
The Role of APOE and NF-κB in Alzheimer’s Disease


{kind=link}
Abstract
:1. Introduction
2. Neuroinflammation and Alzheimer’s Disease
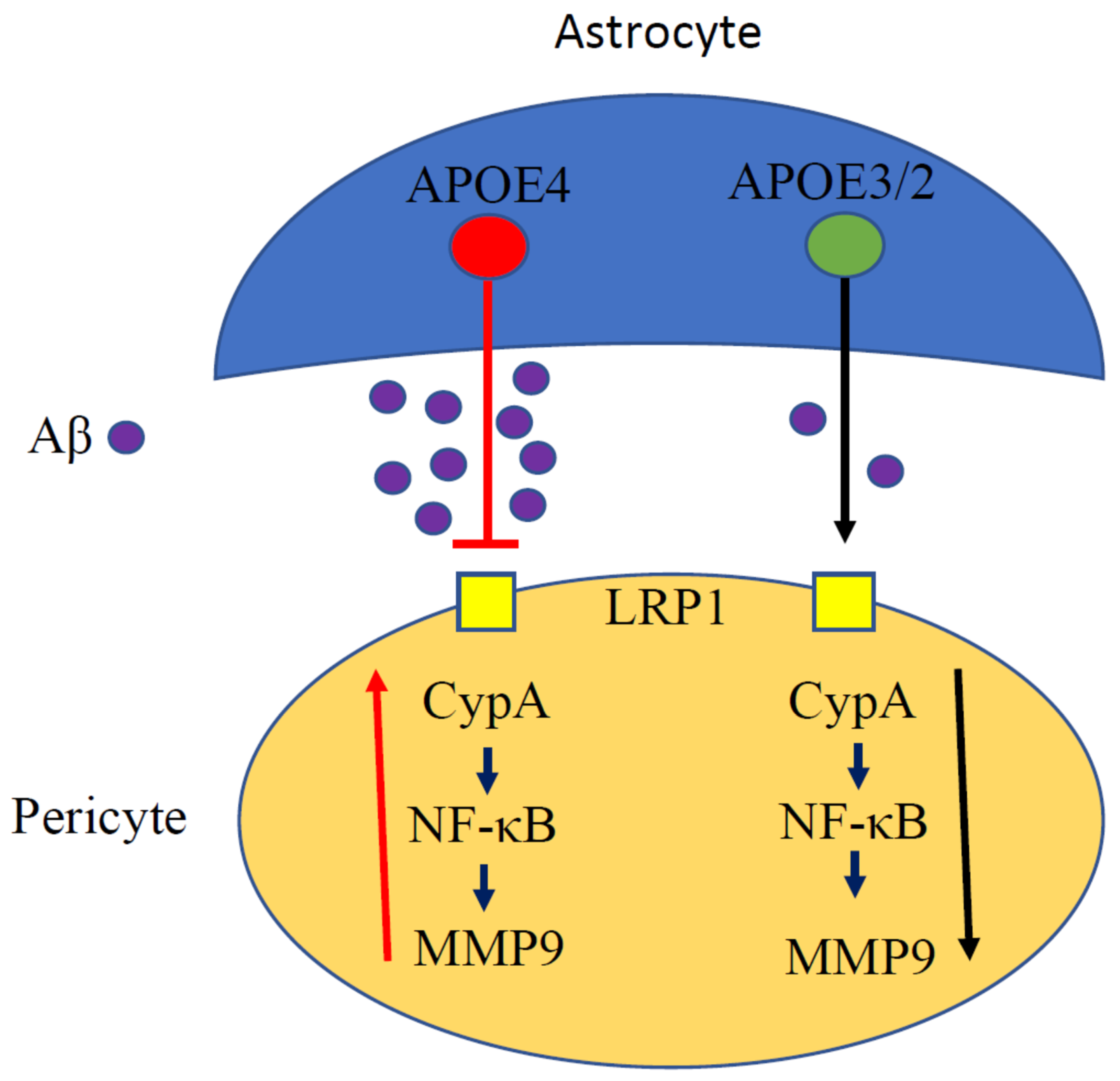
3. APOE and Inflammation
4. APOE and NF-κB
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Glabe, C.C. Amyloid accumulation and pathogensis of Alzheimer’s disease: Significance of monomeric, oligomeric and fibrillar Abeta. Subcell. Biochem. 2005, 38, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020, 163, 1599–1617. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Albensi, B.C. The nuclear factor kappa B (NF-kappaB) signaling pathway is involved in ammonia-induced mitochondrial dysfunction. Mitochondrion 2020, 57, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Banerjee, D. A primer on the evolution of aducanumab: The first antibody approved for treatment of alzheimer’s disease. J. Alzheimers Dis. 2021, 83, 1537–1552. [Google Scholar] [CrossRef]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and alzheimer disease meta analysis consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.P.; Clark, I.A.; Zinn, R.; Vissel, B. Microglia: A new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol. Learn. Mem. 2013, 105, 40–53. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural. Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-kappaB) doing in and to the mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. NF-kappa B as a therapeutic target in neurodegenerative diseases. Expert Opin. Ther. Targets 2007, 11, 123–132. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Baeuerle, P.A.; Kaltschmidt, C. Potential involvement of the transcription factor NF-kappa B in neurological disorders. Mol. Aspects Med. 1993, 14, 171–190. [Google Scholar] [CrossRef]
- Singh, S.; Singh, T.G. Role of nuclear factor kappa B (NF-kappaB) signalling in neurodegenerative diseases: An Mechanistic approach. Curr. Neuropharmacol. 2020, 18, 918–935. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kaltschmidt, C. NF-kappa B: A crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997, 20, 252–258. [Google Scholar] [CrossRef]
- Zhou, Y.; Cui, C.; Ma, X.; Luo, W.; Zheng, S.G.; Qiu, W. Nuclear Factor kappaB (NF-kappaB)-mediated inflammation in multiple sclerosis. Front. Immunol. 2020, 11, 391. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Buggia-Prevot, V.; Sevalle, J.; Rossner, S.; Checler, F. NFkappaB-dependent control of BACE1 promoter transactivation by Abeta42. J. Biol. Chem. 2008, 283, 10037–10047. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Bourne, K.Z.; Ferrari, D.C.; Lange-Dohna, C.; Rossner, S.; Wood, T.G.; Perez-Polo, J.R. Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J. Neurosci. Res. 2007, 85, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.R.; Lyra, E.S.N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazao, R.; et al. Alzheimer-associated Abeta oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Kounatidis, I.; Chtarbanova, S.; Cao, Y.; Hayne, M.; Jayanth, D.; Ganetzky, B.; Ligoxygakis, P. NF-kappaB immunity in the brain determines fly lifespan in healthy aging and age-related neurodegeneration. Cell Rep. 2017, 19, 836–848. [Google Scholar] [CrossRef]
