Abstract
Virus-infected cells trigger a robust innate immune response and facilitate virus replication. Here, we review the role of autophagy in virus infection, focusing on both pro-viral and anti-viral host responses using a select group of viruses. Autophagy is a cellular degradation pathway operated at the basal level to maintain homeostasis and is induced by external stimuli for specific functions. The degradative function of autophagy is considered a cellular anti-viral immune response. However, autophagy is a double-edged sword in viral infection; viruses often benefit from it, and the infected cells can also use it to inhibit viral replication. In addition to viral regulation, autophagy pathway proteins also function in autophagy-independent manners to regulate immune responses. Since viruses have co-evolved with hosts, they have developed ways to evade the anti-viral autophagic responses of the cells. Some of these mechanisms are also covered in our review. Lastly, we conclude with the thought that autophagy can be targeted for therapeutic interventions against viral diseases.
1. Introduction
Virus infection triggers a robust cellular immune response via activating the pattern recognition receptors [1,2,3]. The innate immune response is critical to ensure an immediate anti-viral response and also to help shape the adaptive immune responses [4,5]. In addition to triggering the anti-viral signaling pathways, the virus-infected cells can induce autophagy in the infected cells [6,7]. Autophagy, which can be translated literally as “self-eating”, is a cellular catabolic process required to regulate many cellular functions [8]. Basal level autophagy is required for maintaining cellular homeostasis, and autophagy is often induced in various disease conditions and infections [9]. The catabolic property of autophagy is primarily utilized as a cellular immune response to inhibit viral replication by degrading viral proteins. However, many viruses often exploit autophagy to replicate effectively within infected cells [10]. Therefore, a careful balance between the pro-viral and anti-viral functions of autophagy is important. It is also evident that viruses that do not rely on the machinery of autophagy for replication can still benefit from the components of the pathway. Moreover, autophagy pathway components can participate in autophagy-independent cellular functions. The therapeutic targeting of autophagy pathway components may provide clinical options for the treatment of viral and non-viral diseases [11,12].
2. Autophagy: Activation and Various Forms
2.1. Cellular Autophagy Pathway
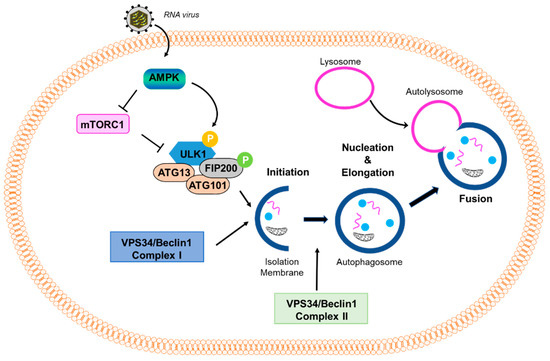
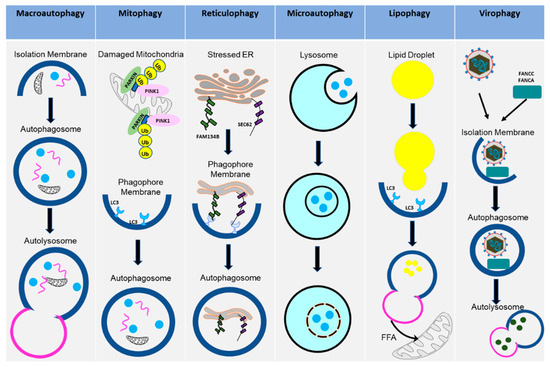
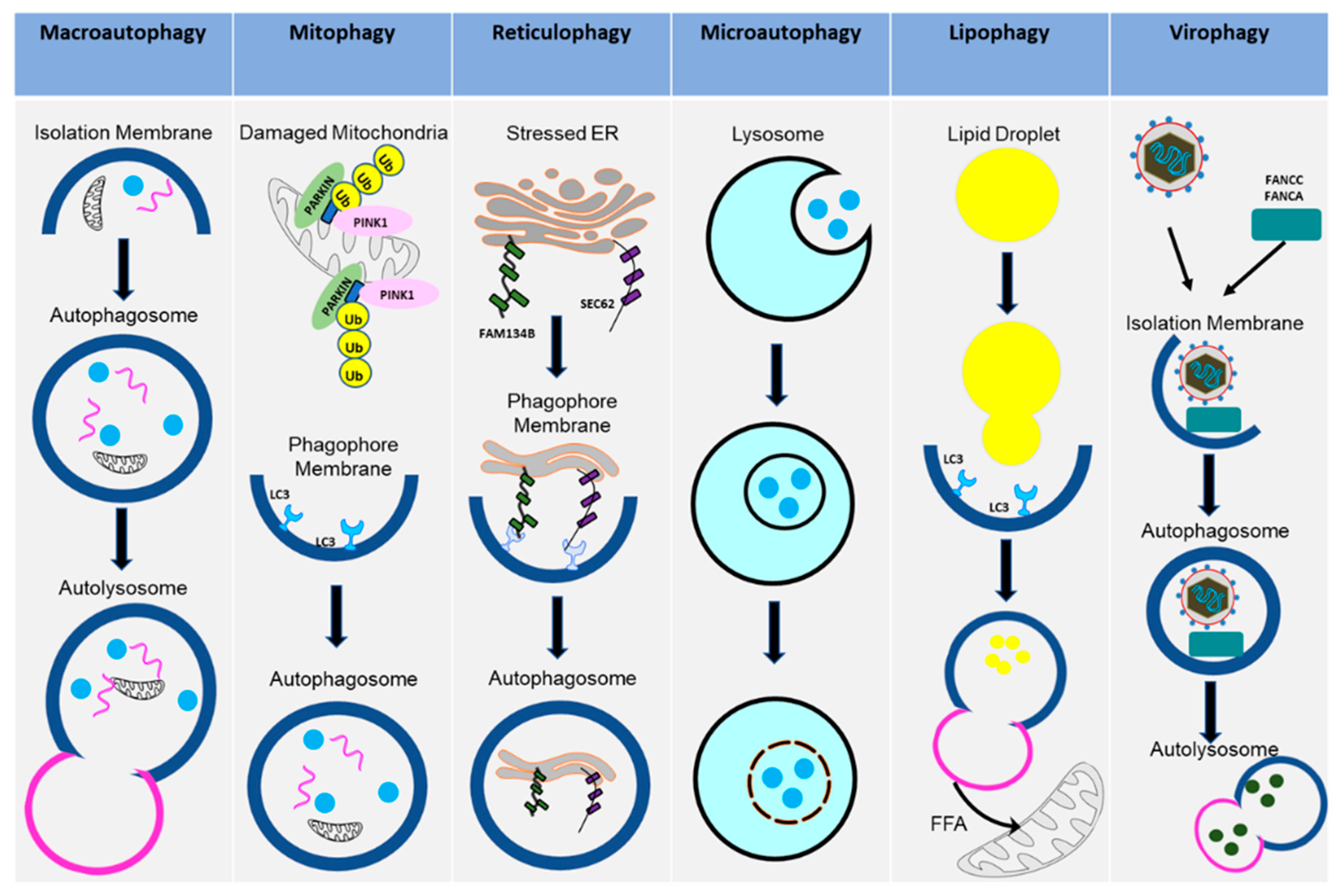
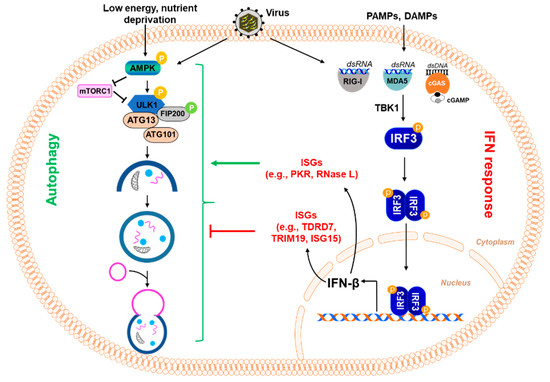
Macroautophagy (referred to in this paper as autophagy from this point on), as the name suggests, signifies self- (auto) eating (phago), and serves as a cellular degradative process. Autophagy occurs at a basal level in healthy cells but is also activated in response to stressors, such as nutrient deprivation, organelle damage, oxidative burst, and viral infection. Autophagy is a cellular pathway that involves four major steps: initiation, nucleation, elongation, and fusion, as depicted in Figure 1. The various forms of autophagy, as detailed below, are shown in Figure 2.
