic analysis of thousands of biodosimetry samples requires many computational tasks to be performed simultaneously. These tasks need to be scheduled to maximize concurrent use of all available processor resources to analyze all metaphase images in each sample [
26]. The algorithm implemented was non-preemptive, since we assumed equivalent priority for processing all samples, and was dynamic, since the resources required depends on the number of samples and size of each sample to be processed (which cannot be known in advance).
A quantitative analysis of image processing performed by ADCI-HT used test samples from Health Canada (n = 6; 540–1136 images each) and Canadian Nuclear Laboratories (n = 7; 500–1527 images each). Performance of the scheduler was correlated with sample size (numbers of images analyzed). The time consumption to load and read a sample was correlated with the size of the compressed archive file (r = 0.95) and to a lesser extent, with the number of images in the sample (r = 0.65).
A data flow diagram apportions the time required to perform the tasks that ADCI performs to process metaphase images (
Supplementary Figure S1). The segmentation task first takes an input image and identifies the regions containing connected objects based on pixel intensities. The chromosome analysis (CA) task identifies valid chromosomes through machine learning (ML)-based recognition of the centromere candidates and other features in each object. The identified chromosomes are then analyzed by another ML task to distinguish the dicentric chromosomes (DCs) from monocentric chromosomes (MCs). The results are then processed by the filtering task to remove false positive DCs. Finally, overall statistics are calculated for each image before the results are saved. The chromosome analysis task was found to be a primary bottleneck among all the tasks, with the highest standard deviation for the time to process images, meaning that the requirements for this task were the most variable among all tasks. This results in CPU load imbalance, whereby some images require substantially more computing resources than others to analyze. In fact, the number of objects in each image was only weakly correlated with chromosome analysis time (r = 0.07). One caveat, however, was that certain datasets created using replicate images with nearly identical sample sizes could impact the independence, and thereby the accuracy of the statistics drawn from those samples.
Within the chromosome analysis task, individual chromosomes are segmented by local thresholding and Gradient Vector Flow (GVF; [
29]) active contours. Upon extraction based on the GVF boundaries, the contour of the chromosome is partitioned using a polygonal shape simplification algorithm known as Discrete Curve Evolution (DCE) which iteratively deletes vertices based on their importance to the overall shape of the object. A Support Vector Machine (SVM) classifier selects the best set of points to isolate the telomeric regions, i.e., at the ends of each chromosome. The segmented telomere regions are then tested for evidence of sister chromatid separation using a second trained SVM classifier designed to capture shape characteristics of the telomere regions and then corrected for that artifact. Afterwards, the chromosome is split into two partitions along the axis of symmetry and a modified Laplacian-based thickness measurement algorithm (called Intensity Integrated Laplacian, or IIL [
30]) is used to calculate the width profile of the chromosome. This profile is then used to identify a possible set of candidates for centromere location(s) and features are calculated for each of those locations. Next, another classifier is trained on expert-classified chromosomes to detect centromere locations in chromosomes. In most instances, each chromosome will contain at least one centromere. The correct centromere is generally present among the candidates. The distance from the separating hyperplane is used as an indicator for the goodness of fit of a given candidate and thereafter used to select the best candidate from the pool of candidates. Finally, we apply a machine learning method that uses image features to distinguish mono- from dicentric chromosomes. The most time-consuming sub-tasks (GVF and IIL) depend on the length of the chromosomes (correlation is 0.89), which is indicated by the area of the region of interest for each chromosome. The maximum value for GVF was high, but the average time was low, suggesting that the high value is an outlier and was not representative of all images. IIL was the actual processing bottleneck, since it required a minimum of 3 s each to perform this step for each of the images analyzed. Dose accuracy relies on metaphase image selection criteria, which depends on thresholding and, in some instances, sorting images by quality and are optimized according to data generated by each biodosimetry laboratory [
24].
To handle the simultaneous analysis of many samples, we developed a three-layer scheduling architecture consisting of a general manager, which directs several managers, each of which are dynamically assigned by the program to multiple worker processors (
Figure 2A). The scheduler minimizes compute load imbalances between different processors. Each manager sends asynchronous requests for analysis of individual images to each worker (based on the value of a variable defined as “extraload”). Once the worker completes the analysis and submits its results, it queries managers for any outstanding requests. If a request has not been fulfilled, the worker receives that message, determines if the value of “extraload” is non-zero, then requests another image from the manager. The manager responds by sending an image while reducing the value of “extraload”. Without “extraload”, this communication would not take place at all. It is conceivable that multiple workers may receive the message before the value of the “extraload” variable is updated; however, the resources used by the resulting unnecessary processes are negligible.
2.3. User Interface and Outputs
The ADCI-HT batch queue allocates shared compute resources to ADCI once they are available. Tasks and CPU resource requests submitted to this queue use a command line interface which provides similar functionality to the graphical user interface of the MS-Windows version. This interface specifies a configuration file that can assign different image selection models and calibration curves to process particular sets of samples, for example, originating from different biodosimetry laboratories. Upon completion, ADCI processed sample files are written in the same directory where unprocessed samples are located and have the same file name as the sample, appended with the suffix “.adcisample.” Dose Estimate reports are formatted as CSV files located in the directory of the samples mentioned in it. The report provides the sample name, the curve file, the selection model, the sigma value, the dicentric chromosome (DC) frequency and the estimated dose.