- Bortolotto, V.; Grilli, M. Every cloud has a silver lining: Proneurogenic effects of abeta oligomers and HMGB-1 via activation of the RAGE-NF-kappaB Axis. CNS Neurol. Disord. Drug Targets 2017, 16, 1066–1079. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Aragno, M.; Tamagno, E.; Vercellinatto, I.; Visentin, S.; Medana, C.; Catalano, M.G.; Smith, M.A.; Perry, G.; Danni, O.; et al. AGEs/RAGE complex upregulates BACE1 via NF-kappaB pathway activation. Neurobiol. Aging 2012, 33, e113–e127. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Camandola, S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J. Clin. Investig. 2001, 107, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-kappaB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef]
- Seo, E.J.; Fischer, N.; Efferth, T. Phytochemicals as inhibitors of NF-kappaB for treatment of Alzheimer’s disease. Pharmacol. Res. 2018, 129, 262–273. [Google Scholar] [CrossRef]
- Ellis, R.J.; Olichney, J.M.; Thal, L.J.; Mirra, S.S.; Morris, J.C.; Beekly, D.; Heyman, A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, Part XV. Neurology 1996, 46, 1592–1596. [Google Scholar] [CrossRef]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Polvikoski, T.; Sulkava, R.; Haltia, M.; Kainulainen, K.; Vuorio, A.; Verkkoniemi, A.; Niinisto, L.; Halonen, P.; Kontula, K. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N. Engl. J. Med. 1995, 333, 1242–1247. [Google Scholar] [CrossRef]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef] [Green Version]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergstrom, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Barthel, H.; Gertz, H.J.; Dresel, S.; Peters, O.; Bartenstein, P.; Buerger, K.; Hiemeyer, F.; Wittemer-Rump, S.M.; Seibyl, J.; Reininger, C.; et al. Cerebral amyloid-beta PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: A multicentre phase 2 diagnostic study. Lancet Neurol. 2011, 10, 424–435. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Liu, X.; Bandy, D.; Yu, M.; Lee, W.; Ayutyanont, N.; Keppler, J.; Reeder, S.A.; Langbaum, J.B.; et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 6820–6825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleisher, A.S.; Chen, K.; Liu, X.; Ayutyanont, N.; Roontiva, A.; Thiyyagura, P.; Protas, H.; Joshi, A.D.; Sabbagh, M.; Sadowsky, C.H.; et al. Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol. Aging 2013, 34, 1–12. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Balu, D.; Karstens, A.J.; Loukenas, E.; Maldonado Weng, J.; York, J.M.; Valencia-Olvera, A.C.; LaDu, M.J. The role of APOE in transgenic mouse models of AD. Neurosci. Lett. 2019, 707, 134285. [Google Scholar] [CrossRef]
