
Dysfunctional Astrocyte Metabolism: A Driver of Imbalanced Excitatory/Inhibitory Tone and Support for Therapeutic Intervention Targets

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Astrocyte Metabolism: The Foundation of E/I Balance
2.1. Glucose Metabolism
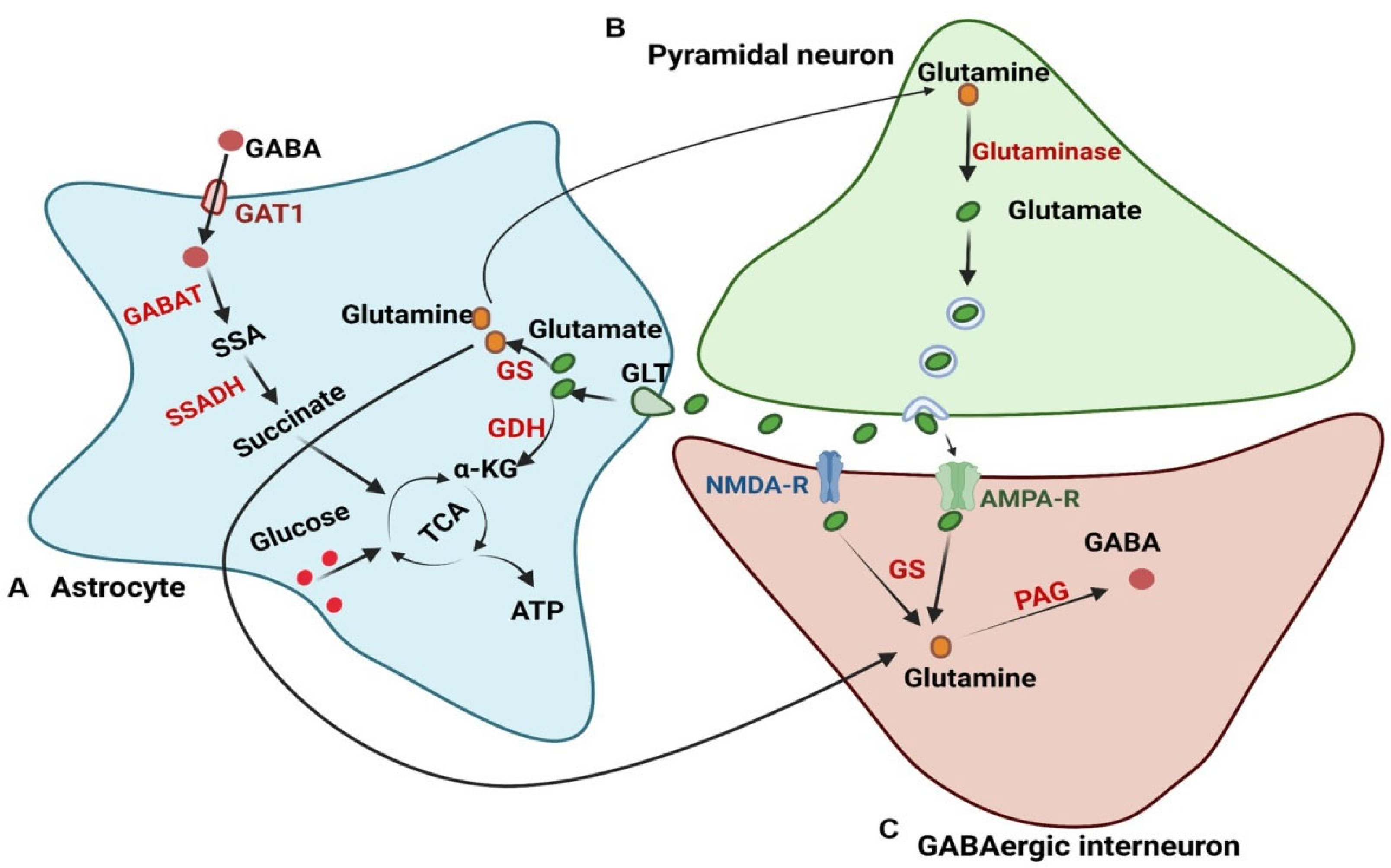
2.2. Glutamate–Glutamine Cycle
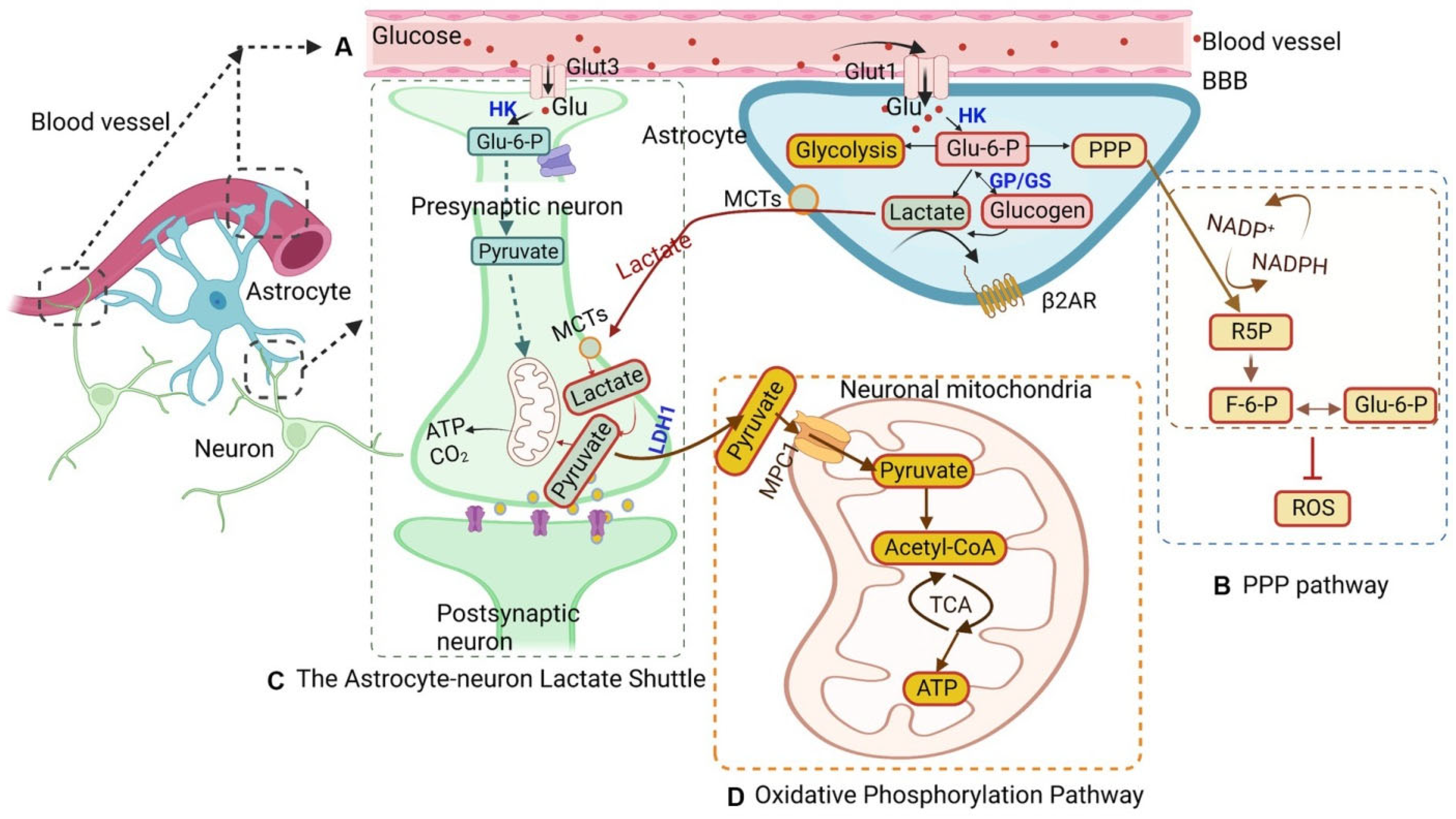
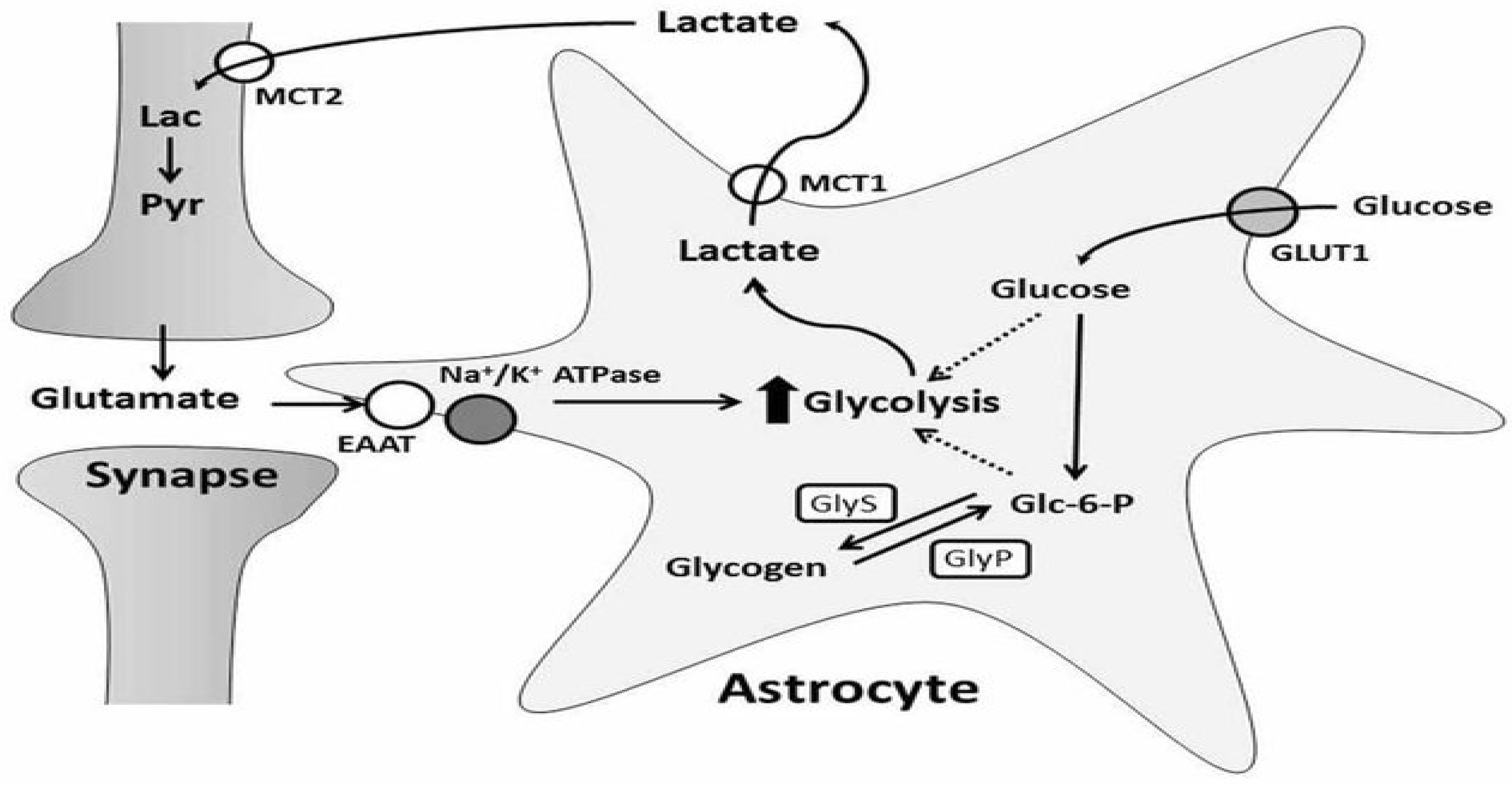
2.3. Lactate Shuttle
2.4. Other Metabolic Pathways
3. Mechanisms Linking Astrocyte Metabolic Dysfunction to E/I Imbalance
3.1. Altered Glutamate Homeostasis
3.2. Disrupted GABA Synthesis and Transport
3.3. Energy Deprivation and Neuronal Vulnerability
3.4. Neuroinflammation and Metabolic Crosstalk
3.5. Impact on Synaptic Function and Plasticity
4. Astrocyte Metabolic Dysfunction and E/I Imbalance in Neurological Disorders
4.1. Epilepsy
4.2. Alzheimer’s Disease
4.3. Autism Spectrum Disorder (ASD)
4.4. Traumatic Brain Injury (TBI)
4.5. Other Neurological Disorders
5. Therapeutic Strategies Targeting Astrocyte Metabolism to Restore E/I Balance
5.1. Modulation of Glutamate Homeostasis
5.2. Enhancement of GABAergic Signaling
5.3. Targeting Neuroinflammation
5.4. Metabolic Support and Energy Restoration
5.5. Emerging Therapeutic Targets
5.6. Nanoparticle Drug Delivery
6. Astrocyte Heterogeneity and Disease
7. Future Directions and Challenges
7.1. Development of Astrocyte-Specific Tools and Techniques
7.2. Longitudinal Studies and Biomarker Discovery
7.3. Clinical Translation
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiareli, R.A.; Carvalho, G.A.; Marques, B.L.; Mota, L.S.; Oliveira-Lima, O.C.; Gomes, R.M.; Birbrair, A.; Gomez, R.S.; Simão, F.; Klempin, F.; et al. The role of astrocytes in the neurorepair process. Front. Cell Dev. Biol. 2021, 9, 665795. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Eilam, R.; Arnon, R. Astrocytes in multiple sclerosis—Essential constituents with diverse multifaceted functions. Int. J. Mol. Sci. 2021, 22, 5904. [Google Scholar] [CrossRef] [PubMed]
- Soares, É.N.; Costa, A.C.; Ferrolho, G.D.; Ureshino, R.P.; Getachew, B.; Costa, S.L.; Da Silva, V.D.; Tizabi, Y. Nicotinic Acetylcholine Receptors in Glial Cells as Molecular Target for Parkinson’s Disease. Cells 2024, 13, 474. [Google Scholar] [CrossRef]
- Sufleţel, R.T.; Mihu, C.M.; Boşca, A.B.; Melincovici, C.S.; Mărginean, M.V.; Jianu, E.M.; Onofrei, M.M.; Constantin, A.M.; Moldovan, I.M.; Coneac, A.; et al. Short histological kaleidoscope–recent findings in histology. Part IV Rom. J. Morphol. Embryol. 2024, 65, 377. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.Q.; Huang, W.P.; Jiang, Y.C.; Xu, H.; Duan, C.S.; Chen, N.H.; Liu, Y.J.; Fu, X.M. The function of astrocytes and their role in neurological diseases. Eur. J. Neurosci. 2023, 58, 3932–3961. [Google Scholar] [CrossRef]
- Theparambil, S.M.; Begum, G.; Rose, C.R. pH regulating mechanisms of astrocytes: A critical component in physiology and disease of the brain. Cell Calcium 2024, 120, 102882. [Google Scholar] [CrossRef] [PubMed]
- Walz, W. The Brain as an Organ. In The Gliocentric Brain: Phenotype Plasticity of the Damaged Brain; Springer International Publishing: Cham, Switzerland, 2024; pp. 7–27. [Google Scholar]
- Syvänen, V.; Koistinaho, J.; Lehtonen, Š. Identification of the abnormalities in astrocytic functions as potential drug targets for neurodegenerative disease. Expert Opin. Drug Discov. 2024, 19, 603–616. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, R.; Mao, K.; Deng, M.; Li, Z. The role of glial cells in synaptic dysfunction: Insights into Alzheimer’s disease mechanisms. Aging Dis. 2024, 15, 459. [Google Scholar] [CrossRef]