2.4. Estimating Affected Population Size in Nuclear Incidents
We estimated the size of the population exposed to different levels of radiation by intersecting geographic contours of minimum radiation exposure levels computed by HPAC v4.02 software (Hazard Prediction and Assessment Capability; developed by the Defense Threat Reduction Agency [DTRA]) with the census in these regions. Keyhole Markup Language (KMP)-based boundary location files for counties and subdivisions from the US census bureau website were downloaded in XML format. Census boundaries that overlap radiation contour plumes were obtained by transforming the polygons of each boundary to Google Maps-encoded polygons and creating javascript (‘plume-census.js’) that draws all the polygons in a map in HTML. The US Census API (
api.census.gov/data/2016/pep/population; Application Program Interface; accessed on 23 February 2018) was interrogated with the intersecting subdivisions to estimate populations residing in the plume and the sum of all populations within a contour estimates the affected population exceeding a particular exposure level (e.g., >3, >2, >1 Gy). Applying this strategy to several large US cities, which are designated as “incorporated places”, had limited success as the populations of these regions are not directly accessible through the API.
2.5. Sample Set Creation
The performance of ADCI-HT was evaluated by creating and processing replicate sample sets programmatically, comprised of fixed and variable numbers of distinct metaphase images and complete samples. While the scheduler is capable of handling samples of different sizes, sets of synthetic samples consisting of 500 images each that were randomly selected from a larger pool of images exposed to the same radiation dose to estimate average processing speed per sample. This fulfills one of the minimum criteria for dose estimation using the DCA [
28]. To ensure that all images were processed, duplicate samples were also created by splitting consecutive subsets of 500 images into new samples. Residual subsets with fewer than 500 images were added to the last sample. Finally, samples of intermediate exposure levels were constructed by mixing sample pairs at different exposure levels from the same laboratory.
A large multi-laboratory exercise (40,000 samples) was simulated by duplicating 10 different exercise samples from five dosimetry laboratories in equal proportions (Canadian Nuclear Laboratories [CNL]: S02 (3.1 Gy), S05 (4.0 Gy), S09 (2.3 Gy); Health Canada [HC]: S01 (3.1 Gy), S05 (2.8 Gy), S07 (3.4 Gy), S08 (2.3 Gy); Radiation Protection Center, Lithuania [LT]: - (0.4 Gy); Public Health England [PHE]: B (4 Gy); Dalat Nuclear Research Institute, Vietnam [VN]: - (2.3 Gy)). These duplicates were dynamically split into sample sets based on the criterion that each set contains at most 25,000 metaphase images. Dose estimation for samples was performed using the ADCI-derived biodosimetry curve corresponding to the respective laboratory from which the samples were derived. For each run, the number of images processed in a sample set is related to the amount of available computer memory, as the output for processing all images is maintained in random access memory until the batch process is completed.
2.6. ADCI-HT Resource Allocation
In general, the CPU resources allocated were limited to 4 node boards by the default job queuing system. A node board contains 32 nodes and a node is a “system-on-a-chip” compute node containing a 16 core 1.6 GHz PowerPC A2 CPU and 16 Gb of RAM. Priority scheduling was approved by system administrators to determine performance in a machine-optimized environment. Higher priority runs maximized the number of processors that could be simultaneously allocated in the supercomputer (though processes could still be delayed due to the BG/Q queuing system). These included 1 and 4 node board runs of sample sets (100 runs with 400 samples each), with each node board containing 512 cores (4 threads per core).
2.7. Comparison with Other Systems
The performance of the high performance, ADCI-HT interpretation of sets of metaphase images was compared to fully automated (ADCI-Windows) and semi-automated analyses (DCScore [Metasystems]) on single computer systems. The DCScore estimates were based on Romm et al. 2013 [
22], with a modification to include a missing processing step of performing thumbnail gallery review and selection of 500 optimal images. The time to perform this step was determined for 3 samples, averaged, and added to the time reported for the other review and processing steps.
3. Results
This study attempts to determine the volume and capacity required to interpret biodosimetry tests by simulating provision of biodosimetry results for affected populations in high yield nuclear incidents. We used ADCI to carry out this task, which has been demonstrated to provide accurate dose estimates for samples of unknown exposure, based on IAEA compliant triage criteria. ADCI estimates radiation exposures using a fully automated process based on a calibration curve derived from the same biodosimetry laboratory [
23,
24,
25]. The Windows-based Desktop version of ADCI was migrated to the PowerPC operating system of IBM BlueGene/Q (BGQ) as ADCI-HT, which significantly improved the throughput of these analyses.
Initially, we determined the actual clock time (ACT) to perform analyses of datasets containing multiple samples, with each consisting of 500 randomly-selected metaphase images. To gain perspective, we processed 50 samples of 500 images each from CNL on both the Desktop and BGQ versions of ADCI. The processing time on Windows was 11 h 10 m, while on BGQ, 30 min 52 s was required, which is equivalent to ADCI-HT being 21.7-fold faster than the Desktop version.