- Chen, X.F.; Zhang, Y.W.; Xu, H.; Bu, G. Transcriptional regulation and its misregulation in Alzheimer’s disease. Mol. Brain 2013, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar-Colucci, S.; Landreth, G.E. Nuclear receptors as therapeutic targets for Alzheimer’s disease. Expert Opin. Ther. Targets 2011, 15, 1085–1097. [Google Scholar] [CrossRef]
- Zannis, V.I.; Kan, H.Y.; Kritis, A.; Zanni, E.; Kardassis, D. Transcriptional regulation of the human apolipoprotein genes. Front. Biosci. 2001, 6, D456–D504. [Google Scholar] [CrossRef] [PubMed]
- Bales, K.R.; Verina, T.; Dodel, R.C.; Du, Y.; Altstiel, L.; Bender, M.; Hyslop, P.; Johnstone, E.M.; Little, S.P.; Cummins, D.J.; et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 1997, 17, 263–264. [Google Scholar] [CrossRef]
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid. Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Nwabuisi-Heath, E.; Dumanis, S.B.; Tai, L.M.; Yu, C.; Rebeck, G.W.; LaDu, M.J. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 2012, 60, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.A.; Tai, L.M.; LaDu, M.J.; Rebeck, G.W. Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J. Neuroinflamm. 2014, 11, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, J.L.; Zhao, C.B.; Vollmer, T.; Coons, S.; Lin, H.J.; Marsh, S.; Treiman, D.M.; Shi, J. APOE 4 polymorphism results in early cognitive deficits in an EAE model. Biochem. Biophys. Res. Commun. 2009, 384, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Tang, W.; Wang, H.; Vitek, M.P.; Bennett, E.R.; Sullivan, P.M.; Warner, D.S.; Laskowitz, D.T. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 2003, 278, 48529–48533. [Google Scholar] [CrossRef] [Green Version]
- Pocivavsek, A.; Burns, M.P.; Rebeck, G.W. Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia 2009, 57, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Lynch, J.R.; Wang, H.; Mace, B.; Leinenweber, S.; Warner, D.S.; Bennett, E.R.; Vitek, M.P.; McKenna, S.; Laskowitz, D.T. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp. Neurol. 2005, 192, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; LaDu, M.J.; Van Eldik, L.J. A dual role for apolipoprotein e in neuroinflammation: Anti-and pro-inflammatory activity. J. Mol. Neurosci. 2004, 23, 205–212. [Google Scholar] [CrossRef]
- Baitsch, D.; Bock, H.H.; Engel, T.; Telgmann, R.; Muller-Tidow, C.; Varga, G.; Bot, M.; Herz, J.; Robenek, H.; von Eckardstein, A.; et al. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1160–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowitz, D.T.; Matthew, W.D.; Bennett, E.R.; Schmechel, D.; Herbstreith, M.H.; Goel, S.; McMillian, M.K. Endogenous apolipoprotein E suppresses LPS-stimulated microglial nitric oxide production. Neuroreport 1998, 9, 615–618. [Google Scholar] [CrossRef]