Figure 1.
Virus-induced autophagy pathway. A scheme of the autophagy pathway: mTOR inhibits the autophagy pathway by negatively regulating the downstream effector protein ULK1 through phosphorylation. Stressful conditions, such as virus infection, activate AMPK, which inhibits mTOR and promotes the activation of ULK1. Activation of downstream effectors, such as ULK1 and VPS34/Beclin1 Complex I, induce the formation of an isolation membrane and initiate the formation and maturation of the autophagosome. VPS34/Beclin1 Complex II is important in the formation of the autophagosome. The autophagosome then fuses with the lysosome to form the autolysosome.
Figure 2.
Various types of autophagy pathways. The diagram shows how the cells activate different types of autophagy pathways in the uninfected or the virus-infected cells. These pathways contribute to cellular homeostasis or disease states.
Autophagy is induced when AMP-dependent kinase (AMPK) is activated in response to stressors, leading to the downstream phosphorylation and activation of ULK1 as well as the inhibition of mTOR [13]. ULK1 functions in a complex with the additional proteins Fip200, Atg101, and Atg13. This complex leads to the activation of the PI3K complex, which comprises Beclin-1, Vps34, and Vps15. The activated PI3K complex converts PIP2 to PIP3, which results in the recruitment of human WD-repeat protein interacting with the phosphoinositide (WIPI) proteins, WIPI1 and WIPI2. The WIPI proteins play an essential role in recognizing and reading the PtdIns3P signal at the nascent autophagosome. Thus, the nucleation stage of autophagy begins, during which the phagophore starts to form. The phagophore is a double-membraned vacuole that encapsulates the proteins to be recycled or degraded, along with other cytoplasm-derived proteins. Next, the elongation process is an Atg5-Atg12-Atg16-dependent step that results in the formation of an autophagosome, which is marked by the lipidation of LC3-I to LC3-II and involves Atg7 and Atg3. After elongation of LC3, the autophagosome fuses with a lysosome using the additional proteins, SNARE and Rab7, which form a structure called the autophagolysosome. Lastly, acidification of the contents within the autophagolysosome occurs, resulting in the degradation of the protein content. This step is marked by the degradation of p62, also known as sequestome-like receptor (SLR), the autophagic adaptor, which binds and packages the cargo into the autophagolysosome [14]. After the degradation of the proteins, the broken-down cargo is available for recycling.
2.2. Mitophagy
There are also selective forms of autophagy that target specific cellular components, such as particular proteins or intracellular organelles. These selective forms of autophagy rely on receptor proteins that determine target-specificity [15]. The first selective process of autophagy to be discussed is mitophagy, which removes the damaged mitochondria for degradation. This process has been implicated in the pathophysiology of neurodegenerative diseases and in maintaining critical physiologic functions, such as the differentiation of red blood cells (RBCs). Mitophagy serves as a quality control mechanism for mitochondria in various tissues and as a process to remove mitochondrial contents. Interest in mitophagy has rapidly grown due to various proteins, such as Parkin and PINK1, which are involved in Alzheimer’s and Parkinson’s diseases [16]. Additional proteins, e.g., BNIP3, FUNDC1, and MURF1, have also been identified as receptors that mediate mitophagy in both physiological and stressed conditions [17]. A greater understanding of this process would reveal vital information regarding the deleterious nature of terminal disease progression.
2.3. Reticulophagy
Like mitophagy, reticulophagy leads to the selective degradation of the endoplasmic reticulum (ER) upon ER stress, nutrient deprivation, and nitrogen deficiency [18]. Four ER-resident proteins, FAM134B, RTN3, SEC62, and CCPG1, have been identified as selective autophagy receptors in mammals. Viruses have evolved strategies to destroy FAM134B, avoiding reticulophagy and escaping elimination [19]. Reticulophagy has been involved in many illnesses caused by the Ebola, Dengue, and Zika viruses. Given the diverse functions of the ER and its complexity, additional proteins may be involved in reticulophagy. Studying this process in detail will help to determine potential points of intervention to treat these harmful viral infections.
2.4. Microautophagy
Unlike macroautophagy, which utilizes an autophagophore to contain the cytoplasm-derived proteins for degradation, microautophagy involves directly internalizing cytosolic contents into lysosomal vacuoles by engulfing the invaginations of the lysosomal membrane [20]. Microautophagy can selectively uptake either whole organelles or soluble components in the cytosol. The process is upregulated under conditions of cellular stress, such as nutrient deprivation, but studies of this pathway have been limited to yeasts and cell-free systems. Such studies have described four stages of microautophagy: stages I–III comprise the invagination of the lysosomal membrane into its lumen, while stage IV completes the process of uptake by fusing the membrane of the budding vesicle. The four stages are distinguished by their sensitivities to various inhibitors and temperatures. For example, stage I is characterized by its sensitivity to GTPγS, aristolochic acid, and nystatin. Stage II is characterized by its sensitivity to cooling. Stage III is characterized by its sensitivity to FCCP/valinomycin. Lastly, stage IV is characterized by its sensitivity to K252a and rapamycin. Nystatin and a temperature of 0 °C directly interfere with phospholipid membrane invagination and scission, while valinomycin and rapamycin disrupt the membrane electrochemical gradient and cell proliferation, respectively [21,22].