We also created and processed parallel sets of 120 samples, comprising 15,970 images with ADCI-HT, by randomizing and splitting original calibration and exercise samples from four different laboratories. This involved submission of 54 computer runs of sample sets containing 120 samples, with each requesting one processing block (or 1024 CPUs). With dedicated or high priority access to ADCI-HT and all runs ending successfully, the ACT would be equal to the maximum processing time of each run in parallel. In fact, the ACT for all of these jobs ranged from 38 min 37 s to 1 h 18 min 2 s, with an average of 1 h 18 s. As these jobs were submitted to the general queue (which is shared between all users of the system), the latency or wait for available CPU resources was variable, with the slowest set of samples to finish requiring 6 h 6 m 46 s. The largest test involved 164 sets of samples, each set consisting of 100 samples (16,400 samples total), with each sample set submitted as a separate run. The maximum ACT to process one of these jobs was 1 h 09 min 58 s, while the minimum was 30 min 24 s, averaging 45 min 45 s per run. The cumulative processing time without parallelization was 125 h 2 min 20 s. We also compared these results with those from larger sized sample sets allocated with proportionally increased CPU resources. When requesting an allocation of four processing blocks (or 4096 total CPUs; use of added resources required special permission), no discernable difference in ACTs was observed (maximum and minimum ACT per run were, respectively, 1 h 5 min 52 s and 34 min 9 s, with an average time of 48 min 48 s). This was primarily due added queuing times when requesting more resources. Constraints on hardware architecture meant that allocation of proportionately increased CPU resources did not confer any advantage for processing larger sample sets.
3.1. Benchmark of Population-Scale Nuclear Incident Simulations
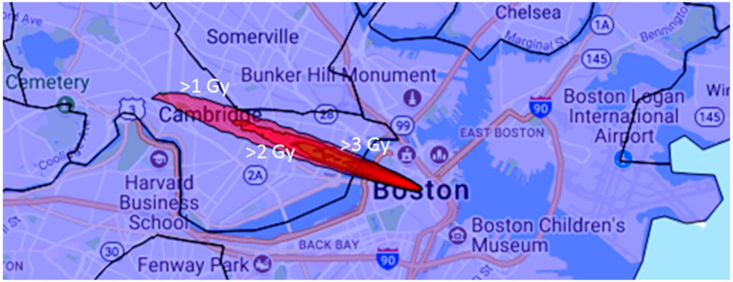
HPAC-derived plumes were generated across 15 populated regions within the United States. The HPAC plume is represented as a series of topological contours (ovals) representing various levels of radiation (ranging from 1.0 (the outermost ring) to 7.0 Gy (the innermost ring)).
Figure 3 shows the radiation plume derived for the Boston scenario. Using U.S. sub-division boundary files and census information, we computed the overall population expected to overlap the >1 and >2 Gy contours of the HPAC plume (
Table 1; column 2 and 3, respectively). We then used sample data to simulate ADCI-HT analysis of all persons expected to obtain >2 Gy exposure (500 images per patient). With four nodes with 16,384 total cores, the overall processing time ranged from 0.6 to 7.4 days (Burlington VT and Boston MA, respectively;
Table 1) depending on population density.
3.2. Performance of ADCI-HT on Sample Sets from Multiple Laboratories
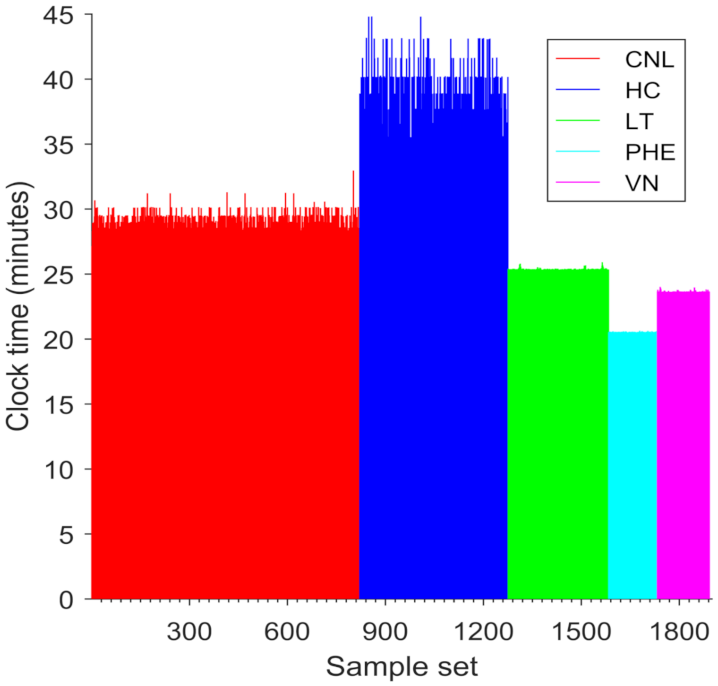
The volume of samples in a large-scale nuclear incident would likely exceed the capacity of any individual biodosimetry laboratory to process. We simulated processing and dose estimation of 40,000 samples (1892 sample sets) by five laboratories, each of which has a distinct dose calibration curve. ADCI-HT took 25 h 8 m 5 s to finish processing the simulated samples, i.e., each duplicated from the same set of metaphase images. These cumulative set of samples contained 46,196,000 metaphase cell images in total and were processed with 37,888 CPUs on average (
Figure 4). The respective durations for processing of the sample sets from the different laboratories (CNL: 14 h 6 m 22 s; HC: 6 h 27 m 22 s; LT: 3 h 16 m 21 s; PHE: 1 h 28 m 40 s; VN: 1 h 44 m 2 s) were proportionate to the total image counts in each set of samples. The dose estimates for individual samples produced by the ADCI-HT version were identical to those produced by the Desktop version on all these samples (not shown), except those from HC. The deviation is due to differences between the order of images selected by the ‘Group Bin Distance’ filter used in the HC dataset (which selected the top 250 images; [
23]), where multiple images had the same rank, resulting in the insignificant discrepancies in the dose estimates generated by the Desktop and HT versions of ADCI.