- Escalada, P.; Ezkurdia, A.; Ramírez, M.J.; Solas, M. Essential role of astrocytes in learning and memory. Int. J. Mol. Sci. 2024, 25, 1899. [Google Scholar] [CrossRef]
- Squadrani, L.; Wert-Carvajal, C.; Müller-Komorowska, D.; Bohmbach, K.; Henneberger, C.; Verzelli, P.; Tchumatchenko, T. Astrocytes enhance plasticity response during reversal learning. Commun. Biol. 2024, 7, 852. [Google Scholar] [CrossRef]
- Chen, T.; Dai, Y.; Hu, C.; Lin, Z.; Wang, S.; Yang, J.; Zeng, L.; Li, S.; Li, W. Cellular and molecular mechanisms of the blood–brain barrier dysfunction in neurodegenerative diseases. Fluids Barriers CNS 2024, 21, 60. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Sun, Y.; Deng, Y.; Zhang, J. Blood-brain barrier disruption: A culprit of cognitive decline? Fluids Barriers CNS 2024, 21, 63. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Schousboe, A.; McKenna, M.C.; Rothman, D.L. A tribute to Leif Hertz: The historical context of his pioneering studies of the roles of astrocytes in brain energy metabolism, neurotransmission, cognitive functions, and pharmacology identifies important, unresolved topics for future studies. J. Neurochem. 2024, 168, 461–495. [Google Scholar] [CrossRef]
- Calì, C.; Cantando, I.; Veloz Castillo, M.F.; Gonzalez, L.; Bezzi, P. Metabolic reprogramming of astrocytes in pathological conditions: Implications for neurodegenerative diseases. Int. J. Mol. Sci. 2024, 25, 8922. [Google Scholar] [CrossRef]
- Vivi, E.; Di Benedetto, B. Brain stars take the lead during critical periods of early postnatal brain development: Relevance of astrocytes in health and mental disorders. Mol. Psychiatry 2024, 29, 2821–2833. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, C.; Li, Q.; Liu, G.; Song, D.; Quan, Z.; Yan, Y.; Qing, H. Neural Network Excitation/Inhibition: A Key to Empathy and Empathy Impairment. Neuroscientist 2024, 30, 644–665. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, C.; Pozo Garcia, V.; García-Adán, B.; Ameen, A.O.; Gegelashvili, G.; Waagepetersen, H.S.; Freude, K.K.; Aldana, B.I. Increased glucose metabolism and impaired glutamate transport in human astrocytes are potential early triggers of abnormal extracellular glutamate accumulation in hiPSC-derived models of Alzheimer’s disease. J. Neurochem. 2024, 168, 822–840. [Google Scholar] [CrossRef]
- Koh, W.; Lee, C.J. Diagnostic and therapeutic potential of tonic gamma-aminobutyric acid from reactive astrocytes in brain diseases. Clin. Transl. Med. 2024, 14, e1642. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, Y.Y.; Long, C.; Peng, X.; Tao, J.; Pu, Y.; Yue, R. Bridging metabolic syndrome and cognitive dysfunction: Role of astrocytes. Front. Endocrinol. 2024, 15, 1393253. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Qi, Y.B.; Gao, Y.N.; Chen, W.G.; Zhou, T.; Zang, Y.; Li, J. Astrocyte metabolism and signaling pathways in the CNS. Front. Neurosci. 2023, 17, 1217451. [Google Scholar] [CrossRef]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as key regulators of brain energy metabolism: New therapeutic perspectives. Front. Physiol. 2022, 12, 825816. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Tapadia, M.G. Dysregulated Peripheral Metabolism in Neurodegenerative Disorders. In Altered Metabolism: A Major Contributor of Comorbidities in Neurodegenerative Diseases; Springer Nature: Singapore, 2024; pp. 157–172. [Google Scholar]
- Brooks, G.A.; Arevalo, J.A.; Osmond, A.D.; Leija, R.G.; Curl, C.C.; Tovar, A.P. Lactate in contemporary biology: A phoenix risen. J. Physiol. 2022, 600, 1229–1251. [Google Scholar] [CrossRef]