2.5. Lipophagy
This is another way by which autophagy contributes to cellular energy stores. The contribution of autophagy to cellular energy was thought to only come from free amino acids (FAAs). However, autophagy can also provide energy through free fatty acids (FFAs). Lipid droplets (LDs) are dynamic and not inert stores, leading to their classification as an intracellular organelle [23,24]. One study showed that when hepatocytes were knocked down for Atg5 and then subjected to an acute oleic acid challenge, there was an overall increase in the size and number of LDs [25]. As was established through biochemical analysis, defective macroautophagy was the sole explanation for this phenomenon [25]. However, lipophagy initiation is poorly understood. The aggregation of ubiquitinated proteins along the surface of LDs, in the presence of proteosomes, has been observed in Atg7-deficient mice [26,27]. These aggregates were eliminated by autophagy, suggesting the targeted activation of lipophagy [28]. Defective lipophagy is characteristic of a variety of pathologies, such as obesity, steatosis, and metabolic syndrome in aging. Animals exposed to high-fat diets for extended periods of time showed an increase in autophagy for the first two weeks, followed by a gradual decrease in autophagic activity. Therefore, this decrease in autophagy predisposes cells to an expansion of the LD compartment, potentially causing hepatotoxicity and steatosis [28,29,30]. Studies have shown that the pharmacological upregulation of autophagy reduced hepatotoxicity in a fatty liver disease model [31]. Decreased autophagy has been associated with increased age (65) and with decreased lipophagy, which would contribute to an overall increase in LD content; this would serve to further reduce autophagy and cause a vicious circle for the development of hypercholesterolemia and lipid deposits in organs [28]. Lipophagy also has a positive effect on Flavivirus production through unmodified AUP1 [32]. Further studies are needed to characterize how lipophagy is initiated and regulated in both normal and diseased states.
2.6. Virophagy (Viral Xenophagy)
Although not entirely understood, virophagy protects against viral infection by linking viral subunits to autophagosomes [33]. Virophagy entails the engulfment of the entire virion, which has been reported in the case of SINV [34]. Additionally, this mechanism could be relevant in anti-viral action against other viruses and warrants further study. Another factor that is thought to be critical in virophagy is the type of cell involved [35]. The contribution of cell type to autophagy is another aspect that needs to be better understood. It is known that this mechanism exists in most cells, but the importance of the activation of this phenomenon in certain cell types over others, and its role in anti-viral activity, could improve our understanding of inflammatory response and innate immunity.
3. Autophagy as an Anti-Viral Immune Response
Autophagy is often considered to be an innate immune response, one that is used to control the early stages of viral infection and form a bridge between innate and adaptive responses in which the degradative process is required for antigen processing [36]. Virophagy is an autophagic process that directly degrades intact virions or specific viral proteins. It is triggered by various mechanisms, depending on the virus at hand, some of which will be briefly discussed in Table 1.
Table 1.
Anti-viral action of autophagy against specific viruses.
For example, upon Sindbis virus (SINV) infection, p62 binds to the viral capsid to activate virophagy [39]. The p62-mediated clearance of SINV in neurons is a host anti-viral response, and the absence of autophagy results in increased neurovirulence. In a hepatitis C virus (HCV) infection, a host ER protein, SHISA5, interacts with the viral nonstructural protein, NS5A, to transport it into the autophagosomes [37]. Vesicular stomatitis virus (VSV) infection triggers a decrease in the cellular PI3K-Akt signaling pathway, which increases autophagy and limits viral replication at the initiation and nucleation stages [40]. During poliovirus infection, the release of viral genome galactosides into the cytoplasm triggers Galectin 8, which initiates autophagy. The initiation step of autophagy, especially, exhibits an anti-viral effect against human immunodeficiency virus-1 (HIV-1), in which the cellular protein, BST2, restricts the viral genome by anchoring it to the cell membrane [41]. Upon norovirus infection, IFN-ɣ induction and the upregulation of GTPases result in the recruitment of the Atg5-Atg12-Atg16L1 complex, which restricts viral replication complexes through a non-degradative mechanism [38]. Plasmacytoid dendritic cells (pDCs) can also detect the presence of a virus without being infected themselves by recognizing viral genomes within lysosomes that have undergone autophagy. When autophagy is inhibited in these cells, interferon (IFN)-α production is also inhibited in response to a range of viruses, such as VSV, Sendai virus (SeV), Influenza virus, HIV-1, and Herpes simplex virus (HSV). This indicates that autophagy is a key process in sensing viruses and initiating an anti-viral response [42].
Autophagy, in addition to its direct anti-viral properties, also plays a role in antigen presentation. Specifically, the autophagy pathway delivers antigens to MHC class I and II molecules, bridging innate and adaptive immune responses. Antigen-presenting cells (APCs) use the transporter associated with antigen processing (TAP) to internalize proteolyzed endogenous antigens, whether viral, tumor, or self, into ER phagosomes containing MHC-I, for presentation to CD8+ T cells. However, studies also implicate autophagy as another mechanism for exogenous antigen internalization and cross-presentation by MHC-I. Treating melanoma cells with specific inhibitors of macroautophagy reduced the MHC-I cross-presentation of the tumor antigen gp100 to corresponding CD8+ cells in vivo [43]. In cases of HIV-1, HSV, or human cytomegalovirus (HCMV) infection, the viral proteins Gag, Gb, and UL138, respectively, are presented through MHC-I and are likely aided by autophagy. Likewise, knocking down Atg12 in EBV-infected cells results in a hampered MHC-II uptake of the viral protein antigen EBNA1, thus blocking T-cell activation. Autophagy also appears to be involved in MHC-II-dependent antigen presentation, even in the absence of viral infection [14]. The induction of autophagy by nutrient deprivation alters the types of peptides expressed on MHC-II, such that the displayed peptides are increasingly derived from intracellular or lysosomal proteins, as opposed to membrane proteins. The increased occurrence of autophagy in APCs, in turn, alters specificity in the subsequent activation of T cells [43].