3.3. Comparison of Speed of ADCI-HT with Other Systems
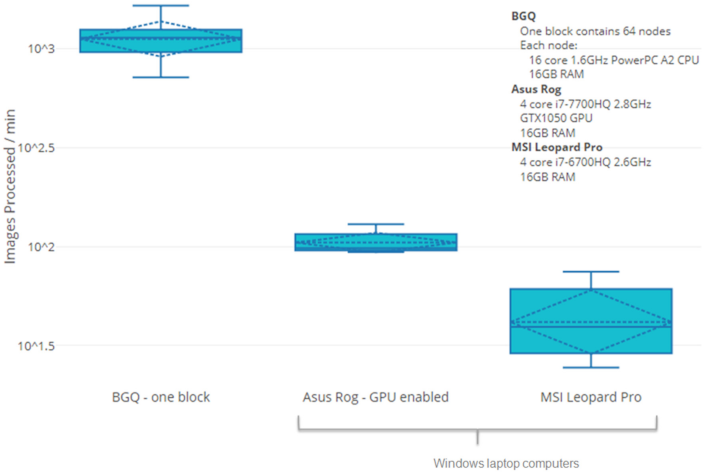
DC identification, the bottleneck for interpretation of DCA results requires substantially less time with ADCI-HQ and ADCI-Windows than with an alternative software product, DCScore (MetaSystems). This is because ADCI does not require either manual curation of images or user confirmation of candidate DCs in order to accurately determine radiation exposures. To determine whether ADCI-HT could provide timely dose estimates with available cytogenetic data for a moderate sized population of potentially exposed individuals, we compared the processing requirements for samples by ADCI-Windows and HT versions with DCScore. Results for individual samples were extrapolated to a population consisting of 1000 samples (
Table 2). The differences in time to process a single sample were negligible between these platforms. Only the HT version was able to estimate exposures with sufficient speed for population-scale triage of all patients to make effective, informed decisions on whether to treat individuals. However, multiple instances of ADCI-Windows running on separate laptop computers (
n = 2) would be required to fulfill assessment of a population of this size in 1 day (
Figure 5). DCScore is unsuitable for this task [
31], as it would require nearly a month to analyze a this population, due to the requirements of manual review and the selection of metaphase cells in each sample and confirmation of candidate dicentric chromosomes.
4. Discussion
ADCI-HT has the capacity to carry out population-scale analysis of cytogenetic biodosimetry data from the DCA after a large radiation event. This capability will be essential for triaging those patients with symptoms of acute radiation syndrome for treatment. With a distributed testing model, it would be feasible to generate different sets of metaphase image samples in multiple laboratories, each customized to use their own image selection models. It is unlikely that all of the images would be generated at a single location based on the capacity limitations of individual laboratories. More likely, a network consisting of multiple commercial and academic laboratories would distribute and process the samples, then the results for each sample would be combined into multiple sample archives from the same laboratory and calibration samples would be assembled into sample sets. These would be uploaded from different laboratories to a federated cloud storage repository, where it would be accessed by the compute cluster running ADCI-HT. ADCI-HT dearchives the sample sets in random access memory and processes the samples. There, processing of each sample set would first generate an image selection-optimized, laboratory-specific calibration curve and then would evaluate doses for all samples of unknown exposure from that laboratory using their curve. We suggest that leveraging existing laboratory infrastructure and supplanting limitations in any of these elements with existing automation at large commercial cytogenetic laboratories may be able to fulfill future testing demands for biodosimetry testing in a population scale nuclear event or accident.