- Pocivavsek, A.; Rebeck, G.W. Inhibition of c-Jun N-terminal kinase increases apoE expression in vitro and in vivo. Biochem. Biophys. Res. Commun. 2009, 387, 516–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldklang, M.; Golovatch, P.; Zelonina, T.; Trischler, J.; Rabinowitz, D.; Lemaitre, V.; D’Armiento, J. Activation of the TLR4 signaling pathway and abnormal cholesterol efflux lead to emphysema in ApoE-deficient mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L1200–L1208. [Google Scholar] [CrossRef] [Green Version]
- Michelsen, K.S.; Doherty, T.M.; Shah, P.K.; Arditi, M. TLR signaling: An emerging bridge from innate immunity to atherogenesis. J. Immunol. 2004, 173, 5901–5907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; LaDu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Ophir, G.; Amariglio, N.; Jacob-Hirsch, J.; Elkon, R.; Rechavi, G.; Michaelson, D.M. Apolipoprotein E4 enhances brain inflammation by modulation of the NF-kappaB signaling cascade. Neurobiol. Dis. 2005, 20, 709–718. [Google Scholar] [CrossRef]
- Noguchi, T.; Ebina, K.; Hirao, M.; Otsuru, S.; Guess, A.J.; Kawase, R.; Ohama, T.; Yamashita, S.; Etani, Y.; Okamura, G.; et al. Apolipoprotein E plays crucial roles in maintaining bone mass by promoting osteoblast differentiation via ERK1/2 pathway and by suppressing osteoclast differentiation via c-Fos, NFATc1, and NF-kappaB pathway. Biochem. Biophys. Res. Commun. 2018, 503, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, S.; Shao, Z.; Li, Y.; Wu, H.; Li, X.; Mao, L.; Zhou, Z.; Bai, L.; Mei, X.; et al. Apolipoprotein E deficiency exacerbates spinal cord injury in mice: Inflammatory response and oxidative stress mediated by NF-kappaB signaling pathway. Front. Cell Neurosci. 2018, 12, 142. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, X.; Ma, Z.; Niu, J.; Ma, S.; Wenjie, W.; Chen, J. Combination of tanshinone IIA and astragaloside IV attenuate atherosclerotic plaque vulnerability in ApoE(−/−) mice by activating PI3K/AKT signaling and suppressing TRL4/NF-kappaB signaling. Biomed. Pharmacother. 2020, 123, 109729. [Google Scholar] [CrossRef]
- Ophir, G.; Mizrahi, L.; Michaelson, D.M. Enhanced activation of NF-kB signaling by apolipoprotein E4. In Advances in Alzheimer’s and Parkinson’s Disease; Springer: Boston, MA, USA, 2008. [Google Scholar]
- Teng, Z.; Guo, Z.; Zhong, J.; Cheng, C.; Huang, Z.; Wu, Y.; Tang, S.; Luo, C.; Peng, X.; Wu, H.; et al. ApoE influences the blood-brain barrier through the NF-kappaB/MMP-9 pathway after traumatic brain injury. Sci. Rep. 2017, 7, 6649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.J.; Zhang, H.L.; Zhang, X.M.; Zheng, X.Y.; Quezada, H.C.; Zhang, D.; Zhu, J. Apolipoprotein E isoform-specific effects on cytokine and nitric oxide production from mouse Schwann cells after inflammatory stimulation. Neurosci. Lett. 2011, 499, 175–180. [Google Scholar] [CrossRef]