4. Autophagy and the Interferon System
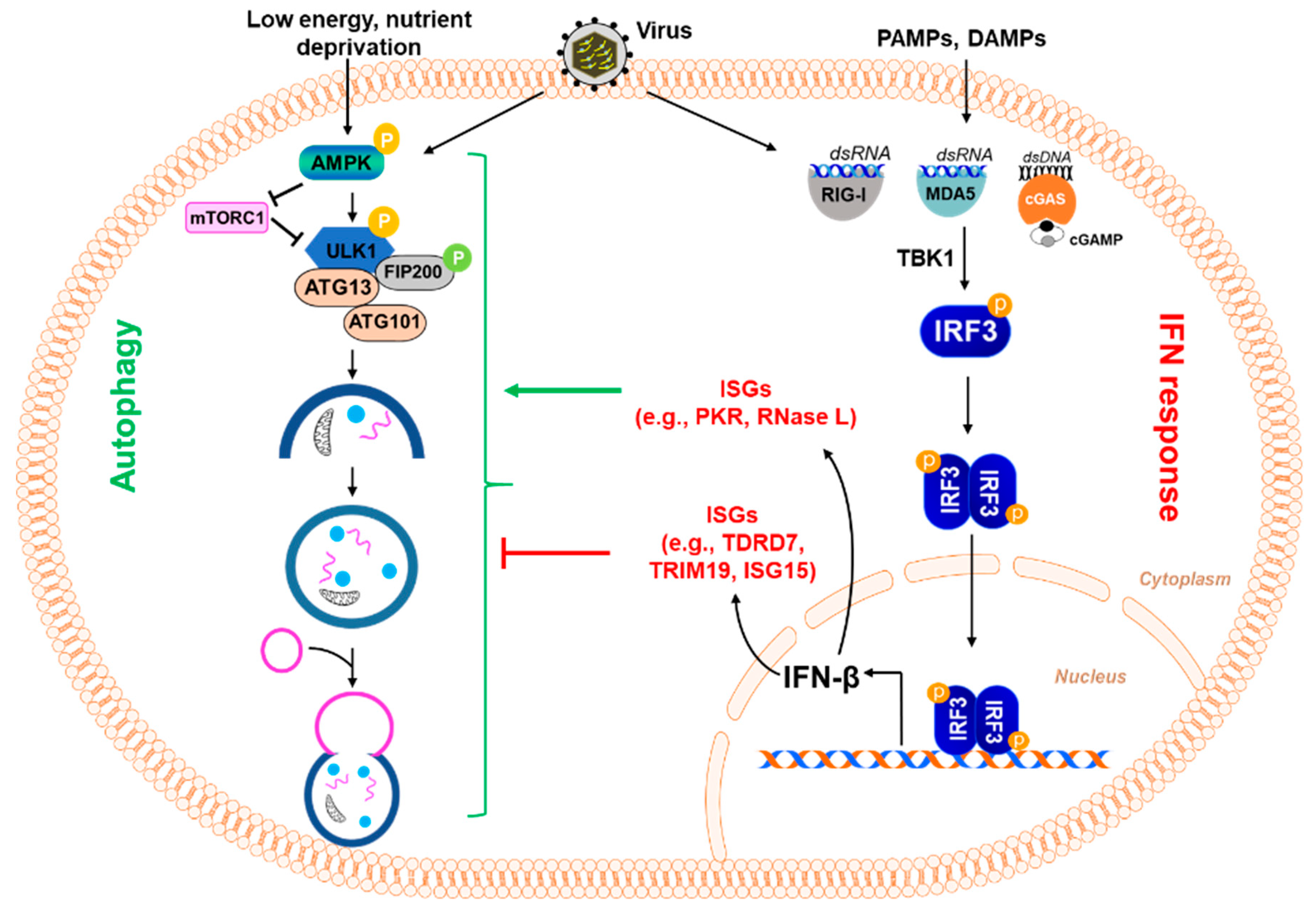
Virus infection triggers a robust host immune response, and the interferon (IFN) system is the first line of defense against a wide range of viral infections [44]. Virus-infected cells rapidly induce the production of IFNs, which are secreted and act on neighboring cells via the JAK-STAT signaling pathway. The IFN-induced proteins, also known as IFN-stimulated genes (ISGs), execute the anti-viral action of IFN. ISGs, in addition to directly interfering with the viral life cycle, also regulate the cellular autophagy pathway to control viral replication (Figure 3). The IFN-induced protein kinase R (PKR) inhibits HSV-1 replication in neurons by inducing autophagy; the viral protein ICP34.5 evades PKR-induced autophagy to facilitate replication [45]. PKR is widely known for phosphorylating elf2α to inhibit viral protein translation and form intracellular stress granules that enhance anti-viral signaling. RNase L, another anti-viral ISG known to cleave viral and cellular RNA, induces autophagy by mediating the role of PKR and amplifies the production of IFN [46,47].
Figure 3.
Regulation of the autophagy pathway by interferon-stimulated genes. Virus infection and exposure to PAMPs and DAMPs result in the activation of IRF3, which causes the induction of type-I interferon (e.g., IFNβ) and other IFN-stimulated genes (ISGs) (e.g., TDRD7, TRIM19, ISG15, etc.). These ISGs can inhibit viral replication in different ways, including their ability to inhibit autophagy, which some viruses induce for their own benefit. In this figure, some ISGs are shown because of their ability to inhibit autophagy. TDRD7 inhibits autophagy by blocking the activation of AMPK, the kinase that initiates the autophagy pathway. TRIM19 acts by suppressing virus-induced LC3-I to LC3-II conversion, which is essential for autophagy. Another ISG, ISG15, ISGylates BECN1, critical for autophagy, resulting in its loss of activity and thus inhibiting the induction of autophagy. In contrast, other ISGs, e.g., PKR, RNase L, can activate the autophagy pathway.
Viperin, the virus-inhibitory protein, which is endoplasmic reticulum-associated and interferon-inducible, inhibits the replication of many viruses, such as flaviviruses. The amphipathic nature of viperin allows it to interact with the ER and lipids, diversifying its anti-viral effects [48]. Keeping this in mind, HCV replicates its RNA on lipid droplets by interacting with NS5A, a similar viral amphipathic protein. Further along the autophagy pathway against HCV, viral NS5A is bound by SCOTIN/SHIAS5, another host ISG localized in the ER, leading to its autophagosomal degradation and the inhibition of HCV RNA replication [37]. Both points are proposed mechanistic targets of the anti-HCV action of viperin. Although viperin has been implicated as a therapeutic target against many other viruses, such as DENV, SINV, influenza A, SeV, VSV, HCMV, WNV, and HIV-1, whether there is a unified mechanism for its anti-viral action against all these viruses is not known [48,49]. While we have discussed various ISGs triggered by the presence of viral genomes, there are also IFN-related proteins that are generated by inflammation in response to viruses. As modeled by the Drosophila adult brain, the Zika virus (ZIKV) infection of neurons is modulated by an inflammation-induced protein called a Drosophila stimulator of IFN genes (dSTING). ZIKV, a flavivirus, initially triggers an NF-kB-dependent inflammatory cascade in the brain, leading to the production of dSTING, which subsequently stimulates IFN-regulatory transcription factor 3 (IRF3) and type I IFN production to induce autophagy and attenuate ZIKV infection in the brain [50].
Another family of immune modulators, known as tripartite motif (TRIM) proteins, often induced by IFN, are ubiquitin E3 ligases that carry out diverse anti-viral functions by either directly inhibiting a viral life cycle or by helping to induce the anti-viral autophagy of virus-infected cells. For instance, TRIM5α interacts with the HIV-1 capsid protein to induce the disassembly of HIV-1 progeny. TRIM proteins play various roles in autophagy induction by upregulating IFN transcription and regulating pattern-recognition receptors with anti-viral signaling cascades. HSV-1, SINV, and adenovirus activate TRIM23’s E3 enzymatic activity, to ubiquitinate cellular proteins linked to viral cargo for their eventual degradation within autophagosomes. TRIM23 is unique in that it also serves as a GTPase to regulate TBK1’s phosphorylation of p62, to enhance cargo recognition in autophagy [51].
In contrast to the various ISGs that induce autophagy to fight against viruses, we have recently discovered an anti-viral ISG, TDRD7, using a high-throughput screen from a library of ISGs. We showed that TDRD7 inhibits virus-induced autophagy to block paramyxovirus replication [6]. Since SeV, RSV, and HPIV3 utilize AMPK, the initiator of the autophagy pathway, we asked whether TDRD7 interferes with AMPK activity. Indeed, our results showed that TDRD7 inhibits AMPK activity in terms of its anti-autophagic and anti-viral functions (Figure 3). We further demonstrated that TDRD7 inhibits HSV-1 replication by its anti-AMPK activity [52]. HSV-1 replication is dependent on AMPK but not the autophagy pathway, and TDRD7 takes advantage of this to inhibit viral replication. We further showed that the TDRD7 anti-viral function is dependent on its ability to inhibit AMPK. The anti-viral function of TDRD7 is diminished in the AMPK inhibitor-treated cells.
Like TDRD7, promyelocytic leukemia (PML), also known as TRIM19, is another ISG in mammalian cells that inhibits autophagy by decreasing viral protein expression. TRIM19 forms aggregates into PML nuclear bodies, which also incorporate other cellular anti-viral restriction factors. TRIM19 has been shown to negatively regulate autophagy in response to an enterovirus 71 (EV71) infection of HeLa cells, as shown by a decrease in the number of autophagosomes and a decrease in the LC3-II/LC3-I ratio when compared to TRIM19 knockout cells [53].