Rapid triaging using fewer metaphase images or other assays (micronuclei, biomarkers in urine, miRNA, or gene expression) have been proposed as alternatives to the DCA, however these approaches may not be sufficiently accurate to robustly distinguish individuals eligible for treatment (>2 Gy exposures; [
32]). Rapid bioassays have been developed for triage evaluation of potentially exposed populations; however specific frameworks have not been demonstrated by large scale implementation of these tests. Throughput estimates have largely been theoretical and based on the performance of the assays themselves, extrapolated to large populations. The details regarding equipment capacity, availability of sufficient quantities of critical reagents and trained personnel to perform and interpret these assays have not been addressed in the Concept of Operations [
33].
HPAC provides the distribution of the physical dose in a certain region. However, the physical doses may not be identical to the biological doses but may, in some instances, be correlated [
34]. Spectral clustering [
35,
36,
37] and geostatistics [
38] can be used to determine the minimum number of data points (e.g., locations in the map) that are needed to construct similar distribution. However, the complexity of the calculations needed for spectral clustering become increasingly prohibitive with large datasets. Our efforts have turned to geostatistical estimation of radiation exposures.
ADCI-HT could be an instrumental resource in the rapid identification of patients requiring treatment in population-scale nuclear incidents. Depending on population-density, ADCI-HT software can identify DCs and estimate dose of a population within an irradiated zone in 0.6–7.4 days (
Table 1;
Figure 3). ADCI-HT by itself, however, only accelerates image processing and DC identification. The acquisition of said samples, the preparation of metaphase cells and the capture of DC images makes the testing of entire populations logistically impossible. Sampling and testing requirements can be reduced via geostatistical sampling [
39]; a method of estimating the spatial boundaries of a region using a small subset of samples at various locations. Applying such methods can limit the number of patients requiring testing while reducing sampling time required for first responders, expediting identification of those requiring treatment in scenarios where time and resources are limited.
Despite this significant reduction in processing time using ADCI software, there are still potential improvements that may increase the feasibility of implementing this software as a routine biodosimetry laboratory resource. Although ADCI-HT was benchmarked as faster than Desktop ADCI, access to supercomputers may not always be feasible, and therefore increasing the rate of analysis via conventional ADCI is still crucial. We have recently implemented GPU acceleration on the Desktop platform [
40]. Preliminary results showed a ~8× increase in sample processing speed, which may not be as fast as ADCI-HT but may be adequate in some population-scale scenarios. Since most environmental radiation exposures are inhomogeneous, we have implemented the contaminated Poisson method to calculate the mean DC frequency within the irradiated fraction of partially irradiated samples [
41].
We have presented a high-throughput implementation of ADCI, which is designed to rapidly process metaphase cell images and assess radiation exposures of populations of individuals. This increased throughput might provide sufficient statistical power to stratify clinical exposures based on underlying properties of the testing protocol or the populations themselves. Previous laboratory intercomparisons have suggested that differences in dose estimates may be associated with laboratory source, transportation delay, cell quality, radiation type, age, or gender [
42,
43]. The dose estimate results are concordant with those generated by the Desktop version of this software using intercomparison exercise sample data from five different biodosimetry laboratories. This level of dedicated computing resources available for the 40,000-sample run made a significant difference in obtaining timely dose estimates for the entire population, as a single processing node would have been insufficient (i.e., 37 days of ACT) for timely estimation of these exposures. ADCI-HT therefore achieves adequate performance to deliver timely and actionable dose estimates in a potential mass casualty radiation event.
Supplementary Materials
The following are available online at
https://www.mdpi.com/article/10.3390/radiation1020008/s1, Figure S1: Adapting ADCI Windows Desktop Graphical User Interface to ADCI-HT. Data flow diagram illustrating the steps required to perform metaphase image processing tasks using: A) MS-Windows ADCI, and B) IBM BG/Q ADCI-HT software platforms.
Author Contributions
Conceptualization (P.K.R.), Data curation (P.K.R., B.C.S., Y.L.), Formal analysis (P.K.R., E.J.M., B.C.S.), Funding (P.K.R., E.W., J.H.M.K.), Investigation (P.K.R., J.H.M.K.), Methodology (P.K.R., E.J.M., B.C.S.), Project administration (P.K.R., E.W., J.H.M.K.), Resources (R.C.W., F.N., O.S., N.-D.P.), Software (B.C.S., Y.L.), Supervision (P.K.R., E.W., J.H.M.K.), Validation (E.J.M.), Visualization (E.J.M., B.C.S.), Writing (P.K.R., E.J.M., J.H.M.K.). All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful to the SOSCIP Consortium (J.H.M.K., P.K.R.), Natural Sciences and Engineering Research Council of Canada (Engage Program; E.W., P.K.R.), Ontario Centres of Excellence (Talent Edge Postdoctoral Fellowship Program; J.H.M.K., P.K.R.), and CytoGnomix (P.K.R., J.H.M.K.) for support of this project. Certain data used in this study were obtained as part of Coordinated Research Project E35010: Applications of Biological Dosimetry Methods in Radiation Oncology, Nuclear Medicine, and Diagnostic and Interventional Radiology (MEDBIODOSE), which was carried out under the sponsorship of the International Atomic Energy Agency.
Institutional Review Board Statement
Ethical review and approval were waived for this study due to minimal risk to subjects. Data were obtained from anonymized samples that were exposed to radiation ex vivo prior to cytogenetic analysis.