- Vavřička, J.; Pavel, B.R.; Follprecht, D.; Novák, J.; Kroužeckŷ, A. Modern perspective of lactate metabolism. Physiol. Res. 2024, 73, 499. [Google Scholar] [CrossRef]
- Ben-Azu, B.; Aderibigbe, A.O.; Ajayi, A.M.; Eneni, A.O.; Omogbiya, I.A.; Owoeye, O.; Umukoro, S.; Iwalewa, E.O. Morin decreases cortical pyramidal neuron degeneration via inhibition of neuroinflammation in mouse model of schizophrenia. Int. Immunopharmacol. 2019, 70, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Markussen, K.H.; Corti, M.; Byrne, B.J.; Vander Kooi, C.W.; Sun, R.C.; Gentry, M.S. The multifaceted roles of the brain glycogen. J. Neurochem. 2024, 168, 728–743. [Google Scholar] [CrossRef]
- Beltran-Velasco, A.I. Brain Glycogen—Its Metabolic Role in Neuronal Health and Neurological Disorders—An Extensive Narrative Review. Metabolites 2025, 15, 128. [Google Scholar] [CrossRef] [PubMed]
- Shichkova, P.; Coggan, J.S.; Markram, H.; Keller, D. Brain Metabolism in Health and Neurodegeneration: The Interplay Among Neurons and Astrocytes. Cells 2024, 13, 1714. [Google Scholar] [CrossRef]
- Prasad, S.K.; Acharjee, A.; Singh, V.V.; Trigun, S.K.; Acharjee, P. Modulation of brain energy metabolism in hepatic encephalopathy: Impact of glucose metabolic dysfunction. Metab. Brain Dis. 2024, 39, 1649–1665. [Google Scholar] [CrossRef]
- Colpaert, M.; Singh, P.K.; Donohue, K.J.; Pires, N.T.; Fuller, D.D.; Corti, M.; Byrne, B.J.; Sun, R.C.; Vander Kooi, C.W.; Gentry, M.S. Neurological glycogen storage diseases and emerging therapeutics. Neurotherapeutics 2024, 21, e00446. [Google Scholar] [CrossRef]
- Hertz, L.; Rothman, D.L. Glutamine-glutamate cycle flux is similar in cultured astrocytes and brain and both glutamate production and oxidation are mainly catalyzed by aspartate aminotransferase. Biology 2017, 6, 17. [Google Scholar] [CrossRef]
- Limón, I.D.; Angulo-Cruz, I.; Sánchez-Abdon, L.; Patricio-Martínez, A. Disturbance of the glutamate-glutamine cycle, secondary to hepatic damage, compromises memory function. Front. Neurosci. 2021, 15, 578922. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk-Węgrzynowicz, M.; Adamiak, K.; Strużyńska, L. Astrocyte–Neuron Interaction via the Glutamate–Glutamine Cycle and Its Dysfunction in Tau-Dependent Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 3050. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hua, Z.; Li, Z. The role of glutamate and glutamine metabolism and related transporters in nerve cells. CNS Neurosci. Ther. 2024, 30, e14617. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Bojja, S.L.; Gurram, P.C.; Mudgal, J.; Arora, D.; Nampoothiri, M. Astrocytic glutamatergic transmission and its implications in neurodegenerative disorders. Cells 2022, 11, 1139. [Google Scholar] [CrossRef]
- Onaolapo, A.Y.; Onaolapo, O.J. Peripheral and central glutamate dyshomeostasis in neurodegenerative disorders. Curr. Neuropharmacol. 2021, 19, 1069–1089. [Google Scholar] [CrossRef]
- Liang, S.L.; Carlson, G.C.; Coulter, D.A. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J. Neurosci. 2006, 26, 8537–8548. [Google Scholar] [CrossRef]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Kann, O.; Söder, L.; Khodaie, B. Lactate is a potentially harmful substitute for brain glucose fuel: Consequences for metabolic restoration of neurotransmission. Neural Regen. Res. 2025, 20, 1403–1404. [Google Scholar] [CrossRef]
- Erlichman, J.S.; Hewitt, A.; Damon, T.L.; Hart, M.; Kurascz, J.; Li, A.; Leiter, J.C. Inhibition of monocarboxylate transporter 2 in the retrotrapezoid nucleus in rats: A test of the astrocyte–neuron lactate-shuttle hypothesis. J. Neurosci. 2008, 28, 4888–4896. [Google Scholar] [CrossRef]