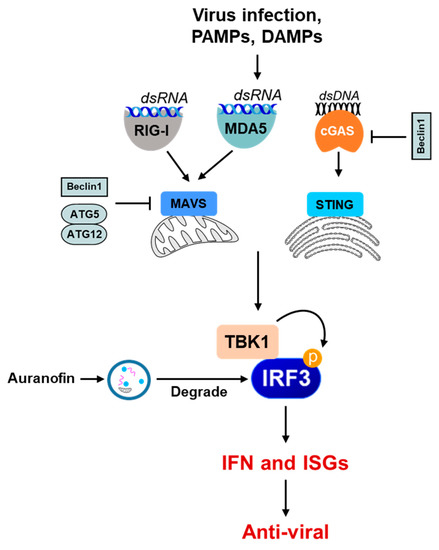
Autophagy-dependent degradation mechanisms can be harmful to IFN-mediated cellular responses. We have recently conducted a high-throughput screen to isolate novel small molecules that regulate viral apoptosis [54]. The screen also isolated auranofin, which inhibited the cellular IFN response [55]. We showed that auranofin activates the cellular autophagy pathway, degrading the IRF3 protein to block the IFN response (Figure 4). The degradation of IRF3 not only inhibits the transcriptional function but also the pro-apoptotic function of IRF3. Therefore, autophagy degrades viral components, as well as causing the degradation of IFN-regulatory proteins [56,57,58,59]. The cytoplasmic DNA-sensing pathway by cGAS/STING depends on the cellular autophagy pathway [60,61,62]. TBK1, a kinase critical for inducing IFN, can also act in the autophagy pathway [63,64], thereby regulating the IFN and autophagy arms of the innate response. Some autophagy pathway proteins can antagonize the innate immune signaling pathways to further regulate the interferon response (Figure 4). Therefore, crosstalk between the IFN and autophagy pathways can be a key regulator of the host response.
Figure 4.
The role of autophagy in the negative regulation of the innate immune signaling pathway. Some examples show that the autophagy machinery or its components inhibit innate immune-sensing pathways (e.g., RIG-I, MDA5, cGAS-STING) in various ways. Beclin1 interacts with MAVS and interferes with the RIG-I signaling pathway, thereby downregulating IFN production. Beclin1 also interacts with cGAS, interfering with the cGAMP synthesis and downstream stimulation of IFN production. The ATG5–ATG12 complex binds to RIG-I CARD and MAVS CARD and negatively regulates IFN production. Selective autophagy also directly degrades the PRRs, such as cGAS, and the adaptor proteins critical in the IFN-I production signaling pathways, such as MAVS, and thus inhibits IFN-I production. Small molecules, e.g., auranofin, can cause the autophagic degradation of the IRF3 protein to inhibit IRF3 activation and IFN induction.
5. Autophagy as a Pro-Viral Cellular Response
Several viruses often exploit autophagy to facilitate virus replication. The viruses use either a specific structure or a component of the autophagy machinery to promote their replication. HCV uses autophagy in several steps of the viral life cycle; HCV infection activates cellular autophagy, and the inhibition of this process results in decreased viral production. While autophagy inhibition does not affect viral entry, it is necessary to replicate viral RNA and translation [65]. HCV generates autophagosomes, using caveolin 1, caveolin 2, and annexin A2 to generate energy for its replication and promote virion assembly. Additionally, it uses the exocytosis pathway to release progeny virions [36]. As demonstrated, HCV serves as an excellent model of autophagy as a pro-viral mechanism, but countless other viruses similarly utilize parts of the process, many of which are mentioned in this review (Table 2).
Table 2.
Virus-induced autophagy as a pro-viral response.
Picornaviruses facilitate the formation of autophagosomes to facilitate viral replication, but they block the binding of lysosomes to these autophagosomes. During poliovirus infection, the use of rapamycin, an activator of autophagy, results in increased viral replication. Encephalomyocarditis virus (EMCV) uses its non-structural proteins 2C and 3D to generate ER stress, inducing autophagy and promoting its replication [70]. In Coxsackievirus B3 (CVB3) infection, the inhibition of autophagy by the deletion of Atg5 results in the inhibition of virus replication, specifically in pancreatic acinar cells [73]. We have shown that the Sendai virus (SeV), a paramyxovirus, also depends on cellular autophagy for viral replication [6]. The pharmacological or genetic inhibition of autophagy blocks SeV replication in vitro. SeV uses AMPK, the kinase required to initiate the autophagy pathway, to facilitate virus replication. The inhibition of AMPK by chemicals or genetic approaches blocks SeV replication. We have also shown that AMPK inhibition attenuates human parainfluenza virus (HPIV3) and respiratory syncytial virus (RSV) replication. The inhibition of autophagy and AMPK also attenuate RSV replication and pathogenesis in mice [78]. HPIV3 also triggers mitophagy to inhibit cellular anti-viral responses to IFN, thus advancing its replication [66,67]. During measles virus (MeV) infection, autophagy prevents host-cell death, thereby activating pro-survival pathways that favor viral replication [68]. Coronaviruses also use this autophagic membrane remodeling mechanism to generate viral membranes, as a crucial step in their life cycle [36]. Dengue virus (DENV) induces autophagy by the activation of AMPK, ultimately inducing the formation of a double-membrane vesicle that encloses lipid droplets. This form of autophagy is called lipophagy, which leads to the degradation of lipid droplets via β-oxidation to generate ATP for viral replication [36,79]. Lipophagy, a subset of autophagy where lipid droplets are mobilized within membraned vesicles, serves as a source of energy during infection with flaviviruses such as DENV and WNV. DENV infection leads to the induction of several innate immune responses. However, Dengue virus evades recognition by the host PRRs by replicating within the endoplasmic reticulum, while still using lipids through autophagy and delaying apoptosis to prevent its clearance [71].
The role of autophagy in herpesvirus replication is complex. Varicella-Zoster virus (VZV) has been shown to initiate autophagy upon infection; the chemical inhibition of autophagy or knocking down ATG5 results in decreased viral titer and viral proteins. Autophagy is thus required for the VZV life cycle, and this virus-induced autophagy results in enhanced viral protein synthesis [76]. In contrast to other viruses, HSV-2 does not induce autophagy but instead uses a basal level of autophagy for its own benefit [75]. The inhibition of autophagy in the setting of HSV-2 infection has been shown to hamper viral replication; however, the process is not needed for viral entry into cells. The formation of a double-membrane autophagosome may serve as a protected environment for the generation of progeny virions. HSV-1 replication does not depend on the autophagy pathway. We showed that HSV-1 replication remains unaltered in ATG5-knockdown cells [52]. However, the chemical or genetic inhibition of AMPK attenuates HSV-1 replication. AMPK inhibition in ATG5 knockdown cells also blocks HSV-1 replication. In murine gammaherpesvirus 68 (MHV68) infection, autophagy supports viral reactivation within macrophages. The induction of systemic inflammation, mediated by macrophagic IFN-ɣ, results in the suppression of autophagy genes. Thus, specifically in myeloid cells, Atg genes not only support MHV68 reactivation but also dampen systemic inflammation to curb autophagy inhibition [80].