Informed Consent Statement
Patient consent was waived due to due to minimal risk to subjects. Data were obtained from anonymized samples that were exposed to radiation ex vivo prior to cytogenetic analysis.
Data Availability Statement
Source data available from the authors whose laboratories generated these images (Ruth Wilkins, Farrah Norton, Olga Sevriukova, Ngoc Duy Pham).
Acknowledgments
The authors acknowledge Ruipeng Lu, Shaimaa Ali, Ryan Cooke, Trent Peerlaproulx, Jayne Moquet and Elizabeth Ainsbury for their contributions at the early stage of this work.
Conflicts of Interest
Ben C. Shirley and Yanxin Li are employees and Peter K. Rogan and Joan H.M. Knoll are cofounders of CytoGnomix Inc. The company developed commercial software which incorporates the methods presented.
References
- Lloyd, D.C.; Purrott, R.J.; Reeder, E.J. The incidence of unstable chromosome aberrations in peripheral blood lymphocytes from unirradiated and occupationally exposed people. Mutat. Res. Mol. Mech. Mutagen. 1980, 72, 523–532. [Google Scholar] [CrossRef]
- Nugis, V.Y.; Filushkin, I.V.; Chistopolskij, A.S. Retrospective dose estimation using the dicentric distribution in human peripheral lymphocytes. Appl. Radiat. Isot. 2000, 52, 1139–1144. [Google Scholar] [CrossRef]
- Sevan’kaev, A.V.; Khvostunov, I.K.; Mikhailova, G.F.; Golub, E.V.; Potetnya, O.I.; Shepel, N.N.; Nugis, V.Y.; Nadejina, N.M. Novel data set for retrospective biodosimetry using both conventional and FISH chromosome analysis after high accidental overexposure. Appl. Radiat. Isot. 2000, 52, 1149–1152. [Google Scholar] [CrossRef]
- Sevan’kaev, A.V.; Lloyd, D.C.; Edwards, A.A.; Khvostunov, I.K.; Mikhailova, G.F.; Golub, E.V.; Shepel, N.N.; Nadejina, N.M.; Galstian, I.A.; Nugis, V.Y.; et al. A cytogenetic follow-up of some highly irradiated victims of the Chernobyl accident. Radiat. Prot. Dosim. 2005, 113, 152–161. [Google Scholar] [CrossRef]
- Natarajan, A.T.; Ramalho, A.T.; Vyas, R.C.; Bernini, L.F.; Tates, A.D.; Ploem, J.S.; Nascimento, A.C.; Curado, M.P. Goiania radiation accident: Results of initial dose estimation and follow up studies. Prog. Clin. Biol. Res. 1991, 372, 145–153. [Google Scholar]
- Natarajan, A.T.; Vyas, R.C.; Wiegant, J.; Curado, M.P. A cytogenetic follow-up study of the victims of a radiation accident in Goiania (Brazil). Mutat. Res. Mol. Mech. Mutagen. 1991, 247, 103–111. [Google Scholar] [CrossRef]
- Ramalho, A.T.; Nascimento, A.C.; Littlefield, L.G.; Natarajan, A.T.; Sasaki, M.S. Frequency of chromosomal aberrations in a subject accidentally exposed to 137Cs in the Goiania (Brazil) radiation accident: Intercomparison among four laboratories. Mutat. Res. Mutagen. Relat. Subj. 1991, 252, 157–160. [Google Scholar] [CrossRef]
- Lee, J.K.; Han, E.A.; Lee, S.S.; Ha, W.H.; Barquinero, J.F.; Lee, H.R.; Cho, M.S. Cytogenetic biodosimetry for Fukushima travelers after the nuclear power plant accident: No evidence of enhanced yield of dicentrics. J. Radiat. Res. 2012, 53, 876–881. [Google Scholar] [CrossRef]
- Suto, Y. Review of Cytogenetic analysis of restoration workers for Fukushima Daiichi nuclear power station accident. Radiat. Prot. Dosim. 2016, 171, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Suto, Y.; Hirai, M.; Akiyama, M.; Kobashi, G.; Itokawa, M.; Akashi, M.; Sugiura, N. Biodosimetry of restoration workers for the Tokyo Electric Power Company (TEPCO) Fukushima Daiichi nuclear power station accident. Health Phys. 2013, 105, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Moquet, J.; Barnard, S.; Rothkamm, K. Gamma-H2AX biodosimetry for use in large scale radiation incidents: Comparison of a rapid ’96 well lyse/fix’ protocol with a routine method. Peer J. 2014, 2, e282. [Google Scholar] [CrossRef]
- Balajee, A.S.; Smith, T.; Ryan, T.; Escalona, M.; Dainiak, N. DEVELOPMENT OF A MINIATURIZED VERSION OF DICENTRIC CHROMOSOME ASSAY TOOL FOR RADIOLOGICAL TRIAGE. Radiat. Prot. Dosim. 2018, 182, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Romm, H.; Wilkins, R.C.; Coleman, C.N.; Lillis-Hearne, P.K.; Pellmar, T.C.; Livingston, G.K.; Awa, A.A.; Jenkins, M.S.; Yoshida, M.A.; Oestreicher, U.; et al. Biological dosimetry by the triage dicentric chromosome assay: Potential implications for treatment of acute radiation syndrome in radiological mass casualties. Radiat Res. 2011, 175, 397–404. [Google Scholar] [CrossRef]