- Bhatti, M.S.; Frostig, R.D. Astrocyte-neuron lactate shuttle plays a pivotal role in sensory-based neuroprotection in a rat model of permanent middle cerebral artery occlusion. Sci. Rep. 2023, 13, 12799. [Google Scholar] [CrossRef]
- Kim, Y.; Dube, S.E.; Park, C.B. Brain energy homeostasis: The evolution of the astrocyte-neuron lactate shuttle hypothesis. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2025, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Chen, S.; Ye, Q. The lactate metabolism and protein lactylation in epilepsy. Front. Cell. Neurosci. 2025, 18, 1464169. [Google Scholar] [CrossRef] [PubMed]
- Mason, S. Lactate shuttles in neuroenergetics—Homeostasis, allostasis and beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef]
- Rose, J.; Brian, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Mitochondrial Metabolism in Astrocytes Regulates Brain Bioenergetics, Neurotransmission and Redox Balance. Front. Neurosci. 2020, 14, 536682. [Google Scholar] [CrossRef] [PubMed]
- Hasel, P.; Aisenberg, W.H.; Bennett, F.C.; Liddelow, S.A. Molecular and metabolic heterogeneity of astrocytes and microglia. Cell. Metab. 2023, 35, 555–570. [Google Scholar] [CrossRef]
- Barber, C.N.; Raben, D.M. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front. Cell. Neurosci. 2019, 13, 212. [Google Scholar] [CrossRef]
- Passlick, S.; Rose, C.R.; Petzold, G.C.; Henneberger, C. Disruption of glutamate transport and homeostasis by acute metabolic stress. Front. Cell. Neurosci. 2021, 15, 637784. [Google Scholar] [CrossRef]
- Bell, S.M.; Wareing, H.; Capriglia, F.; Hughes, R.; Barnes, K.; Hamshaw, A.; Adair, L.; Shaw, A.; Olejnik, A.; De, S.; et al. Increasing hexokinase 1 expression improves mitochondrial and glycolytic functional deficits seen in sporadic Alzheimer’s disease astrocytes. Mol. Psychiatry. 2025, 30, 1369–1382. [Google Scholar] [CrossRef]
- Mulica, P.; Grünewald, A.; Pereira, S.L. Astrocyte-neuron metabolic crosstalk in neurodegeneration: A mitochondrial perspective. Front. Endocrinol. 2021, 12, 668517. [Google Scholar] [CrossRef]
- Andersen, J.V.; Schousboe, A. Milestone Review: Metabolic dynamics of glutamate and GABA mediated neurotransmission—The essential roles of astrocytes. J. Neurochem. 2023, 166, 109–137. [Google Scholar] [CrossRef]
- Andersen, J.V.; Schousboe, A.; Wellendorph, P. Astrocytes regulate inhibitory neurotransmission through GABA uptake, metabolism, and recycling. Essays Biochem. 2023, 67, 77–91. [Google Scholar] [PubMed]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient astrocyte metabolism impairs glutamine synthesis and neurotransmitter homeostasis in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef] [PubMed]
- Chatton, J.Y.; Pellerin, L.; Magistretti, P.J. GABA uptake into astrocytes is not associated with significant metabolic cost: Implications for brain imaging of inhibitory transmission. Proc. Natl. Acad. Sci. USA 2003, 100, 12456–12461. [Google Scholar] [CrossRef]
- Brezolin, É.C.; Gayger-Dias, V.; Da Silva, V.F.; Cigerce, A.; Schultz, B.; Sobottka, T.M.; Nardin, P.; de Assis, A.M.; Leite, M.C.; Quincozes-Santos, A.; et al. Astrocyte dysfunction alters GABAergic communication and ammonia metabolism in the streptozotocin-induced sporadic Alzheimer’s disease model. J. Alzheimer’s Dis. Rep. 2024, 8, 1381–1393. [Google Scholar] [CrossRef]
