
Efficacy of Intravenous Immunoglobulins and Other Immunotherapies in Neurological Disorders and Immunological Mechanisms Involved

 , ,
, ,
Abstract

1. Introduction
Therapeutic and Diagnostic Considerations
- Immune modulation: Therapies that reduce pathogenic autoantibodies (high-dose corticosteroids, B-cell depletion with rituximab or BAFF inhibitors) or remove them (plasmapheresis, IVIg) are mainstays for CNS autoimmunity.
- Glutamate antagonists: NMDA/AMPA receptor blockers (e.g., memantine or experimental AMPA antagonists) may confer neuroprotection; indeed, NBQX treatment reduced demyelination in an MS model [3].
- Antibody screening: Serologic or CSF testing for anti-NMDA and anti-AMPA receptor autoantibodies can aid diagnosis and identify patients for targeted therapy.
2. Methods
2.1. The Biological Basis of Some Neuropsychiatric Diseases
2.2. Review of Neurological Immune-Related Adverse Events (n-irAEs)
2.3. Cytokine Involvement in Neurological and Psychiatric Diseases Such as Guillain–Barré Syndrome and Schizophrenia
3. Immunotherapy Used in Neurological Disorders
3.1. The Use of Intravenous Immunoglobulin (IVIG) in Treating Neurological Disorders
3.2. Abstract Discussion
3.3. How IVIG Works
3.4. Conditions Treated with IVIG
3.5. Conditions Where IVIG Is Not Recommended
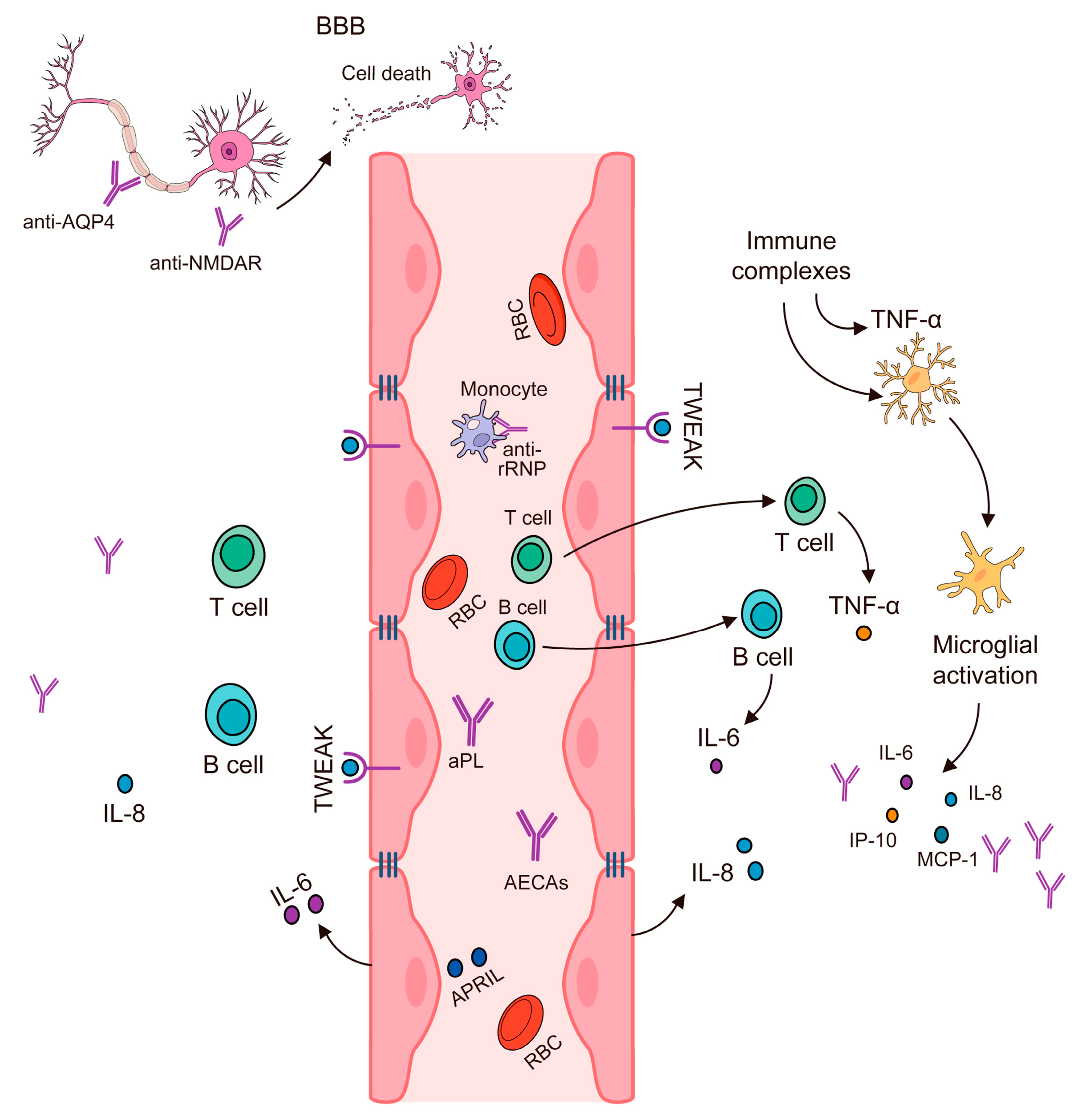
4. Disruption of the Blood–Brain Barrier
5. Cytokines, Antibodies, and Autoimmune Inflammation
6. New Approaches in the Treatment of Neurological Disorders
7. Control Randomized Trials in Neurological Diseases
8. Limitations of IVIG Therapy in Autoimmune Neurological Diseases
8.1. Efficacy and Therapeutic Limitations in Specific Diseases
8.1.1. Multiple Sclerosis (MS)
8.1.2. Neuropsychiatric Systemic Lupus Erythematosus (NPS.LE)
8.1.3. Autoimmune Encephalitis (And Limbic Autoimmune Epilepsy)
8.1.4. Autoimmune “Limbic” Epilepsy (LGI1/CASPR2 Encephalitis) Deserves Special Mention
8.1.5. Myasthenia Gravis (MG)
8.1.6. Guillain–Barré Syndrome (GBS)
8.2. Safety and Practical Constraints of IVIG Therapy
8.2.1. Adverse Effects and Safety Concerns
8.2.2. Cost and Availability Challenges
8.2.3. Patient-Specific and Logistic Factors
8.3. Methodological Limitations in IVIG Research
- Scarcity of High-Quality Trials: For many autoimmune neurological conditions, there is a lack of large RCTs evaluating IVIG. Ethically and logistically, conducting placebo-controlled trials can be difficult in life-threatening or rare diseases, so the evidence often comes from lower levels. In NPSLE, for example, trials are “scarce and most of the data are extracted from case series and case reports”, with virtually no RCTs to guide therapy [129]. Similarly, in autoimmune encephalitis, no randomized controlled treatment trials have been available up to recent years [123]—the field has relied on observational studies and expert consensus. The first-ever RCT in autoimmune epilepsy (LGI1/CASPR2 encephalitis) had only 17 patients, illustrating how rare such trials have been [124]. The absence of robust trials means that many purported benefits of IVIG (or lack thereof) rest on uncontrolled observations that are prone to bias. Publication bias is a concern: positive case reports are more likely to be published than negative ones, potentially overstating IVIG efficacy in the literature. Without controlled comparisons, it is hard to determine how much improvement in a given study was due to IVIG versus the natural disease course or concurrent treatments. This limitation is widely acknowledged, and experts consistently call for more rigorous studies—for instance, a review in NPSLE explicitly concludes that “future RCTs are needed” to establish the efficacy, optimal dose, and duration of IVIG [129]. Until more trials are performed, the confidence in IVIG’s effectiveness for many indications remains limited by the quality of evidence.