6. Viral Antagonism to Autophagy
While autophagy is an effective mechanism against viral infections, many viruses have evolved to utilize counter-mechanisms that antagonize this anti-viral cellular response. Influenza A virus (IAV) evades cellular autophagy by using its matrix 2 (M2) proteins [74]. The cytoplasmic tail of viral M2 possesses an interaction motif—the LC3 interaction region (LIR)—through which the protein binds to LC3. This binding causes LC3 to re-localize to the plasma membrane, stabilizing influenza virions and facilitating the filamentous budding of the virion. IAV uses a protein–protein interaction to both inhibit anti-viral autophagy and promote its replication [74]. HSV-1 inhibits Beclin-1, a component of the autophagy machinery, to block autophagy induction. The HSV-1 protein ICP34.5 contains a Beclin-1-binding-domain (BBD), which is critical for inhibiting autophagy. The inhibition of autophagy by ICP34.5 results in the development of HSV-1 encephalitis [45,81]. HCMV has two ICP34.5 homologs called TRS1 and IRS1, both of which have BBDs to inhibit cellular autophagy and evade the anti-viral process [82,83]. Kaposi sarcoma-associated herpesvirus (KSHV) possesses a viral Bcl2 analog called ORF16 that mimics cellular Bcl2 and binds Beclin-1, as one of many immune evasion strategies [84]. Although not a member of the Herpesviridae, HIV has also been shown to use Beclin-1 inhibition as a viral counter-mechanism to autophagy. Several of these viral evasion mechanisms are listed in Table 3.
Table 3.
Viral antagonism to autophagy.
Viral Antagonism by SARS-CoV-2
It would be remiss to discuss the viral antagonism of host-cell defense mechanisms without addressing SARS-CoV-2, the beta-coronavirus responsible for the COVID-19 pandemic. Although much of the exact mechanism by which SARS-CoV-2 affects the autophagic pathway is yet to be discovered, AMPK has been shown to be a primary target. Upon the infection of a host cell by the (+)-sense RNA virus, levels of active phosphorylated AMPK (pAMPK) are reduced. Consequently, there is also a decreased presence of the active form of downstream target ULK (pULK), suggesting the inhibition of autophagy initiation as a key mechanism aiding in the CoV-2 evasion of anti-viral defense [90]. Furthermore, accessory factor ORF3a, encoded by the CoV-2 genome, has been identified as a key antagonist of host-cell autophagy. By binding to VPS39, a crucial facilitator of vesicle fusion, ORF3a inhibits the recruitment of Rab7 and the subsequent assembly of the SNARE complex, preventing autophagosome fusion with the lysosome [89]. This multifaceted inhibition of autophagy by the SARS-CoV-2 virus may explain its persistence upon infection.
7. Therapeutic Application of Autophagy in Virus Infection
The autophagy pathway can be targeted for therapeutic applications in many diseases, including viral infection [92]. However, because autophagy plays a dual role in viral infection and the immune response, it might be complex to target the autophagy pathway for anti-viral therapy. For example, chloroquine and hydroxychloroquine, originally approved as COVID-19 treatments, are inhibitors of the autophagy pathway [93]. Numerous reports suggest that SARS-CoV-2 entry may depend on the autophagy pathway; therefore, these autophagy inhibitors may be used to counteract the replication of the CoV-2 virus. We have shown that auranofin, an anti-rheumatic compound, induces autophagy to degrade IRF3 [55]. Auranofin has also been shown to inhibit SARS-CoV-2 replication [94]; whether auranofin degrades a viral protein or a pro-viral host protein in this context will require future evaluation. Oncolytic viruses, which are often used in anti-cancer therapy, may function by inducing the autophagy pathway [95]. Anti-retroviral drugs have been shown to impact the cellular autophagy pathway [96]. For example, it was shown that autophagy-promoting drugs, such as everolimus (chemotherapy), carbamazepine (anticonvulsant), and rapamycin (immunotherapy) restricted HIV-1 replication in a cell-specific manner [97]. This highlights the importance of developing and testing new drugs that act on autophagy and re-evaluating previously approved drugs that could be repurposed for therapeutic intervention. We recently characterized a screening strategy that would be useful in repurposing FDA-approved small compounds [54]. Virus-specific therapeutics that impact the autophagy pathway will require in-depth investigation before becoming viable treatment options.
8. Conclusions and Future Directions
Autophagy has been studied extensively in virus infection; however, it is not entirely clear how the viruses specifically utilize or are inhibited by autophagy. Similarly, how autophagy regulates the cellular responses to virus infection is under-studied. Since many autophagy-related proteins have roles in regulating ubiquitination, it would be interesting to assess them in autophagy-independent cellular functions. For example, the Atg5-Atg12 complex is involved in RLR signaling, independent of autophagy function [98]. Since viruses have co-evolved along with inhibiting various steps of autophagy, it is considered an anti-viral response of the host. However, viruses may block parts of the autophagy machinery while exploiting other proteins involved in autophagy for their own benefit. Virophagy, the engulfment of the entire virion, although reported in the case of SINV, is not entirely understood. This mechanism could be relevant in anti-viral action against other viruses as well and needs to be studied further. Another factor that is thought to be critical in virophagy is the type of cell involved [99]. The contribution of cell-specific autophagy is, thus, another aspect that needs to be better understood. It is known that this mechanism exists in most cells, but the importance of the activation of this phenomenon in certain cell types over others, and its role in anti-viral activity, could improve our understanding of innate immunity. Several autophagic proteins have been found to interact with viral proteins, ranging across five RNA virus families, as part of an interactome study [100]. These have yet to be studied in terms of a direct role in the restriction of viruses.
Author Contributions
K.C., G.S., S.C. (Saurabh Chattopadhyay) conceptualization; K.C., G.S., T.R., S.F., S.C. (Sukanya Chakravarty), S.G., H.D., R.C., S.C. (Saurabh Chattopadhyay) data curation; S.C. (Saurabh Chattopadhyay); project administration; K.C., G.S., T.R., S.C. (Saurabh Chattopadhyay) writing—original draft; K.C., G.S., T.R., S.G., H.D., R.C., S.C. (Saurabh Chattopadhyay) writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the National Institutes of Health grants AI155545 (S.C. (Saurabh Chattopadhyay)), AA026017 (S.C. (Saurabh Chattopadhyay)), AA027456 (S.C. (Saurabh Chattopadhyay)), AI153496 (S.C. (Saurabh Chattopadhyay)), Medical Research Society (S.C. (Saurabh Chattopadhyay)), Ohio Department of Health (S.C. (Saurabh Chattopadhyay) and R.C.), and the University of Toledo College of Medicine and Life Sciences (S.C. (Saurabh Chattopadhyay) and R.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
We thank Julie Gartland, Bryanna Jay, and Stephanie Fabry for their continued support of this review.
Conflicts of Interest
The authors declare that they have no conflict of interest with the contents of this article.