- Kemp, S.B.; Carpenter, E.S.; Steele, N.G.; Donahue, K.L.; Nwosu, Z.C.; Pacheco, A.; Velez-Delgado, A.; Menjivar, R.E.; Lima, F.; The, S.; et al. Apolipoprotein E promotes immune suppression in pancreatic cancer through NF-kappaB-mediated production of CXCL1. Cancer Res. 2021, 81, 4305–4318. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, J.; Lu, T.; Zhan, Z.; Wei, W.; Lyu, X.; Jiang, Y.; Xue, X. The effect of swimming exercise and diet on the hypothalamic inflammation of ApoE−/− mice based on SIRT1-NF-kappaB-GnRH expression. Aging 2020, 12, 11085–11099. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Tang, C.; Peng, K.; Sun, H.; Yang, Y. Effects of chronic mild stress on the development of atherosclerosis and expression of toll-like receptor 4 signaling pathway in adolescent apolipoprotein E knockout mice. J. Biomed. Biotechnol. 2009, 2009, 613879. [Google Scholar] [CrossRef]
- Tang, Y.L.; Jiang, J.H.; Wang, S.; Liu, Z.; Tang, X.Q.; Peng, J.; Yang, Y.Z.; Gu, H.F. TLR4/NF-kappaB signaling contributes to chronic unpredictable mild stress-induced atherosclerosis in ApoE−/− mice. PLoS ONE 2015, 10, e0123685. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, P.; Feng, B. Effect of atorvastatin on AGEs-induced injury of cerebral cortex via inhibiting NADPH oxidase -NF-kappaB pathway in ApoE(−/−) mice. Mol. Biol. Rep. 2020, 47, 9479–9488. [Google Scholar] [CrossRef]
- Han, J.; Chen, D.; Liu, D.; Zhu, Y. Modafinil attenuates inflammation via inhibiting Akt/NF-kappaB pathway in apoE-deficient mouse model of atherosclerosis. Inflammopharmacology 2018, 26, 385–393. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Cerebrovascular effects of apolipoprotein E: Implications for Alzheimer disease. JAMA Neurol. 2013, 70, 440–444. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.; Spiller, K.J.; Towne, C.; Kanning, K.C.; Choe, G.T.; Geber, A.; Akay, T.; Aebischer, P.; Henderson, C.E. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 2014, 81, 333–348. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Nikolakopoulou, A.M.; Huuskonen, M.T.; Sagare, A.P.; Lawson, E.J.; Lazic, D.; Rege, S.; Grond, A.; Zuniga, E.; Zlokovic, B.V.; et al. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat. Aging 2021, 1, 506–520. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offen, D.; Rabinowitz, R.; Michaelson, D.; Ben-Zur, T. Towards gene-editing treatment for alzheimer’s disease: ApoE4 allele-specific knockout using CRISPR cas9 variant. Cytotherapy 2018, 20, S18. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor-kappa beta as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davies, D.A. The Role of APOE and NF-κB in Alzheimer’s Disease. Immuno 2021, 1, 391-399. https://doi.org/10.3390/immuno1040027
Davies DA. The Role of APOE and NF-κB in Alzheimer’s Disease. Immuno. 2021; 1(4):391-399. https://doi.org/10.3390/immuno1040027
Chicago/Turabian StyleDavies, Don A. 2021. "The Role of APOE and NF-κB in Alzheimer’s Disease" Immuno 1, no. 4: 391-399. https://doi.org/10.3390/immuno1040027
APA StyleDavies, D. A. (2021). The Role of APOE and NF-κB in Alzheimer’s Disease. Immuno, 1(4), 391-399. https://doi.org/10.3390/immuno1040027