- Small Sample Sizes and Power: Even when RCTs or controlled studies have been conducted, they often involve small sample sizes, reducing statistical power. Many trials in rare neuroimmunological conditions have been underpowered to detect anything but very large effects. For example, several MG trials had between 12 and 84 participants and were unable to detect moderate differences between IVIG and comparators, partly due to limited enrolment [125]. Landmark trials in MS with IVIG were relatively small (on the order of a few dozen patients per arm), and meta-analyses had to combine data from just a handful of studies [117]. The IVIG in postpartum MS meta-analysis included only 380 total treated patients across all studies [119]—a modest number considering the question spanned multiple trials. Small studies increase the margin of error and make results less reliable (a single outlier patient can sway outcomes). They also often cannot rigorously assess subgroups or rare adverse events. Null results in underpowered studies cannot be taken as proof of no effect—for instance, a trial of 15 MG patients found no difference between IVIG and placebo at 6 weeks [125], but such a small study could easily miss a true benefit. Thus, the literature may contain false negatives (or false positives) due to sample size limitations. Combining data via systematic reviews helps but can be hampered by study heterogeneity. Overall, the evidence base for IVIG in many of these disorders is built on relatively small cohorts, and this is a key methodological shortcoming.
- Heterogeneity and Inconsistent Outcome Measures: Another challenge is the inconsistency in outcome measures and study designs across published studies. Different trials often use different endpoints to define “response” to IVIG. For example, in MS some studies focused on annual relapse rate, others on MRI lesions, and others on disability progression—outcomes that do not always align, leading to mixed conclusions (IVIG appeared to reduce relapses in RRMS but showed no effect on disability in SPMS) [117]. In NPSLE and autoimmune encephalitis case series, outcomes are typically reported in subjective terms (“improved” vs. “not improved”), without standardized scales. An evidence review by NHS England highlighted that most studies in autoimmune encephalitis did not include a precise definition of patient outcomes, and only a few used a common scale like the modified Rankin Scale [123]. This lack of uniform outcome metrics makes it hard to compare results across studies or perform meta-analyses. Additionally, non-standardized treatment protocols contribute to heterogeneity. Dosing of IVIG (e.g., 0.4 g/kg for 5 days vs. 2 g/kg over 2 days) and timing relative to disease onset vary between studies. According to the same NHS review, “a standardised protocol for the use of IVIG was lacking” in the literature, with many studies not detailing the sequence of therapies used [123]. Some patients received IVIG as first-line, others after steroids or plasma exchange failure, etc., introducing variability. Such heterogeneity in methodologies and patient populations (different disease severities, diagnostic criteria, concomitant treatments) is a serious limitation—it precludes definitive conclusions about efficacy and makes it difficult to generalize results. What works in one context (say, IVIG after steroid failure in one series) might not in another, yet the data are often pooled together. Consistency in study design is improving somewhat (for instance, MG trials now often use the Quantitative Myasthenia Gravis score as a standard outcome), but for many autoimmune neurologic diseases, the literature remains a patchwork of disparate reports. In summary, the lack of standard outcomes and the heterogeneity of study conditions undermine the strength of evidence regarding IVIG.
- Biases and Confounding in Observational Studies: Given the reliance on case series and open-label studies, methodological biases are a major concern. Many reports are retrospective, meaning they rely on chart review and are subject to selection bias (e.g., a clinician might publish on the 5 patients who responded to IVIG, while not reporting on 5 others who did not). Confounding by indication is another issue: sicker patients are more likely to receive treatments like IVIG (especially second-line), which can skew outcomes. As an example, one analysis of autoimmune encephalitis noted that patients who required second-line therapies tended to have worse initial severity, complicating any comparison of those who received only first-line (IVIG/steroids) vs. those who escalated to rituximab [123]. Without randomization, it is difficult to disentangle whether IVIG was truly ineffective in those severe cases or whether their poor outcomes were due to the disease’s aggressiveness. Lack of blinding in open-label studies can also inflate perceived benefits due to placebo effect or observer bias. Some MG studies that were unblinded reported subjective improvement with IVIG, but when tested in blinded trials, the differences narrowed or vanished [125]. Furthermore, outcome reporting bias may exist: studies may emphasize whichever endpoints showed a favorable trend. All these potential biases mean that the current literature likely paints an overly optimistic picture of IVIG in some conditions while also leaving certain risks underreported. High-quality RCTs are the antidote to these biases, but as noted, they are scarce. Until more rigorous data are available, any conclusions about IVIG’s efficacy in conditions like NPSLE or autoimmune encephalitis must be made cautiously, and with the understanding that the evidence is low level (levels 3–4 in the hierarchy) [123].
Summary
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, M.; Wang, Z.; Zhang, S.; Wu, Y.; Zhang, L.; Zhao, J.; Wang, Q.; Tian, X.; Li, M.; Zeng, X. Progress in the pathogenesis and treatment of neuropsychiatric systemic lupus erythematosus. J. Clin. Med. 2022, 11, 4955. [Google Scholar] [CrossRef]
- Taiwo, R.O.; Goldberg, H.S.; Ilouz, N.; Singh, P.K.; Shekh-Ahmad, T.; Levite, M. Enigmatic intractable Epilepsy patients have antibodies that bind glutamate receptor peptides, kill neurons, damage the brain, and cause Generalized Tonic Clonic Seizures. J. Neural Transm. 2025, 132, 663–688. [Google Scholar] [CrossRef]
- Kao, Y.C.; Lin, M.I.; Weng, W.C.; Lee, W.T. Neuropsychiatric disorders due to limbic encephalitis: Immunologic aspect. Int. J. Mol. Sci. 2020, 22, 389. [Google Scholar] [CrossRef]
- Pavlekovics, M.; Engh, M.A.; Lugosi, K.; Szabo, L.; Hegyi, P.; Terebessy, T.; Csukly, G.; Molnar, Z.; Illes, Z.; Lovas, G. Plasma Exchange versus Intravenous Immunoglobulin in Worsening Myasthenia Gravis: A Systematic Review and Meta-Analysis with Special Attention to Faster Relapse Control. Biomedicines 2023, 11, 3180. [Google Scholar] [CrossRef]