References
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Chisari, F.V. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 2001, 19, 65–91. [Google Scholar] [CrossRef]
- Subramanian, G.; Kuzmanovic, T.; Zhang, Y.; Peter, C.B.; Veleeparambil, M.; Chakravarti, R.; Sen, G.C.; Chattopadhyay, S. A new mechanism of interferon’s antiviral action: Induction of autophagy, essential for paramyxovirus replication, is inhibited by the interferon stimulated gene, TDRD7. PLoS Pathog. 2018, 14, e1006877. [Google Scholar] [CrossRef] [Green Version]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Ahmad, L.; Mostowy, S.; Sancho-Shimizu, V. Autophagy-Virus Interplay: From Cell Biology to Human Disease. Front. Cell Dev. Biol. 2018, 6, 155. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Lin, E.; He, L.; Yu, J.; Tan, P.; Zhou, Y. Autophagy and Viral Infection. Adv. Exp. Med. Biol. 2019, 1209, 55–78. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Pereira, G.; Leao, A.; Erustes, A.G.; Morais, I.B.M.; Vrechi, T.A.M.; Zamarioli, L.D.S.; Pereira, C.A.S.; Marchioro, L.O.; Sperandio, L.P.; Lins, I.V.F.; et al. Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19. Int. J. Mol. Sci. 2021, 22, 4067. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent roles of autophagy in virus infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farre, J.C.; Subramani, S. Mechanistic insights into selective autophagy pathways: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2016, 17, 537–552. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrosio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Nakatogawa, H.; Mochida, K. Reticulophagy and nucleophagy: New findings and unsolved issues. Autophagy 2015, 11, 2377–2378. [Google Scholar] [CrossRef] [Green Version]
- Lennemann, N.J.; Coyne, C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Blum, J.S. Autophagy and adaptive immunity. Immunology 2010, 131, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.B.; Schwarz, H.; Mayer, A. Determination of four sequential stages during microautophagy in vitro. J. Biol. Chem. 2004, 279, 9987–9996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.W.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, T.; Parton, R.G. Not just fat: The structure and function of the lipid droplet. Cold Spring Harb. Perspect. Biol. 2011, 3, a004838. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Cheng, J.; Fujita, A.; Tokumoto, T.; Fujimoto, T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol. Biol. Cell 2006, 17, 2674–2683. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010, 24, 3052–3065. [Google Scholar] [CrossRef] [Green Version]
- Mei, S.; Ni, H.M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.X. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Thoen, L.F.; Guimaraes, E.L.; Dolle, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses Exploit the Lipid Droplet Protein AUP1 to Trigger Lipophagy and Drive Virus Production. Cell Host Microbe 2018, 23, 819–831.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, S.K.; Dang, J.W.; Lin, N.; Qin, Y.; Wang, S.; Rana, T.M. Zika virus depletes neural stem cells and evades selective autophagy by suppressing the Fanconi anemia protein FANCC. EMBO Rep. 2020, 21, e49183. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R., Jr.; Sirasanagandla, S.; Fernandez, A.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viret, C.; Rozieres, A.; Faure, M. Novel Insights into NDP52 Autophagy Receptor Functioning. Trends Cell Biol. 2018, 28, 255–257. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, M.J.; Sung, P.S.; Bae, Y.C.; Shin, E.C.; Yoo, J.Y. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 2016, 7, 10631. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Maloney, N.S.; Bruinsma, M.W.; Goel, G.; Duan, E.; Zhang, L.; Shrestha, B.; Diamond, M.S.; Dani, A.; Sosnovtsev, S.V.; et al. Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 2012, 11, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Shelly, S.; Lukinova, N.; Bambina, S.; Berman, A.; Cherry, S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009, 30, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Tokarev, A.; Suarez, M.; Kwan, W.; Fitzpatrick, K.; Singh, R.; Guatelli, J. Stimulation of NF-kappaB activity by the HIV restriction factor BST2. J. Virol. 2013, 87, 2046–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotzer, V.L.; Blum, J.S. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glanz, A.; Chakravarty, S.; Varghese, M.; Kottapalli, A.; Fan, S.; Chakravarti, R.; Chattopadhyay, S. Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus. Viruses 2021, 13, 575. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.A.; Malathi, K. RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 2012, 287, 43651–43664. [Google Scholar] [CrossRef] [Green Version]
- Manivannan, P.; Siddiqui, M.A.; Malathi, K. RNase L Amplifies Interferon Signaling by Inducing Protein Kinase R-Mediated Antiviral Stress Granules. J. Virol. 2020, 94, e00205-20. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.Y.; Yaneva, R.; Cresswell, P. Viperin: A multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe 2011, 10, 534–539. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.Y.; Cresswell, P. Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLoS Pathog. 2013, 9, e1003497. [Google Scholar] [CrossRef]
- Liu, Y.; Gordesky-Gold, B.; Leney-Greene, M.; Weinbren, N.L.; Tudor, M.; Cherry, S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe 2018, 24, 57–68.e3. [Google Scholar] [CrossRef] [Green Version]
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Popli, S.; Chakravarty, S.; Taylor, R.T.; Chakravarti, R.; Chattopadhyay, S. The interferon-inducible protein TDRD7 inhibits AMP-activated protein kinase and thereby restricts autophagy-independent virus replication. J. Biol. Chem. 2020, 295, 6811–6822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Feng, C.; Tian, X.; Zheng, N.; Wu, Z. Promyelocytic Leukemia Restricts Enterovirus 71 Replication by Inhibiting Autophagy. Front. Immunol. 2018, 9, 1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glanz, A.; Chawla, K.; Fabry, S.; Subramanian, G.; Garcia, J.; Jay, B.; Ciricillo, J.; Chakravarti, R.; Taylor, R.T.; Chattopadhyay, S. High Throughput Screening of FDA-Approved Drug Library Reveals the Compounds that Promote IRF3-Mediated Pro-Apoptotic Pathway Inhibit Virus Replication. Viruses 2020, 12, 442. [Google Scholar] [CrossRef] [Green Version]
- Glanz, A.; Chakravarty, S.; Fan, S.; Chawla, K.; Subramanian, G.; Rahman, T.; Walters, D.; Chakravarti, R.; Chattopadhyay, S. Autophagic degradation of IRF3 induced by the small-molecule auranofin inhibits its transcriptional and proapoptotic activities. J. Biol. Chem. 2021, 297, 101274. [Google Scholar] [CrossRef]
- Xie, W.; Jin, S.; Zhang, C.; Yang, S.; Wu, Y.; Zhao, Y.; Songyang, Z.; Cui, J. Selective autophagy controls the stability of TBK1 via NEDD4 to balance host defense. Cell Death Differ. 2021, 29, 40–53. [Google Scholar] [CrossRef]
- Liu, J.; Wu, X.; Wang, H.; Wei, J.; Wu, Q.; Wang, X.; Yan, Y.; Cui, J.; Min, J.; Wang, F.; et al. HFE inhibits type I IFNs signaling by targeting the SQSTM1-mediated MAVS autophagic degradation. Autophagy 2021, 17, 1962–1977. [Google Scholar] [CrossRef]