- Andersen, J.V.; Marian, O.C.; Qvist, F.L.; Westi, E.W.; Aldana, B.I.; Schousboe, A.; Don, A.S.; Skotte, N.H.; Wellendorph, P. Deficient brain GABA metabolism leads to widespread impairments of astrocyte and oligodendrocyte function. Glia 2024, 72, 1821–1839. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Tjia, M.; Thapliyal, S. Homeostatic plasticity and excitation-inhibition balance: The good, the bad, and the ugly. Curr. Opin. Neurobiol. 2022, 75, 102553. [Google Scholar] [CrossRef]
- Sukenik, N.; Vinogradov, O.; Weinreb, E.; Segal, M.; Levina, A.; Moses, E. Neuronal circuits overcome imbalance in excitation and inhibition by adjusting connection numbers. Proc. Natl. Acad. Sci. USA 2021, 118, e2018459118. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, J.; Zhang, Y.; Peng, L.; Li, X.; Li, C.; Wu, X.; Wang, C. Epilepsy therapy beyond neurons: Unveiling astrocytes as cellular targets. Neural Regen. Res. 2026, 21, 23–38. [Google Scholar] [CrossRef]
- Espinelli Amorim, F.; Rye, C.S.; Milton, A.L. Mitochondrial dysfunction in PTSD: A mechanism to understand trauma susceptibility? MedRxiv 2025. [Google Scholar] [CrossRef]
- Ben-Azu, B.; Adebesin, A.; Moke, G.E.; Ojiokor, V.O.; Olusegun, A.; Jarikre, T.A.; Akinluyi, E.T.; Olukemi, O.A.; Omeiza, N.A.; Nkenchor, P.; et al. Alcohol exacerbates psychosocial stress-induced neuropsychiatric symptoms: Attenuation by geraniol. Neurochem. Int. 2024, 177, 105748. [Google Scholar] [CrossRef]
- Frisardi, V. Apolipoprotein E genotype: The innocent bystander or active bridge between metabolic syndrome and cognitive impairment? J. Alzheimer’s Dis. 2012, 30, S283–S304. [Google Scholar] [CrossRef] [PubMed]
- Cerasuolo, M.; Papa, M.; Colangelo, A.M.; Rizzo, M.R. Alzheimer’s disease from the amyloidogenic theory to the puzzling crossroads between vascular, metabolic and energetic maladaptive plasticity. Biomedicines 2023, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef]
- Jabeen, K.; Rehman, K.; Akash, M.S. Genetic mutations of APOEε4 carriers in cardiovascular patients lead to the development of insulin resistance and risk of Alzheimer’s disease. J. Biochem. Mol. Toxicol. 2022, 36, e22953. [Google Scholar] [CrossRef]
- Cho, S.; Lee, H.; Seo, J. Impact of genetic risk factors for Alzheimer’s disease on brain glucose metabolism. Mol. Neurobiol. 2021, 58, 2608–2619. [Google Scholar] [CrossRef]
- Liu, X.; Ying, J.; Wang, X.; Zheng, Q.; Zhao, T.; Yoon, S.; Yu, W.; Yang, D.; Fang, Y.; Hua, F. Astrocytes in neural circuits: Key factors in synaptic regulation and potential targets for neurodevelopmental disorders. Front. Mol. Neurosci. 2021, 14, 729273. [Google Scholar] [CrossRef]
- Gąssowska-Dobrowolska, M.; Chlubek, M.; Kolasa, A.; Tomasiak, P.; Korbecki, J.; Skowrońska, K.; Tarnowski, M.; Masztalewicz, M.; Baranowska-Bosiacka, I. Microglia and astroglia—The potential role in neuroinflammation induced by pre-and neonatal exposure to lead (Pb). Int. J. Mol. Sci. 2023, 24, 9903. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, M.M.; Korshunov, K.S.; Grant, R.A.; Martin, M.E.; Valencia, H.A.; Budinger, G.S.; Radulovic, J.; Prakriya, M. Astrocyte reactivity and inflammation-induced depression-like behaviors are regulated by Orai1 calcium channels. Nat. Commun. 2023, 14, 5500. [Google Scholar] [CrossRef]
- Xiong, Y.; Chen, J.; Li, Y. Microglia and astrocytes underlie neuroinflammation and synaptic susceptibility in autism spectrum disorder. Front. Neurosci. 2023, 17, 1125428. [Google Scholar] [CrossRef]
- Purushotham, S.S.; Buskila, Y. Astrocytic modulation of neuronal signalling. Front. Netw. Physiol. 2023, 3, 1205544. [Google Scholar] [CrossRef]