- Dargahi, N.; Katsara, M.; Tselios, T.; Androutsou, M.E.; de Courten, M.; Matsoukas, J.; Apostolopoulos, V. Multiple Sclerosis: Immunopathology and Treatment Update. Brain Sci. 2017, 7, 78. [Google Scholar] [CrossRef]
- Appenzeller, S.; Andrade, S.D.O.; Bombini, M.F.; Sepresse, S.R.; Reis, F.; França, M.C., Jr. Neuropsychiatric manifestations in primary Sjogren syndrome. Expert Rev. Clin. Immunol. 2022, 18, 1071–1081. [Google Scholar] [CrossRef]
- Dima, A.; Caraiola, S.; Delcea, C.; Ionescu, R.A.; Jurcut, C.; Badea, C. Self-reported disease severity in women with systemic lupus erythematosus. Rheumatol. Int. 2019, 39, 533–539. [Google Scholar] [CrossRef]
- Gremke, N.; Printz, M.; Möller, L.; Ehrenberg, C.; Kostev, K.; Kalder, M. Association between anti-seizure medication and the risk of lower urinary tract infection in patients with epilepsy. Epilepsy Behav. 2022, 135, 108910. [Google Scholar] [CrossRef]
- Kamyshna, I.I.; Pavlovych, L.B.; Kamyshnyi, A.M. Prediction of the cognitive impairment development in patients with autoimmune thyroiditis and hypothyroidism. Endocr. Regul. 2022, 56, 178–189. [Google Scholar] [CrossRef]
- Manocchio, N.; Magro, V.M.; Massaro, L.; Sorbino, A.; Ljoka, C.; Foti, C. Hashimoto’s Encephalopathy: Clinical Features, Therapeutic Strategies, and Rehabilitation Approaches. Biomedicines 2025, 13, 726. [Google Scholar] [CrossRef]
- Shojima, Y.; Nishioka, K.; Watanabe, M.; Jo, T.; Tanaka, K.; Takashima, H.; Nodayes, K.; Okuma, Y.; Urabe, T.; Yokoyama, K.; et al. Clinical characterization of definite autoimmune limbic encephalitis: A 30-case series. Intern. Med. 2019, 58, 3369–3378. [Google Scholar] [CrossRef]
- Mangnus, T.J.; Dirckx, M.; Huygen, F.J. Different types of pain in complex regional pain syndrome require a personalized treatment strategy. J. Pain Res. 2023, 16, 4379–4391. [Google Scholar] [CrossRef]
- Dziadkowiak, E.; Moreira, H.; Buska-Mach, K.; Szmyrka, M.; Budrewicz, S.; Barg, E.; Janik, M.; Pokryszko-Dragan, A. Occult Autoimmune Background for Epilepsy—The Preliminary Study on Antibodies Against Neuronal Surface Antigens. Front. Neurol. 2021, 12, 660126. [Google Scholar] [CrossRef]
- Murashko, A.A.; Pavlov, K.A.; Pavlova, O.V.; Gurina, O.I.; Shmukler, A. Antibodies against N-Methyl D-aspartate receptor in psychotic disorders: A systematic review. Neuropsychobiology 2022, 81, 1–18. [Google Scholar] [CrossRef]
- Kitanosono, H.; Motomura, M.; Tomita, H.; Iwanaga, H.; Iwanaga, N.; Irioka, T.; Shiraishi, H.; Tsujino, A. Paraneoplastic cerebellar degeneration with lambert-eaton myasthenic syndrome: A report of an effectively treated case and systematic review of Japanese cases. Brain Nerve. Shinkei Kenkyu No Shinpo 2019, 71, 167–174. [Google Scholar]
- Takamori, M. Myasthenia gravis: From the viewpoint of pathogenicity focusing on acetylcholine receptor clustering, trans-synaptic homeostasis and synaptic stability. Front. Mol. Neurosci. 2020, 13, 86. [Google Scholar] [CrossRef]
- Hébert, J.; Muccilli, A.; Wennberg, R.A.; Tang-Wai, D.F. Autoimmune encephalitis and autoantibodies: A review of clinical implications. J. Appl. Lab. Med. 2022, 7, 81–98. [Google Scholar] [CrossRef]
- Zammit, F.; Seront, E. Neurological Adverse Events Related to Immune Checkpoint Inhibitors: A Practical Review. Pharmaceuticals 2024, 17, 501. [Google Scholar] [CrossRef]
- Justiz-Vaillant, A.; Soodeen, S.; Gopaul, D.; Arozarena-Fundora, R.; Thompson, R.; Unakal, C.; Akpaka, P.E. Tackling Infectious Diseases in the Caribbean and South America: Epidemiological Insights, Antibiotic Resistance, Associated Infectious Diseases in Immunological Disorders, Global Infection Response, and Experimental Anti-Idiotypic Vaccine Candidates Against Microorganisms of Public Health Importance. Microorganisms 2025, 13, 282. [Google Scholar]
- Ferrazzano, G.; Crisafulli, S.G.; Baione, V.; Tartaglia, M.; Cortese, A.; Frontoni, M.; Altieri, M.; Pauri, F.; Millefiorini, E.; Conte, A. Early diagnosis of secondary progressive multiple sclerosis: Focus on fluid and neurophysiological biomarkers. J. Neurol. 2021, 268, 3626–3645. [Google Scholar] [CrossRef]
- Li, H.; Liu, S.; Han, J.; Li, S.; Gao, X.; Wang, M.; Zhu, J.; Jin, T. Role of toll-like receptors in neuroimmune diseases: Therapeutic targets and problems. Front. Immunol. 2021, 12, 777606. [Google Scholar] [CrossRef]
- Bryll, A.; Skrzypek, J.; Krzyściak, W.; Szelągowska, M.; Śmierciak, N.; Kozicz, T.; Popiela, T. Oxidative-antioxidant imbalance and impaired glucose metabolism in schizophrenia. Biomolecules 2020, 10, 384. [Google Scholar] [CrossRef]
- Gagné, A.M.; Moreau, I.; St-Amour, I.; Marquet, P.; Maziade, M. Retinal function anomalies in young offspring at genetic risk of schizophrenia and mood disorder: The meaning for the illness pathophysiology. Schizophr. Res. 2020, 219, 19–24. [Google Scholar] [CrossRef]
- Slavov, G. Changes in serum cytokine profile and deficit severity in patients with relapsing-remitting multiple sclerosis. Folia Medica 2023, 65, 625–630. [Google Scholar] [CrossRef]
- Velikova, T.; Sekulovski, M.; Bogdanova, S.; Vasilev, G.; Peshevska-Sekulovska, M.; Miteva, D.; Georgiev, T. Intravenous immunoglobulins as immunomodulators in autoimmune diseases and reproductive medicine. Antibodies 2023, 12, 20. [Google Scholar] [CrossRef]
- Conti, F.; Moratti, M.; Leonardi, L.; Catelli, A.; Bortolamedi, E.; Filice, E.; Fetta, A.; Fabi, M.; Facchini, E.; Cantarini, M.E.; et al. Anti-inflammatory and immunomodulatory effect of high-dose immunoglobulins in children: From approved indications to off-label use. Cells 2023, 12, 2417. [Google Scholar] [CrossRef]
- Manganotti, P.; Garascia, G.; Furlanis, G.; Buoite Stella, A. Efficacy of intravenous immunoglobulin (IVIg) on COVID-19-related neurological disorders over the last 2 years: An up-to-date narrative review. Front. Neurosci. 2023, 17, 1159929. [Google Scholar] [CrossRef]
- Bayry, J.; Ahmed, E.A.; Toscano-Rivero, D.; Vonniessen, N.; Genest, G.; Cohen, C.G.; Dembele, M.; Kaveri, S.V.; Mazer, B.D. Intravenous immunoglobulin: Mechanism of action in autoimmune and inflammatory conditions. J. Allergy Clin. Immunol. Pract. 2023, 11, 1688–1697. [Google Scholar] [CrossRef]
- Shock, A.; Humphreys, D.; Nimmerjahn, F. Dissecting the mechanism of action of intravenous immunoglobulin in human autoimmune disease: Lessons from therapeutic modalities targeting Fcγ receptors. J. Allergy Clin. Immunol. 2020, 146, 492–500. [Google Scholar] [CrossRef]
- Ashton, C.; Paramalingam, S.; Stevenson, B.; Brusch, A.; Needham, M. Idiopathic inflammatory myopathies: A review. Intern. Med. J. 2021, 51, 845–852. [Google Scholar] [CrossRef]
- Zeng, R.; Glaubitz, S.; Schmidt, J. Antibody therapies in autoimmune inflammatory myopathies: Promising treatment options. Neurotherapeutics 2022, 19, 911–921. [Google Scholar] [CrossRef]