- Flegal, F.N.; Devantier, Y.; Marro, L.; Wilkins, R.C. Validation of QuickScan dicentric chromosome analysis for high throughput radiation biological dosimetry. Health Phys. 2012, 102, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lue, S.W.; Repin, M.; Mahnke, R.; Brenner, D.J. Development of a High-Throughput and Miniaturized Cytokinesis-Block Micronucleus Assay for Use as a Biological Dosimetry Population Triage Tool. Radiat. Res. 2015, 184, 134–142. [Google Scholar] [CrossRef][Green Version]
- De Amicis, A.; De Sanctis, S.; Di Cristofaro, S.; Franchini, V.; Regalbuto, E.; Mammana, G.; Lista, F. Dose estimation using dicentric chromosome assay and cytokinesis block micronucleus assay: Comparison between manual and automated scoring in triage mode. Health Phys. 2014, 106, 787–797. [Google Scholar] [CrossRef][Green Version]
- Oestreicher, U.; Samaga, D.; Ainsbury, E.; Antunes, A.C.; Baeyens, A.; Barrios, L.; Beinke, C.; Beukes, P.; Blakely, W.F.; Cucu, A.; et al. RENEB intercomparisons applying the conventional Dicentric Chromosome Assay (DCA). Int. J. Radiat. Biol. 2017, 93, 20–29. [Google Scholar] [CrossRef]
- Wilkins, R.C.; Romm, H.; Oestreicher, U.; Marro, L.; Yoshida, M.A.; Suto, Y.; Prasanna, P.G. Biological Dosimetry by the Triage Dicentric Chromosome Assay—Further validation of International Networking. Radiat. Meas. 2011, 46, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, M.; Barquinero, J.F.; Vallerga, M.B.; Radl, A.; Taja, M.R.; Seoane, A.; De Luca, J.; Oliveira, M.S.; Valdivia, P.; Lima, O.G.; et al. Biological dosimetry intercomparison exercise: An evaluation of triage and routine mode results by robust methods. Radiat. Res. 2011, 175, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Ainsbury, E.A.; Livingston, G.K.; Abbott, M.G.; Moquet, J.E.; Hone, P.A.; Jenkins, M.S.; Christensen, D.M.; Lloyd, D.C.; Rothkamm, K. Interlaboratory variation in scoring dicentric chromosomes in a case of partial-body x-ray exposure: Implications for biodosimetry networking and cytogenetic “triage mode” scoring. Radiat. Res. 2009, 172, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Beinke, C.; Barnard, S.; Boulay-Greene, H.; De Amicis, A.; De Sanctis, S.; Herodin, F.; Jones, A.; Kulka, U.; Lista, F.; Lloyd, D.; et al. Laboratory intercomparison of the dicentric chromosome analysis assay. Radiat. Res. 2013, 180, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Maznyk, N.A.; Wilkins, R.C.; Carr, Z.; Lloyd, D.C. The capacity, capabilities and needs of the WHO BioDoseNet member laboratories. Radiat. Prot. Dosim. 2012, 151, 611–620. [Google Scholar] [CrossRef]
- Rogan, P.K.; Li, Y.; Wilkins, R.C.; Flegal, F.N.; Knoll, J.H. Radiation Dose Estimation by Automated Cytogenetic Biodosimetry. Radiat. Prot. Dosim. 2016, 172, 207–217. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Wilkins, R.; Flegal, F.; Knoll, J.H.M.; Rogan, P.K. Accurate cytogenetic biodosimetry through automated dicentric chromosome curation and metaphase cell selection. F1000 Res. 2017, 6, 1396. [Google Scholar] [CrossRef] [PubMed]
- Rogan, P.K.; Li, Y.; Wickramasinghe, A.; Subasinghe, A.; Caminsky, N.; Khan, W.; Samarabandu, J.; Wilkins, R.; Flegal, F.; Knoll, J.H. Automating dicentric chromosome detection from cytogenetic biodosimetry data. Radiat. Prot. Dosim. 2014, 159, 95–104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Y.; Wickramasinghe, A.; Subasinghe, A.; Samarabandu, J.; Knoll, J.H.M.; Wilkins, R.; Flegal, F.; Rogan, P.K. Towards large scale automated interpretation of cytogenetic biodosimetry data. In Proceedings of the 2012 IEEE 6th International Conference on Information and Automation for Sustainability, Beijing, China, 27–29 September 2012; pp. 30–35. [Google Scholar]
- Li, Y.; Knoll, J.H.; Wilkins, R.C.; Flegal, F.N.; Rogan, P.K. Automated discrimination of dicentric and monocentric chromosomes by machine learning-based image processing. Microsc. Res. Tech. 2016, 79, 393–402. [Google Scholar] [CrossRef]