- Goenaga, J.; Araque, A.; Kofuji, P.; Chao, D.H.M. Calcium signaling in astrocytes and gliotransmitter release. Front. Synaptic Neurosci. 2023, 15, 1138577. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Baldwin, K.T.; Allen, N.J. Astrocyte regulation of synapse formation, maturation, and elimination. Cold Spring Harb. Perspect. Biol. 2024, 16, a041352. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Cuartero, M.I.; Palenzuela, R.; Draffin, J.E.; Konomi, A.; Serra, I.; Colié, S.; Castaño-Castaño, S.; Hasan, M.T.; Nebreda, Á.R.; et al. Astrocytic p38α MAPK drives NMDA receptor-dependent long-term depression and modulates long-term memory. Nat. Commun. 2019, 10, 2968. [Google Scholar] [CrossRef]
- Manninen, T.; Saudargiene, A.; Linne, M.L. Astrocyte-mediated spike-timing-dependent long-term depression modulates synaptic properties in the developing cortex. PLoS Comput. Biol. 2020, 16, e1008360. [Google Scholar] [CrossRef]
- Pereira, M.F.; Amaral, I.M.; Lopes, C.; Leitão, C.; Madeira, D.; Lopes, J.P.; Gonçalves, F.Q.; Canas, P.M.; Cunha, R.A.; Agostinho, P. l-α-aminoadipate causes astrocyte pathology with negative impact on mouse hippocampal synaptic plasticity and memory. FASEB J. 2021, 35, e21726. [Google Scholar] [CrossRef]
- Veiga, A.; Abreu, D.S.; Dias, J.D.; Azenha, P.; Barsanti, S.; Oliveira, J.F. Calcium-Dependent Signaling in Astrocytes: Downstream Mechanisms and Implications for Cognition. J. Neurochem. 2025, 169, e70019. [Google Scholar] [CrossRef]
- Mathure, D.; Kumar, D.; Ranpise, H.; Awasthi, R. Reactive Astrogliosis and Neuronal Functions of Astrocytes in Neurological Disorders. Ind. J. Pharm. Edu. Res. 2024, 58, 386–400. [Google Scholar] [CrossRef]
- Boison, D.; Steinhäuser, C. Epilepsy and astrocyte energy metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef]
- Banerjee, S.; Jirsa, V. A review of epileptic markers: From ion channels, astrocytes, synaptic imbalance to whole brain network dynamics. Explor. Neurosci. 2024, 3, 478–492. [Google Scholar] [CrossRef]
- Çarçak, N.; Onat, F.; Sitnikova, E. Astrocytes as a target for therapeutic strategies in epilepsy: Current insights. Front. Mol. Neurosci. 2023, 16, 1183775. [Google Scholar] [CrossRef]
- Chen, P.; Chen, F.; Zhou, B. Understanding the role of glia-neuron communication in the pathophysiology of epilepsy: A review. J. Integr. Neurosci. 2022, 21, 102. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Ravizza, T.; Bedner, P.; Aronica, E.; Steinhäuser, C.; Boison, D. Astrocytes in the initiation and progression of epilepsy. Nat. Rev. Neurol. 2022, 18, 707–722. [Google Scholar] [CrossRef]
- Yang, F.; Sun, X.; Ding, Y.; Ma, H.; Yang, T.O.; Ma, Y.; Wei, D.; Li, W.; Xu, T.; Jiang, W. Astrocytic Acid-Sensing Ion Channel 1a Contributes to the Development of Chronic Epileptogenesis. Sci. Rep. 2016, 6, 31581. [Google Scholar] [CrossRef]
- Cai, Z.; Wan, C.Q.; Liu, Z. Astrocyte and Alzheimer’s disease. J. Neurol. 2017, 264, 2068–2074. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Giraldo, M.; González-Reyes, R.E.; Ramírez-Guerrero, S.; Bonilla-Trilleras, C.E.; Guardo-Maya, S.; Nava-Mesa, M.O. Astrocytes as a therapeutic target in alzheimer’s disease–comprehensive review and recent developments. Int. J. Mol. Sci. 2022, 23, 13630. [Google Scholar] [CrossRef]
- Thal, D.R. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp. Neurol. 2012, 236, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Botella Lucena, P.; Heneka, M.T. Inflammatory aspects of Alzheimer’s disease. Acta Neuropathol. 2024, 148, 31. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Zulfiqar, S.; Garg, P.; Nieweg, K. Contribution of astrocytes to metabolic dysfunction in the Alzheimer’s disease brain. Biol. Chem. 2019, 400, 1113–1127. [Google Scholar] [CrossRef]