- Gandiga, P.C.; Ghetie, D.; Anderson, E.; Aggrawal, R. Intravenous immunoglobulin in idiopathic inflammatory myopathies: A practical guide for clinical use. Curr. Rheumatol. Rep. 2023, 25, 152–168. [Google Scholar] [CrossRef]
- Sanchis, P.; Fernández-Gayol, O.; Comes, G.; Escrig, A.; Giralt, M.; Palmiter, R.D.; Hidalgo, J. Interleukin-6 derived from the central nervous system may influence the pathogenesis of experimental autoimmune encephalomyelitis in a cell-dependent manner. Cells 2020, 9, 330. [Google Scholar] [CrossRef]
- Danieli, M.G.; Antonelli, E.; Auria, S.; Buti, E.; Shoenfeld, Y. Low-dose intravenous immunoglobulin (IVIg) in different immune-mediated conditions. Autoimmun. Rev. 2023, 22, 103451. [Google Scholar] [CrossRef]
- Gillespie, E.R.; Ruitenberg, M.J. Neuroinflammation after SCI: Current insights and therapeutic potential of intravenous immunoglobulin. J. Neurotrauma 2022, 39, 320–332. [Google Scholar] [CrossRef]
- Mroué, M.; Bessaguet, F.; Nizou, A.; Richard, L.; Sturtz, F.; Magy, L.; Bourthoumieu, S.; Danigo, A.; Demiot, C. Neuroprotective Effect of Polyvalent Immunoglobulins on Mouse Models of Chemotherapy-Induced Peripheral Neuropathy. Pharmaceutics 2024, 16, 139. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, M.; Zhang, X.; Zhao, P.; Zhai, W. Can low-dose intravenous immunoglobulin be an alternative to high-dose intravenous immunoglobulin in the treatment of children with newly diagnosed immune thrombocytopenia: A systematic review and meta-analysis. BMC Pediatr. 2024, 24, 199. [Google Scholar] [CrossRef]
- Hoffmann, J.H.; Enk, A.H. High-dose intravenous immunoglobulin in skin autoimmune disease. Front. Immunol. 2019, 10, 1090. [Google Scholar] [CrossRef]
- Nadig, P.L.; Joshi, V.; Pilania, R.K.; Kumrah, R.; Kabeerdoss, J.; Sharma, S.; Suri, D.; Rawat, A.; Singh, S. Intravenous immunoglobulin in Kawasaki disease—Evolution and pathogenic mechanisms. Diagnostics 2023, 13, 2338. [Google Scholar] [CrossRef]
- de Carvalho, J.F.; Skare, T.L. Rituximab combined with intravenous immunoglobulin in autoimmune diseases: A systematic review. Adv. Rheumatol. 2025, 65, 19. [Google Scholar] [CrossRef]
- N’kaoua, E.; Attarian, S.; Delmont, E.; Campana-Salort, E.; Verschueren, A.; Grapperon, A.-M.; Mestivier, E.; Roche, M. Immunoglobulin shortage: Practice modifications and clinical outcomes in a reference centre. Rev. Neurol. 2022, 178, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Barthel, C.; Musquer, M.; Veyrac, G.; Bernier, C. Delayed eczematous skin reaction as an adverse drug reaction to immunoglobulin infusions: A case series. In Annales de Dermatologie et de Vénéréologie; Elsevier Masson: Amsterdam, The Netherlands, 2022; Volume 149, pp. 264–270. [Google Scholar]
- Ozen, S.; Esenboga, S. Alternative Therapies for Cytokine Storm Syndromes. In Cytokine Storm Syndromes; Cron, R.Q., Behrens, E.M., Eds.; Springer: Cham, Switzerland, 2019; pp. 581–593. [Google Scholar]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Justiz-Vaillant, A.; Gopaul, D.; Soodeen, S.; Unakal, C.; Thompson, R.; Pooransingh, S.; Arozarena-Fundora, R.; Asin-Milan, O.; Akpaka, P.E. Advancements in Immunology and Microbiology Research: A Comprehensive Exploration of Key Areas. Microorganisms 2024, 12, 1672. [Google Scholar] [CrossRef] [PubMed]
- Justiz-Vaillant, A.A.; Gopaul, D.; Soodeen, S.; Arozarena-Fundora, R.; Barbosa, O.A.; Unakal, C.; Thompson, R.; Pandit, B.; Umakanthan, S.; Akpaka, P.E. Neuropsychiatric Systemic Lupus Erythematosus: Molecules Involved in Its Imunopathogenesis, Clinical Features, and Treatment. Molecules 2024, 29, 747. [Google Scholar] [CrossRef]
- Wesselingh, R.; Butzkueven, H.; Buzzard, K.; Tarlinton, D.; O’Brien, T.J.; Monif, M. Innate immunity in the central nervous system: A missing piece of the autoimmune encephalitis puzzle? Front. Immunol. 2019, 10, 2066. [Google Scholar] [CrossRef]
- Radetz, A.; Groppa, S. White Matter Pathology. In Translational Methods for Multiple Sclerosis Research; Springer: New York, NY, USA, 2021; pp. 29–46. [Google Scholar]
- Dhaiban, S.; Al-Ani, M.; Elemam, N.M.; Al-Aawad, M.H.; Al-Rawi, Z.; Maghazachi, A.A. Role of peripheral immune cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Science 2021, 3, 12. [Google Scholar] [CrossRef]
- Bordet, R.; Camu, W.; De Seze, J.; Laplaud, D.A.; Ouallet, J.C.; Thouvenot, E. Mechanism of action of s1p receptor modulators in multiple sclerosis: The double requirement. Rev. Neurol. 2020, 176, 100–112. [Google Scholar] [CrossRef]
- Yang, T.; Tian, X.; Chen, C.; Ma, L.; Zhou, S.; Li, M.; Wu, Y.; Zhou, Y.; Cui, Y. The efficacy and safety of fingolimod in patients with relapsing multiple sclerosis: A meta-analysis. Br. J. Clin. Pharmacol. 2020, 86, 637–645. [Google Scholar] [CrossRef]
- Stascheit, F.; Li, L.; Mai, K.; Baum, K.; Siebert, E.; Ruprecht, K. Delayed onset hypophysitis after therapy with daclizumab for multiple sclerosis–A report of two cases. J. Neuroimmunol. 2021, 351, 577469. [Google Scholar] [CrossRef]
- Kim, W.; Patsopoulos, N.A. Genetics and functional genomics of multiple sclerosis. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2022; Volume 44, pp. 63–79. [Google Scholar]
- Cohan, S.L.; Lucassen, E.B.; Romba, M.C.; Linch, S.N. Daclizumab: Mechanisms of action, therapeutic efficacy, adverse events and its uncovering the potential role of innate immune system recruitment as a treatment strategy for relapsing multiple sclerosis. Biomedicines 2019, 7, 18. [Google Scholar] [CrossRef]
- Rothhammer, V.; Kenison, J.E.; Li, Z.; Tjon, E.; Takenaka, M.C.; Chao, C.C.; de Lima, K.A.; Borucki, D.M.; Kaye, J.; Quintana, F.J. Aryl hydrocarbon receptor activation in astrocytes by laquinimod ameliorates autoimmune inflammation in the CNS. Neurol. Neuroimmunol. Neuroinflam. 2021, 8, e946. [Google Scholar] [CrossRef]
- Biernacki, T.; Sandi, D.; Bencsik, K.; Vécsei, L. Kynurenines in the Pathogenesis of Multiple Sclerosis: Therapeutic Perspectives. Cells 2020, 9, 1564. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Dadon, Y.; Sasson, N.; Steinerman, J.R.; Knappertz, V.; Vollmer, T.L.; Boyko, A.; Vermersch, P.; Ziemssen, T.; Montalban, X.; et al. CONCERTO: A randomized, placebo-controlled trial of oral laquinimod in relapsing-remitting multiple sclerosis. Mult. Scler. J. 2022, 28, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Ruetsch-Chelli, C.; Bresch, S.; Seitz-Polski, B.; Rosenthal, A.; Desnuelle, C.; Cohen, M.; Brglez, V.; Ticchioni, M.; Lebrun-Frenay, C. Memory B cells predict relapse in rituximab-treated myasthenia gravis. Neurotherapeutics 2021, 18, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, R.; Antozzi, C. When myasthenia gravis is deemed refractory: Clinical signposts and treatment strategies. Ther. Adv. Neurol. Disord. 2018, 11, 1756285617749134. [Google Scholar] [CrossRef]