- International Atomic Energy Agency; Pan American Health Organization; World Health Organization. Cytogenetic Dosimetry: Applications in Preparedness for and Response to Radiation Emergencies; International Atomic Energy Agency: Vienna, Austria, 2011. [Google Scholar]
- Xu, C.; Prince, J.L. Snakes, shapes, and gradient vector flow. IEEE Trans. Image Process. 1998, 7, 359–369. [Google Scholar] [PubMed]
- Arachchige, A.S.; Samarabandu, J.; Knoll, J.H.; Rogan, P.K. Intensity integrated Laplacian-based thickness measurement for detecting human metaphase chromosome centromere location. IEEE Trans. Biomed. Eng. 2013, 60, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Romm, H.; Ainsbury, E.; Barnard, S.; Barrios, L.; Barquinero, J.F.; Beinke, C.; Deperas, M.; Gregoire, E.; Koivistoinen, A.; Lindholm, C.; et al. Automatic scoring of dicentric chromosomes as a tool in large scale radiation accidents. Mutat. Res. Toxicol. Environ. Mutagen. 2013, 756, 174–183. [Google Scholar] [CrossRef]
- Ainsbury, E.A.; Higueras, M.; Puig, P.; Einbeck, J.; Samaga, D.; Barquinero, J.F.; Barrios, L.; Brzozowska, B.; Fattibene, P.; Gregoire, E.; et al. Uncertainty of fast biological radiation dose assessment for emergency response scenarios. Int. J. Radiat. Biol. 2017, 93, 127–135. [Google Scholar] [CrossRef]
- Dainiak, N.; Albanese, J.; Kaushik, M.; Balajee, A.S.; Romanyukha, A.; Sharp, T.J.; Blakely, W.F. CONCEPTS OF OPERATIONS FOR A US DOSIMETRY AND BIODOSIMETRY NETWORK. Radiat. Prot. Dosim. 2019, 186, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.L.; Bailey, S.M.; Beck, H.L.; Boice, J.D.; Bouville, A.; Brill, A.B.; Cornforth, M.N.; Inskip, P.D.; McKenna, M.J.; Mumma, M.T.; et al. Estimation of Radiation Doses to U.S. Military Test Participants from Nuclear Testing: A Comparison of Historical Film-Badge Measurements, Dose Reconstruction and Retrospective Biodosimetry. Radiat. Res. 2019, 191, 297–310. [Google Scholar] [CrossRef]
- Shi, J.; Malik, J. Normalized cuts and image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2000, 22, 888–905. [Google Scholar]
- Wang, L.; Dong, M.; Kotov, A. Multi-level Approximate Spectral Clustering. In Proceedings of the IEEE International Conference on Data Mining, Atlantic City, NJ, USA, 14–17 November 2015; pp. 439–448. [Google Scholar]
- Dhillon, I.S.; Guan, Y.; Kulis, B. Weighted Graph Cuts without eigenvectors a Multilevel Approach. IEEE Trans. Pattern Anal. Mach. Intell. 2007, 29, 1944–1957. [Google Scholar] [CrossRef] [PubMed]
- Leuangthong, O.; Khan, K.D.; Deutsch, C.V. Solved Problems in Geostatistics; Wiley-Interscience: Hoboken, NJ, USA, 2008; pp. 85–101. [Google Scholar]
- Rogan, P.K.; Mucaki, E.J.; Lu, R.; Shirley, B.C.; Waller, E.; Knoll, J.H.M. Meeting radiation dosimetry capacity requirements of population-scale exposures by geostatistical sampling. PLoS ONE 2020, 15, e0232008. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shirley, B.C.; Wilkins, R.C.; Norton, F.; Knoll, J.H.M.; Rogan, P.K. RADIATION DOSE ESTIMATION BY COMPLETELY AUTOMATED INTERPRETATION OF THE DICENTRIC CHROMOSOME ASSAY. Radiat. Prot. Dosim. 2019, 186, 42–47. [Google Scholar] [CrossRef]
- Shirley, B.C.; Knoll, J.H.M.; Moquet, J.; Ainsbury, E.; Pham, N.D.; Norton, F.; Wilkins, R.C.; Rogan, P.K. Estimating partial-body ionizing radiation exposure by automated cytogenetic biodosimetry. Int. J. Radiat. Biol. 2020, 96, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.; Buard, V.; Delbos, M.; Durand, V.; Paillole, N.; Grégoire, E.; Voisin, P. International intercomparison for criticality dosimetry: The case of biological dosimetry. Radiat. Prot. Dosim. 2004, 110, 471–476–476. [Google Scholar]
- Wilkins, R.C.; Romm, H.; Kao, T.-C.; Awa, A.A.; Yoshida, M.A.; Livingston, G.K.; Jenkins, M.S.; Oestreicher, U.; Pellmar, T.C.; Prasanna, P.G.S. Interlaboratory Comparison of the Dicentric Chromosome Assay for Radiation Biodosimetry in Mass Casualty Events. Radiat. Res. 2008, 169, 551–560. [Google Scholar]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}