- Uzunova, G.; Pallanti, S.; Hollander, E. Excitatory/inhibitory imbalance in autism spectrum disorders: Implications for interventions and therapeutics. World J. Biol. Psychiatry 2016, 17, 174–186. [Google Scholar] [CrossRef]
- Canitano, R.; Palumbi, R. Excitation/inhibition modulators in autism spectrum disorder: Current clinical research. Front. Neurosci. 2021, 15, 753274. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Huang, B.S.; Notaras, M.J.; Lodhi, A.; Barrio-Alonso, E.; Lituma, P.J.; Wolujewicz, P.; Witztum, J.; Longo, F.; Chen, M.; et al. Astrocytes derived from ASD individuals alter behavior and destabilize neuronal activity through aberrant Ca2+ signaling. Mol. Psychiatry. 2022, 27, 2470–2484. [Google Scholar] [CrossRef]
- Cano, A.C.; Santos, D.; Beltrão-Braga, P.C. The Interplay of Astrocytes and Neurons in Autism Spectrum Disorder. Adv. Neurobiol. 2024, 269–284. [Google Scholar]
- Novakovic, M.M.; Prakriya, M. Calcium signaling at the interface between astrocytes and brain inflammation. Curr. Opin. Neurobiol. 2025, 90, 102940. [Google Scholar] [CrossRef] [PubMed]
- Colomar, L.; San José Cáceres, A.; Álvarez-Linera, J.; González-Peñas, J.; Huertas Patón, A.; Martín de Blas, D.; Polo Arrondo, A.P.; Solís, A.; Jones, E.; Parellada, M. Role of cortical excitatory/inhibitory imbalance in autism spectrum disorders from a symptom severity trajectories framework: A study protocol. BMC Psychiatry 2023, 23, 213. [Google Scholar] [CrossRef]
- Cantando, I.; Centofanti, C.; D’Alessandro, G.; Limatola, C.; Bezzi, P. Metabolic dynamics in astrocytes and microglia during post-natal development and their implications for autism spectrum disorders. Front. Cell. Neurosci. 2024, 18, 1354259. [Google Scholar] [CrossRef]
- Jalloh, I.; Carpenter, K.L.; Helmy, A.; Carpenter, T.A.; Menon, D.K.; Hutchinson, P.J. Glucose metabolism following human traumatic brain injury: Methods of assessment and pathophysiological findings. Metab. Brain Dis. 2015, 30, 615–632. [Google Scholar] [CrossRef]
- Ben-Azu, B.; Oritsemuelebi, B.; Oghorodi, A.M.; Adebesin, A.; Isibor, H.; Eduviere, A.T.; Otuacha, O.S.; Akudo, M.; Ekereya, S.; Maidoh, I.F.; et al. Psychopharmacological interaction of alcohol and posttraumatic stress disorder: Effective action of naringin. Eur. J. Pharmacol. 2024, 978, 176791. [Google Scholar] [CrossRef]
- Kurtz, P.; Rocha, E.E. Nutrition therapy, glucose control, and brain metabolism in traumatic brain injury: A multimodal monitoring approach. Front. Neurosci. 2020, 14, 190. [Google Scholar] [CrossRef]
- Freire, M.A.; Rocha, G.S.; Bittencourt, L.O.; Falcao, D.; Lima, R.R.; Cavalcanti, J.R. Cellular and molecular pathophysiology of traumatic brain injury: What have we learned so far? Biology 2023, 12, 1139. [Google Scholar] [CrossRef]
- Hovda, D.A.; Lee, S.M.; Smith, M.L.; Von Stuck, S.; Bergsneider, M.; Kelly, D.; Shalmon, E.; Martin, N.; Caron, M.; Mazziotta, J.; et al. The neurochemical and metabolic cascade following brain injury: Moving from animal models to man. J. Neurotrauma 1995, 12, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Jurcău, M.C.; Andronie-Cioara, F.L.; Jurcău, A.; Marcu, F.; Ţiț, D.M.; Pașcalău, N.; Nistor-Cseppentö, D.C. The Link between Oxidative Stress, Mitochondrial Dysfunction and Neuroinflammation in the Pathophysiology of Alzheimer’s Disease: Therapeutic Implications and Future Perspectives. Antioxidants 2022, 11, 2167. [Google Scholar] [CrossRef] [PubMed]
- Fesharaki-Zadeh, A.; Datta, D. An overview of preclinical models of traumatic brain injury (TBI): Relevance to pathophysiological mechanisms. Front. Cell. Neurosci. 2024, 18, 1371213. [Google Scholar] [CrossRef] [PubMed]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2016, 173, 692–702. [Google Scholar] [CrossRef]