- Chan, A.M.; Baehring, J.M. Paraneoplastic neurological syndromes: A single institution 10-year case series. J. Neurooncol. 2019, 141, 431–439. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Dalmau, J. Paraneoplastic neurologic syndromes. Neurol. Clin. 2018, 36, 675–685. [Google Scholar] [CrossRef]
- Mladinich, M.C.; Himmler, G.E.; Conde, J.N.; Gorbunova, E.E.; Schutt, W.R.; Sarkar, S.; Tsirka, S.-A.E.; Kim, H.K.; Mackow, E.R. Age-dependent Powassan virus lethality is linked to glial cell activation and divergent neuroinflammatory cytokine responses in a murine model. J. Virol. 2024, 98, e0056024. [Google Scholar] [CrossRef]
- Alakhras, N.S.; Zhang, W.; Barros, N.; Sharma, A.; Ropa, J.; Priya, R.; Yang, X.F.; Kaplan, M.H. An IL-23-STAT4 pathway is required for the proinflammatory function of classical dendritic cells during CNS inflammation. Proc. Natl. Acad. Sci. USA 2024, 121, e2400153121. [Google Scholar] [CrossRef]
- Ruan, Z.; Tang, Y.; Gao, T.; Li, C.; Guo, R.; Sun, C.; Huang, X.; Li, Z.; Chang, T. Efficacy and safety of tocilizumab in patients with refractory generalized myasthenia gravis. CNS Neurosci. Ther. 2024, 30, e14793. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Heitmann, H.; Andlauer, T.F.; Korn, T.; Mühlau, M.; Henningsen, P.; Hemmer, B.; Ploner, M. Fatigue, depression, and pain in multiple sclerosis: How neuroinflammation translates into dysfunctional reward processing and anhedonic symptoms. Mult. Scler. J. 2022, 28, 1020–1027. [Google Scholar] [CrossRef]
- Zarghami, A.; Li, Y.; Claflin, S.B.; van der Mei, I.; Taylor, B.V. Role of environmental factors in multiple sclerosis. Expert Rev. Neurother. 2021, 21, 1389–1408. [Google Scholar] [CrossRef]
- Cruciani, C.; Puthenparampil, M.; Tomas-Ojer, P.; Jelcic, I.; Docampo, M.J.; Planas, R.; Manogaran, P.; Opfer, R.; Wicki, C.; Reindl, M.; et al. T-cell specificity influences disease heterogeneity in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1075. [Google Scholar] [CrossRef]
- Long, H.M.; Meckiff, B.J.; Taylor, G.S. The T-cell response to Epstein-Barr virus–new tricks from an old dog. Front. Immunol. 2019, 10, 2193. [Google Scholar] [CrossRef]
- Bjornevik, K.; Münz, C.; Cohen, J.I.; Ascherio, A. Epstein–Barr virus as a leading cause of multiple sclerosis: Mechanisms and implications. Nat. Rev. Neurol. 2023, 19, 160–171. [Google Scholar] [CrossRef]
- Deeba, E.; Koptides, D.; Gaglia, E.; Constantinou, A.; Lambrianides, A.; Pantzaris, M.; Krashias, G.; Christodoulou, C. Evaluation of Epstein-Barr virus-specific antibodies in Cypriot multiple sclerosis patients. Mol. Immunol. 2019, 105, 270–275. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Huang, J.; Tengvall, K.; Lima, I.B.; Hedström, A.K.; Butt, J.; Brenner, N.; Gyllenberg, A.; Stridh, P.; Khademi, M.; Ernberg, I.; et al. Genetics of immune response to Epstein-Barr virus: Prospects for multiple sclerosis pathogenesis. Brain 2024, 147, 3573–3582. [Google Scholar] [CrossRef]
- Jog, N.R.; McClain, M.T.; Heinlen, L.D.; Gross, T.; Towner, R.; Guthridge, J.M.; Axtell, R.C.; Pardo, G.; Harley, J.B.; James, J.A. Epstein Barr virus nuclear antigen 1 (EBNA-1) peptides recognized by adult multiple sclerosis patient sera induce neurologic symptoms in a murine model. J. Autoimmun. 2020, 106, 102332. [Google Scholar] [CrossRef]
- Robinson, W.H.; Steinman, L. Epstein-Barr virus and multiple sclerosis. Science 2022, 375, 264–265. [Google Scholar] [CrossRef]
- Tengvall, K.; Huang, J.; Hellström, C.; Kammer, P.; Biström, M.; Ayoglu, B.; Bomfim, I.L.; Stridh, P.; Butt, J.; Brenner, N.; et al. Molecular mimicry between Anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc. Natl. Acad. Sci. USA 2019, 116, 16955–16960. [Google Scholar] [CrossRef]
- Wang, Z.; Kennedy, P.G.; Dupree, C.; Wang, M.; Lee, C.; Pointon, T.; Langford, T.D.; Graner, M.W.; Yu, X. Antibodies from Multiple Sclerosis Brain Identified Epstein-Barr Virus Nuclear Antigen 1 & 2 Epitopes which Are Recognized by Oligoclonal Bands. J. Neuroimmune Pharmacol. 2021, 16, 567–580. [Google Scholar]
- Neves, M.; Marinho-Dias, J.; Ribeiro, J.; Sousa, H. Epstein-Barr virus strains and variations: Geographic or disease-specific variants? J. Med. Virol. 2017, 89, 373–387. [Google Scholar] [CrossRef]
- Thomas, O.G.; Bronge, M.; Tengvall, K.; Akpinar, B.; Nilsson, O.B.; Holmgren, E.; Hessa, T.; Gafvelin, G.; Khademi, M.; Alfredsson, L.; et al. Cross-reactive EBNA1 immunity targets alpha-crystallin B and is associated with multiple sclerosis. Sci. Adv. 2023, 9, eadg3032. [Google Scholar] [CrossRef]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.-S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef]
- Telford, M.; Hughes, D.A.; Juan, D.; Stoneking, M.; Navarro, A.; Santpere, G. Expanding the geographic characterisation of Epstein–Barr virus variation through gene-based approaches. Microorganisms 2020, 8, 1686. [Google Scholar] [CrossRef]
- Romero, C.; Quijada, A.; Abudinén, G.; Céspedes, C.; Aguilera, L. Opercular myoclonic-anarthric status (OMASE) secondary to anti-Hu paraneoplastic neurological syndrome. Epilepsy Behav. Rep. 2024, 27, 100703. [Google Scholar] [CrossRef]
- Zhu, F.; Shan, W.; Lv, R.; Li, Z.; Wang, Q. Clinical characteristics of anti-GABA-B receptor encephalitis. Front. Neurol. 2020, 11, 403. [Google Scholar] [CrossRef]
- Tsang-Shan, C.; Ming-Chi, L.; Huang, H.Y.I.; Chin-Wei, H. Immunity, Ion Channels and Epilepsy. Int. J. Mol. Sci. 2022, 23, 6446. [Google Scholar]
- Gilligan, M.; McGuigan, C.; McKeon, A. Paraneoplastic neurologic disorders. Curr. Neurol. Neurosci. Rep. 2023, 23, 67–82. [Google Scholar] [CrossRef]
- Malvaso, A.; Cerne, D.; Bernini, S.; Bottiroli, S.; Marchioni, E.; Businaro, P.; Masciocchi, S.; Morandi, C.; Scaranzin, S.; Mobilia, E.M.; et al. Retrograde Amnesia in LGI1 and CASPR2 Limbic Encephalitis: Two Case Reports and a Systematic Literature Review. Eur. J. Neurol. 2025, 32, e70113. [Google Scholar] [CrossRef]
- Ricken, G.; Schwaiger, C.; De Simoni, D.; Pichler, V.; Lang, J.; Glatter, S.; Macher, S.; Rommer, P.S.; Scholze, P.; Kubista, H.; et al. Detection methods for autoantibodies in suspected autoimmune encephalitis. Front. Neurol. 2018, 9, 841. [Google Scholar] [CrossRef]
- Tanaka, K.; Kawamura, M.; Sakimura, K.; Kato, N. Significance of Autoantibodies in Autoimmune Encephalitis in Relation to Antigen Localization: An Outline of Frequently Reported Autoantibodies with a Non-Systematic Review. Int. J. Mol. Sci. 2020, 21, 4941. [Google Scholar] [CrossRef]
- Orozco, E.; Valencia-Sanchez, C.; Britton, J.; Dubey, D.; Flanagan, E.P.; Lopez-Chiriboga, A.S.; Zalewski, N.; Zekeridou, A.; Pittock, S.J.; McKeon, A. Autoimmune encephalitis criteria in clinical practice. Neurol. Clin. Pract. 2023, 13, e200151. [Google Scholar] [CrossRef]
- Abide, Z.; Nasr, K.S.; Kaddouri, S.; Edderai, M.; Elfenni, J.; Salaheddine, T. Bickerstaff brainstem encephalitis: A case report. Radiol. Case Rep. 2023, 18, 2704–2706. [Google Scholar] [CrossRef]