- Wu, Y.; Jin, S.; Liu, Q.; Zhang, Y.; Ma, L.; Zhao, Z.; Yang, S.; Li, Y.P.; Cui, J. Selective autophagy controls the stability of transcription factor IRF3 to balance type I interferon production and immune suppression. Autophagy 2021, 17, 1379–1392. [Google Scholar] [CrossRef]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef] [Green Version]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef]
- Prantner, D.; Perkins, D.J.; Vogel, S.N. AMP-activated Kinase (AMPK) Promotes Innate Immunity and Antiviral Defense through Modulation of Stimulator of Interferon Genes (STING) Signaling. J. Biol. Chem. 2017, 292, 292–304. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Ge, J.; Rodgers, M.A.; Shi, M.; Jung, J.U. Autophagy side of MB21D1/cGAS DNA sensor. Autophagy 2014, 10, 1146–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herhaus, L.; Bhaskara, R.M.; Lystad, A.H.; Gestal-Mato, U.; Covarrubias-Pinto, A.; Bonn, F.; Simonsen, A.; Hummer, G.; Dikic, I. TBK1-mediated phosphorylation of LC3C and GABARAP-L2 controls autophagosome shedding by ATG4 protease. EMBO Rep. 2020, 21, e48317. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Elazar, Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 2011, 4, pe39. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Qin, Y.; Chen, M. Viral strategies for triggering and manipulating mitophagy. Autophagy 2018, 14, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.; Zhang, L.; Li, Z.; Zhong, Y.; Tang, Q.; Qin, Y.; Chen, M. The Matrix Protein of Human Parainfluenza Virus Type 3 Induces Mitophagy that Suppresses Interferon Responses. Cell Host Microbe 2017, 21, 538–547.e4. [Google Scholar] [CrossRef] [Green Version]
- Lennemann, N.J.; Coyne, C.B. Catch me if you can: The link between autophagy and viruses. PLoS Pathog. 2015, 11, e1004685. [Google Scholar] [CrossRef]
- Richetta, C.; Gregoire, I.P.; Verlhac, P.; Azocar, O.; Baguet, J.; Flacher, M.; Tangy, F.; Rabourdin-Combe, C.; Faure, M. Sustained autophagy contributes to measles virus infectivity. PLoS Pathog. 2013, 9, e1003599. [Google Scholar] [CrossRef]
- Hou, L.; Ge, X.; Xin, L.; Zhou, L.; Guo, X.; Yang, H. Nonstructural proteins 2C and 3D are involved in autophagy as induced by the encephalomyocarditis virus. Virol. J. 2014, 11, 156. [Google Scholar] [CrossRef] [Green Version]
- Green, A.M.; Beatty, P.R.; Hadjilaou, A.; Harris, E. Innate immunity to dengue virus infection and subversion of antiviral responses. J. Mol. Biol. 2014, 426, 1148–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, V.; Lai, C.K.; Chao, T.C.; Jeng, K.S.; Lai, M.M. Annexin A2 is involved in the formation of hepatitis C virus replication complex on the lipid raft. J. Virol. 2012, 86, 4139–4150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Whitton, J.L. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell Host Microbe 2012, 11, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Yakoub, A.M.; Shukla, D. Basal Autophagy Is Required for Herpes simplex Virus-2 Infection. Sci. Rep. 2015, 5, 12985. [Google Scholar] [CrossRef]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Grose, C. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J. Virol. 2014, 88, 890–902. [Google Scholar] [CrossRef] [Green Version]
- Lukhele, S.; Cohen, E.A. Conserved residues within the HIV-1 Vpu transmembrane-proximal hinge region modulate BST2 binding and antagonism. Retrovirology 2017, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, J.; Zeng, R.; Yang, J.; Liu, J.; Zhang, Z.; Song, X.; Yao, Z.; Ma, C.; Li, W.; et al. Respiratory Syncytial Virus Replication Is Promoted by Autophagy-Mediated Inhibition of Apoptosis. J. Virol. 2018, 92, e02193-17. [Google Scholar] [CrossRef] [Green Version]
- Jordan, T.X.; Randall, G. Dengue Virus Activates the AMP Kinase-mTOR Axis To Stimulate a Proviral Lipophagy. J. Virol. 2017, 91, e02020-16. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Buck, M.D.; Desai, C.; Zhang, X.; Loginicheva, E.; Martinez, J.; Freeman, M.L.; Saitoh, T.; Akira, S.; Guan, J.L.; et al. Autophagy Genes Enhance Murine Gammaherpesvirus 68 Reactivation from Latency by Preventing Virus-Induced Systemic Inflammation. Cell Host Microbe 2016, 19, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Yordy, B.; Iijima, N.; Huttner, A.; Leib, D.; Iwasaki, A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 2012, 12, 334–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaumorcel, M.; Lussignol, M.; Mouna, L.; Cavignac, Y.; Fahie, K.; Cotte-Laffitte, J.; Geballe, A.; Brune, W.; Beau, I.; Codogno, P.; et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 2012, 86, 2571–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouna, L.; Hernandez, E.; Bonte, D.; Brost, R.; Amazit, L.; Delgui, L.R.; Brune, W.; Geballe, A.P.; Beau, I.; Esclatine, A. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 2016, 12, 327–342. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Wei, D.; Chung, B.; Brulois, K.F.; Guo, C.; Dong, S.; Gao, S.J.; Feng, P.; Liang, C.; Jung, J.U. Novel Role of vBcl2 in the Virion Assembly of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2018, 92, e00914-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [Green Version]
- Staring, J.; von Castelmur, E.; Blomen, V.A.; van den Hengel, L.G.; Brockmann, M.; Baggen, J.; Thibaut, H.J.; Nieuwenhuis, J.; Janssen, H.; van Kuppeveld, F.J.; et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature 2017, 541, 412–416. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.; Pacheco, J.M.; LaRocco, M.; Burrage, T.; Jackson, W.; Rodriguez, L.L.; Borca, M.V.; Baxt, B. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 2011, 410, 142–150. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, S.; Saha, A. Therapeutic Potential of Exploiting Autophagy Cascade Against Coronavirus Infection. Front. Microbiol. 2021, 12, 675419. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Stone, S.; Natekar, J.; Kumari, P.; Arora, K.; Kumar, M. The FDA-approved gold drug auranofin inhibits novel coronavirus (SARS-CoV-2) replication and attenuates inflammation in human cells. Virology 2020, 547, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Jiang, K.; Ding, C.; Meng, S. Targeting Autophagy for Oncolytic Immunotherapy. Biomedicines 2017, 5, 5. [Google Scholar] [CrossRef]
- Cheney, L.; Barbaro, J.M.; Berman, J.W. Antiretroviral Drugs Impact Autophagy with Toxic Outcomes. Cells 2021, 10, 909. [Google Scholar] [CrossRef]
- Cloherty, A.P.M.; van Teijlingen, N.H.; Eisden, T.; van Hamme, J.L.; Rader, A.G.; Geijtenbeek, T.B.H.; Schreurs, R.; Ribeiro, C.M.S. Autophagy-enhancing drugs limit mucosal HIV-1 acquisition and suppress viral replication ex vivo. Sci. Rep. 2021, 11, 4767. [Google Scholar] [CrossRef]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [Green Version]
- Viret, C.; Rozieres, A.; Faure, M. Autophagy during Early Virus-Host Cell Interactions. J. Mol. Biol. 2018, 430, 1696–1713. [Google Scholar] [CrossRef]
- Gregoire, I.P.; Rabourdin-Combe, C.; Faure, M. Autophagy and RNA virus interactomes reveal IRGM as a common target. Autophagy 2012, 8, 1136–1137. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).