- Orozco, E.; Guo, Y.; Chen, J.J.; Dubey, D.; Howell, B.; Moutvic, M.; Louis, E.K.S.; McKeon, A. Clinical reasoning: A 43-year-old man with subacute onset of vision disturbances, jaw spasms, and balance and sleep difficulties. Neurology 2022, 99, 387–392. [Google Scholar] [CrossRef]
- Tisavipat, N.; Chang, B.K.; Ali, F.; Pittock, S.J.; Kammeyer, R.; Declusin, A.; Cohn, S.J.; Flanagan, E.P. Subacute horizontal diplopia, jaw dystonia, and laryngospasm. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200128. [Google Scholar] [CrossRef]
- Lana-Peixoto, M.A.; Talim, N. Neuromyelitis optica spectrum disorder and anti-MOG syndromes. Biomedicines 2019, 7, 42. [Google Scholar] [CrossRef]
- Blattner, M.S.; Day, G.S. Sleep disturbances in patients with autoimmune encephalitis. Curr. Neurol. Neurosci. Rep. 2020, 20, 28. [Google Scholar] [CrossRef]
- O’Connor, K.; Waters, P.; Komorowski, L.; Zekeridou, A.; Guo, C.Y.; Mgbachi, V.C.; Probst, C.; Mindorf, S.; Teegen, B.; Gelfand, J.M.; et al. GABA(A) receptor autoimmunity: A multicenter experience. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e552. [Google Scholar] [CrossRef]
- Gravier-Dumonceau, A.; Ameli, R.; Rogemond, V.; Ruiz, A.; Joubert, B.; Muniz-Castrillo, S.; Vogrig, A.; Picard, G.; Ambati, A.; Benaiteau, M.; et al. Glial fibrillary acidic protein autoimmunity: A French cohort study. Neurology 2022, 98, e653–e668. [Google Scholar] [CrossRef]
- Flanagan, E.P.; Hinson, S.R.; Lennon, V.A.; Fang, B.; Aksamit, A.J.; Morris, P.P.; Basal, E.; Honorat, J.A.; Alfugham, N.B.; Linnoila, J.J.; et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: Analysis of 102 patients. Ann. Neurol. 2017, 81, 298–309. [Google Scholar] [CrossRef]
- Guasp, M.; Dalmau, J. Encephalitis associated with antibodies against the NMDA receptor. Med. Clin. 2018, 151, 71–79. [Google Scholar] [CrossRef]
- Budhram, A.; Sharma, M.; Young, G.B. Seizures in anti-hu-associated extra-limbic encephalitis: Characterization of a unique disease manifestation. Epilepsia 2022, 63, e172–e177. [Google Scholar] [CrossRef]
- Liu, M.; Ren, H.; Wang, L.; Fan, S.; Bai, L.; Guan, H. Prognostic and relapsing factors of primary autoimmune cerebellar ataxia: A prospective cohort study. J. Neurol. 2024, 271, 1072–1079. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Graus, F.; Honnorat, J.; Jarius, S.; Titulaer, M.; Manto, M.; Hoggard, N.; Sarrigiannis, P.; Mitoma, H. Diagnostic criteria for primary autoimmune cerebellar ataxia-guidelines from an international task force on immune-mediated cerebellar ataxias. Cerebellum 2020, 19, 605–610. [Google Scholar] [CrossRef]
- Banwell, B.; Bennett, J.L.; Marignier, R.; Kim, H.J.; Brilot, F.; Flanagan, E.P.; Ramanathan, S.; Waters, P.; Tenembaum, S.; Graves, J.S.; et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD panel proposed criteria. Lancet Neurol. 2023, 22, 268–282. [Google Scholar] [CrossRef]
- Valencia-Sanchez, C.; Flanagan, E.P. Uncommon inflammatory/immune-related myelopathies. J. Neuroimmunol. 2021, 361, 577750. [Google Scholar] [CrossRef]
- Chiriboga, S.L.; Flanagan, E.P. Myelitis and other autoimmune myelopathies. Contin. Lifelong Learn. Neurol. 2021, 27, 62–92. [Google Scholar] [CrossRef]
- Banks, S.A.; Morris, P.P.; Chen, J.J.; Pittock, S.J.; Sechi, E.; Kunchok, A.; Tillema, J.-M.; Fryer, J.P.; Weinshenker, B.G.; Krecke, K.N.; et al. Brainstem and cerebellar involvement in MOG-IgG-associated disorder versus aquaporin-4-IgG and MS. J. Neurol. Neurosurg. Psychiatry 2021, 92, 384–390. [Google Scholar] [CrossRef]
- Ciron, J.; Cobo-Calvo, A.; Audoin, B.; Bourre, B.; Brassat, D.; Cohen, M.; Collongues, N.; Deschamps, R.; Durand-Dubief, F.; Laplaud, D.; et al. Frequency and characteristics of short versus longitudinally extensive myelitis in adults with MOG antibodies: A retrospective multicentric study. Mult. Scler. J. 2020, 26, 936–944. [Google Scholar] [CrossRef]
- Cacciaguerra, L.; Sechi, E.; Rocca, M.A.; Filippi, M.; Pittock, S.J.; Flanagan, E.P. Neuroimaging features in inflammatory myelopathies: A review. Front. Neurol. 2022, 13, 993645. [Google Scholar] [CrossRef]
- McKeon, A.; Lesnick, C.; Vorasoot, N.; Buckley, M.W.; Dasari, S.; Flanagan, E.P.; Gilligan, M.; Lafrance-Corey, R.; Miske, R.; Pittock, S.J.; et al. Utility of protein microarrays for detection of classified and novel antibodies in autoimmune neurologic disease. Neurol. Neuroimmunol. Neuroinflam. 2023, 10, e200145. [Google Scholar] [CrossRef]
- Fonseca, E.; Varas, R.; Godoy-Santín, J.; Valenzuela, R.; Sandoval, P. Opsoclonus-myoclonus syndrome associated with anti Kelch-like protein-11 antibodies in a young female patient without cancer. J. Neuroimmunol. 2021, 355, 577570. [Google Scholar] [CrossRef]
- Qin, C.; Zhang, M.; Mou, D.P.; Zhou, L.Q.; Dong, M.H.; Huang, L.; Wang, W.; Cai, S.B.; You, Y.F.; Shang, K.; et al. Single-cell analysis of anti-BCMA CAR T cell therapy in patients with central nervous system autoimmunity. Sci. Immunol. 2024, 9, eadj9730. [Google Scholar] [CrossRef]
- Vukovic, J.; Abazovic, D.; Vucetic, D.; Medenica, S. CAR-engineered T cell therapy as an emerging strategy for treating autoimmune diseases. Front Med. 2024, 11, 1447147. [Google Scholar] [CrossRef]
- Li, Y.R.; Lyu, Z.; Chen, Y.; Fang, Y.; Yang, L. Frontiers in CAR-T cell therapy for autoimmune diseases. Trends Pharmacol. Sci. 2024, 45, 839–857. [Google Scholar] [CrossRef]
- Bien, C.G.; Tiemeier, H.; Sassen, R.; Kuczaty, S.; Urbach, H.; von Lehe, M.; Becker, A.J.; Bast, T.; Herkenrath, P.; Karenfort, M.; et al. Rasmussen encephalitis: Incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia 2013, 54, 543–550. [Google Scholar] [CrossRef]
- Hommes, O.R.; Sørensen, P.S.; Fazekas, F.; Enriquez, M.M.; Koelmel, H.W.; Fernandez, O.; Pozzilli, C.; O’Connor, P. Intravenous immunoglobulin in secondary progressive multiple sclerosis: Randomised placebo-controlled trial. Lancet 2004, 364, 1149–1156. [Google Scholar] [CrossRef]
- Sandoglobulin Guillain-Barre Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet 1997, 349, 225–230. [Google Scholar] [CrossRef]
- Wolfe, G.I.; Barohn, R.J.; Foster, B.M.; Jackson, C.E.; Kissel, J.T.; Day, J.W.; Thornton, C.A.; Nations, S.P.; Bryan, W.W.; Amato, A.; et al. Randomized, controlled trial of intravenous immunoglobulin in myasthenia gravis. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2002, 26, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Gray, O.; McDonnell, G.V.; Forbes, R.B. Intravenous immunoglobulins for multiple sclerosis. Cochrane Database Syst Rev. 2003, 2003, CD002936. [Google Scholar] [CrossRef] [PubMed]
- Multiple Sclerosis—(MS) [Relapsing Remitting Multiple Sclerosis (RRMS)]. Available online: https://www.criteria.blood.gov.au/MedicalCondition/View/2553#:~:text=Justification%20for%20Evidence%20Category%20While,For%20more%20information%20see (accessed on 20 May 2025).
- Fragoso, Y.D.; Adoni, T.; Anacleto, A.; Barreira, A.A.; Brooks, J.B.; Carneiro, T.; Damasceno, A.; Ferreira, M.L.B.; Gonçalves, M.V.; Gonçalves, M.V.; et al. There is no benefit in the use of postnatal intravenous immunoglobulin for the prevention of relapses of multiple sclerosis: Findings from a systematic review and meta-analysis. Arq. Neuro-Psiquiatr. 2018, 76, 407–412. [Google Scholar]
- Guo, Y.; Tian, X.; Wang, X.; Xiao, Z. Adverse Effects of Immunoglobulin Therapy. Front Immunol 2018, 9, 1299. [Google Scholar] [CrossRef]
- Toubi, E.; Kessel, A.; Shoenfeld, Y. High-dose intravenous immunoglobulins: An option in the treatment of systemic lupus erythematosus. Hum. Immunol. 2005, 66, 395–402. [Google Scholar] [CrossRef]
- IVIG (Intravenous Immunoglobulin). Available online: https://www.fepblue.org/-/media/PDFs/Medical-Policies/2024/March/Mar-2024-Pharmacy-Policies/Remove---Replace/520003-IVIG-intravenous-immunoglobulin.pdf#:~:text=There%20are%20various%20types%20of,15 (accessed on 19 May 2025).
- Turnkey Clinical Evidence Review Team on Behalf of NHS England Specialised Commissionnig. Intravenous Immunoglobulins for Autoimmune Encephalitis. Available online: https://www.engage.england.nhs.uk/consultation/clinical-commissioning-wave8/user_uploads/f06x05-aie-evidence-rev.pdf#:~:text=The%20review%20does%20not%20include,DPPX (accessed on 20 May 2025).
- Otto, A.M. First Autoimmune Epilepsy RCT Supports IVIG Therapy. Available online: https://www.mdedge9-ma1.mdedge.com/neurology/article/214674/epilepsy-seizures/first-autoimmune-epilepsy-rct-supports-ivig-therapy#:~:text=Although%20the%20numbers%20of%20enrolled,MBBS%2C%20from%20the%20Mayo%20Clinic (accessed on 20 May 2025).
- Gajdos, P.; Chevret, S.; Toyka, K.V. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst. Rev. 2012, 12, CD002277. [Google Scholar] [CrossRef]
- Hughes, R.A.; Swan, A.V.; van Doorn, P.A. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst. Rev. 2014, 2014, CD002063. [Google Scholar] [CrossRef]
- van Doorn, P.A.; Kuitwaard, K.; Walgaard, C.; van Koningsveld, R.; Ruts, L.; Jacobs, B.C. IVIG treatment and prognosis in Guillain-Barré syndrome. J. Clin. Immunol. 2010, 30 (Suppl. S1), S74–S78. [Google Scholar] [CrossRef]
- Tilburg, S.J.V.; Huizinga, R.; Kuitwaard, K.; Sassen, S.D.; Walgaard, C.; van Doorn, P.A.; Jacobs, B.C.; Koch, B.C. If it does not help, it might hurt: Pharmacodynamics of a second IVIG course in Guillain-Barre syndrome. Ann. Clin. Transl. Neurol. 2025, 12, 966–975. [Google Scholar] [CrossRef]
- Magro-Checa , C.; Zirkzee, E.J.; Huizinga, T.W.; Steup-Beekman, G.M. Management of Neuropsychiatric Systemic Lupus Erythematosus: Current Approaches and Future Perspectives. Drugs 2016, 76, 459–483. [Google Scholar] [CrossRef]
- IVIG (Intravenous Immunoglobulin). Available online: https://www.fepblue.org/-/media/PDFs/Medical-Policies/2024/March/Mar-2024-Pharmacy-Policies/Remove---Replace/520003-IVIG-intravenous-immunoglobulin.pdf#:~:text=Immune%20globulin%20use%20is%20associated,administering%20the%20medication%2C%20practitioners (accessed on 20 May 2025).
- Arumugham, V.B.; Rayi, A. Intravenous Immunoglobulin (IVIG); StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Mahadeen, A.Z.; Carlson, A.K.; Cohen, J.A.; Galioto, R.; Abbatemarco, J.R.; Kunchok, A. Review of the Longitudinal Management of Autoimmune Encephalitis, Potential Biomarkers, and Novel Therapeutics. Neurol. Clin. Pr. 2024, 14, e200306. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Disease | Autoantibody | Reference |
|---|---|---|
| Cognitive and affective dysfunctions in autoimmune thyroiditis | Anti-thyroid peroxidase Ab; anti-central nervous system Ab | [8] |
| Hashimoto’s encephalopathy (HE) | Anti-α-enolase Ab; anti-thyroid peroxidase Ab | [8,9] |
| Limbic encephalitis–multiple sclerosis | Anti-N-methyl D-aspartate-type glutamate receptor Ab | [1,10] |
| Complex regional syndrome | Anti-nuclear Ab (ANA); anti-neuronal Ab | [11] |
| Idiopathic and symptomatic epilepsies | Neurotropic Abs to NF-200, GFAP, MBP, and S100β, and to receptors of neuromediators (glutamate, GABA, dopamine, serotonin, and choline receptors) | [12] |
| Schizophrenia | Autoantibodies against glutamate, dopamine, acetylcholine, and serotonin receptors, and antineuronal antibodies against synaptic biomolecules | [13] |
| Lambert–Eaton myasthenic syndrome | Autoantibodies against P/Q-type voltage-gated calcium channels | [14] |
| Myasthenia gravis | Autoantibodies to acetylcholine receptor | [15] |
| Autoimmune encephalitis | Anti-N-methyl-D-aspartate receptor antibody | [16] |
| Multiple sclerosis | Anti-Oligoclonal bands (OCBs) antibodies | [17] |
| Disease | Cytokines Involved | Reference |
|---|---|---|
| Neuropsychiatric systemic lupus erythematosus | Elevated interleukin (IL)-17, IL-2, interferon-gamma (IFN-γ), IL-5, basic fibroblast growth factor (FGF), and IL-15 levels | [19] |
| Relapsing–remitting multiple sclerosis | Elevated IL-17 and INF-gamma and decreased transforming growth factor-beta (TGF-beta 1) levels | [20] |
| Guillain–Barré syndrome | Elevated TNFα and IL-10 levels | [21] |
| Schizophrenia | Increased interleukin (IL)-1, IL-6, and TGF-β appear to be state markers, whereas IL-12, interferon-gamma (IFN-γ), TNF-α, and soluble IL-2 receptor appear to be trait markers | [22,23] |
| Multiple sclerosis (MS) | IL-17 plays an important role in the inflammatory phase of relapsing–remitting MS | [24] |
| Disease | IVIG Outcome | Evidence Source |
|---|---|---|
| Guillain–Barré syndrome | Successful | [25,26,27,28,29] |
| Chronic inflammatory demyelinating polyneuropathy (CIDP) | Successful | [25,26,27,28,29] |
| Multifocal motor neuropathy | Successful | [26,27] |
| Myasthenia gravis | Successful | [26,27,28] |
| Acute disseminated encephalomyelitis (ADEM) | Successful | [27] |
| Diabetic neuropathy | Limited/Off-label use | [27] |
| Lambert–Eaton myasthenic syndrome | Successful | [27] |
| Opsoclonus–myoclonus | Successful | [27] |
| Pediatric autoimmune neuropsychiatric disorders (PANDAS) | Successful/Case-based | [27] |
| Polymyositis | Successful | [27,30,31,32] |
| Rasmussen’s encephalitis | Limited/Experimental | [27] |
| Multiple sclerosis (MS) | Mixed/Experimental | [26,27,33] |
| Disease | Drug Used | Mechanism of Action | References |
|---|---|---|---|
| Multiple sclerosis | Oral fingolimod | Inhibits egress of lymphocytes from lymph nodes and their recirculation | [50,51] |
| Multiple sclerosis | Daclizumab | Humanized neutralizing monoclonal antibody against the α-chain of the interleukin-2 receptor | [52,53,54] |
| Experimental autoimmune encephalomyelitis | Laquinimod | Modulates adaptive T cell immune responses via its effects on cells of the innate immune system and may not directly influence T cells | [55,56,57] |
| Myasthenia gravis | Rituximab | A chimeric IgG k monoclonal antibody that targets CD20 on B cells | [58,59] |
| Guillain–Barré syndrome | Plasma exchange | Depletes pathogenic autoantibodies | [60] |
| Paraneoplastic neurological disorders | IVIG; plasma exchange | Immunomodulator that depletes auto-Abs | [61] |
| Category | |
|---|---|
| Cognitive | Memory loss, confusion, disorientation, impaired attention/concentration |
| Psychiatric | Anxiety, depression, psychosis, hallucinations, agitation, paranoia |
| Seizures | Focal or generalised seizures, status epilepticus |
| Movement disorders | Dyskinesias, chorea, dystonia, catatonia, myoclonus |
| Speech disturbance | Aphasia, mutism, echolalia |
| Autonomic dysfunction | Cardiac arrhythmias, blood pressure fluctuations, hyperthermia, urinary retention |
| Sleep abnormalities | Hypersomnia, insomnia, disrupted circadian rhythm |
| Consciousness | Lethargy, stupor, coma |
| Autoantibody | Target Antigen | Clinical Association |
|---|---|---|
| Anti-NMDAR | NMDA receptor (NR1 subunit) | Young women, ovarian teratoma; psychosis, seizures |
| Anti-AMPAR | AMPA receptor (GluR1, GluR2 subunits) | Limbic encephalitis, memory loss, seizures |
| Anti-LGI1 | Leucine-rich glioma-inactivated protein 1 | Elderly males; faciobrachial dystonic seizures (FBDS) |
| Anti-CASPR2 | Contactin-associated protein-like 2 | Limbic encephalitis, Morvan’s syndrome |
| Anti-GABA_A-R | GABA-A receptor | Refractory seizures, encephalopathy |
| Anti-GABA_B-R | GABA-B receptor | Seizures, associated with small cell lung cancer |
| Anti-GlyR | Glycine receptor | Stiff-person spectrum, brainstem encephalitis |
| Anti-DPPX | Dipeptidyl-peptidase-like protein 6 | Diarrhoea, weight loss, encephalopathy |
| Anti-GFAP | Glial fibrillary acidic protein | Meningoencephalomyelitis, optic involvement |
| Anti-Hu (ANNA-1) | Neuronal nuclear antigen | Paraneoplastic, small cell lung cancer |
| Anti-Ma2 | Ma2/Ta protein | Testicular cancer; diencephalic/brainstem involvement |
| Diseases | Clinical Randomized Trial | Results | References |
|---|---|---|---|
| Rasmussen’s encephalitis (RE) | Germany-wide, patients with suspected recent-onset RE were recruited and if eligible randomized to tacrolimus or intravenous immunoglobulins (IVIGs). | Treatment with tacrolimus or IVIG may slow down tissue and function loss and prevent development of intractable epilepsy. | [113] |
| Multiple sclerosis | “318 patients with clinically definite secondary progressive multiple sclerosis (mean age 44 years [SD 7]) were randomly assigned IVIG 1 g/kg per month (n = 159) or an equivalent volume of placebo (albumin 0.1%; n = 159) for 27 months”. | “Treatment with IVIG in this study did not show any clinical benefit and therefore cannot be recommended for patients with secondary progressive multiple sclerosis”. | [114] |
| Severe Guillain–Barré syndrome | Patients with severe neuropathy onset within 14 days were randomly assigned to plasma exchange, IVIG, or both. Treatments were administered over 8–13 days, with clinical outcomes monitored for 48 weeks post-intervention. | “In treatment of severe Guillain-Barré syndrome during the first 2 weeks after onset of neuropathic symptoms, PE and IVIg had equivalent efficacy. The combination of PE with IVIg did not confer a significant advantage”. | [115] |
| Acute exacerbation of myasthenia gravis | “Randomized double-blind placebo-controlled multicenter trial designed to demonstrate superiority of the 2 g/kg dose over the 1 g/kg dose of IVIG, conducted between 13 November 1996, and 26 October 2002”. | “This trial found no significant superiority of 2 g/kg over 1 g/kg of IVIG in the treatment of myasthenia gravis exacerbation”. | [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Justiz-Vaillant, A.; Soodeen, S.; Asin-Milan, O.; Morales-Esquivel, J.; Arozarena-Fundora, R. Efficacy of Intravenous Immunoglobulins and Other Immunotherapies in Neurological Disorders and Immunological Mechanisms Involved. Immuno 2025, 5, 18. https://doi.org/10.3390/immuno5020018
Justiz-Vaillant A, Soodeen S, Asin-Milan O, Morales-Esquivel J, Arozarena-Fundora R. Efficacy of Intravenous Immunoglobulins and Other Immunotherapies in Neurological Disorders and Immunological Mechanisms Involved. Immuno. 2025; 5(2):18. https://doi.org/10.3390/immuno5020018
Chicago/Turabian StyleJustiz-Vaillant, Angel, Sachin Soodeen, Odalis Asin-Milan, Julio Morales-Esquivel, and Rodolfo Arozarena-Fundora. 2025. "Efficacy of Intravenous Immunoglobulins and Other Immunotherapies in Neurological Disorders and Immunological Mechanisms Involved" Immuno 5, no. 2: 18. https://doi.org/10.3390/immuno5020018
APA StyleJustiz-Vaillant, A., Soodeen, S., Asin-Milan, O., Morales-Esquivel, J., & Arozarena-Fundora, R. (2025). Efficacy of Intravenous Immunoglobulins and Other Immunotherapies in Neurological Disorders and Immunological Mechanisms Involved. Immuno, 5(2), 18. https://doi.org/10.3390/immuno5020018