- Giza, C.C.; Hovda, D.A. The neurometabolic cascade of concussion. J. Athl. Train. 2001, 36, 228. [Google Scholar] [CrossRef]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell. Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Ben-Azu, B.; Fokoua, A.R.; Annafi, O.S.; Adebayo, O.G.; Del Re, E.C.; Okuchukwu, N.; Aregbesola, G.J.; Ejenavi, A.C.; Isiwele, D.M.; Efezino, A.J.; et al. Effective action of silymarin against ketamine-induced schizophrenia in male mice: Insight into the biochemical and molecular mechanisms of action. J. Psychiatr. Res. 2024, 179, 141–155. [Google Scholar] [CrossRef]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Nuszkiewicz, J.; Kukulska-Pawluczuk, B.; Piec, K.; Jarek, D.J.; Motolko, K.; Szewczyk-Golec, K.; Woźniak, A. Intersecting pathways: The role of metabolic dysregulation, gastrointestinal microbiome, and inflammation in acute ischemic stroke pathogenesis and outcomes. J. Clin. Med. 2024, 13, 4258. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell. Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Chang, C.P.; Wu, C.W.; Chern, Y. Metabolic dysregulation in Huntington’s disease: Neuronal and glial perspectives. Neurobiol. Dis. 2024, 201, 106672. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. TINS 2017, 40, 422–437. [Google Scholar] [CrossRef]
- Rawani, N.S.; Chan, A.W.; Dursun, S.M.; Baker, G.B. The underlying neurobiological mechanisms of psychosis: Focus on neurotransmission dysregulation, neuroinflammation, oxidative stress, and mitochondrial dysfunction. Antioxidants 2024, 13, 709. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.K. Astrocyte-Neuron Interactions Contributing to Amyotrophic Lateral Sclerosis Progression. Adv. Neurobiol. 2024, 285–318. [Google Scholar]
- Jagaraj, C.J.; Shadfar, S.; Kashani, S.A.; Saravanabavan, S.; Farzana, F.; Atkin, J.D. Molecular hallmarks of ageing in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2024, 81, 111. [Google Scholar] [CrossRef]
- Pathak, D.; Sriram, K. Neuron-astrocyte omnidirectional signaling in neurological health and disease. Front. Mol. Neurosci. 2023, 16, 1169320. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Todd, A.C.; Hardingham, G.E. The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9607. [Google Scholar] [CrossRef]
- Magdaleno Roman, J.Y.; Chapa González, C. Glutamate and excitotoxicity in central nervous system disorders: Ionotropic glutamate receptors as a target for neuroprotection. Neuroprotection 2024, 2, 137–150. [Google Scholar] [CrossRef]
- Sears, S.M.; Hewett, S.J. Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Exp. Biol. Med. 2021, 246, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.N.; Li, L.; Hu, S.H.; Yang, Y.X.; Ma, Z.Z.; Huang, L.; An, Y.P.; Yuan, Y.Y.; Lin, Y.; Xu, W.; et al. Ketogenic diet-produced β-hydroxybutyric acid accumulates brain GABA and increases GABA/glutamate ratio to inhibit epilepsy. Cell Discov. 2024, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Huang, L.; Cheng, H.; Li, N.; Zhang, B.; Dai, W.; Wu, X.; Zhang, D.; Feng, W.; Li, S.; et al. GABA and its receptors’ mechanisms in the treatment of insomnia. Heliyon 2024, 10, e40665. [Google Scholar] [CrossRef]
- Reyes-Haro, D.; Cisneros-Mejorado, A.; Arellano, R.O. Therapeutic potential of GABAergic signaling in myelin plasticity and repair. Front. Cell Dev. Biol. 2021, 9, 662191. [Google Scholar] [CrossRef]
- Vélez-Fort, M.; Audinat, E.; Angulo, M.C. Central role of GABA in neuron–glia interactions. Neurosci. 2012, 18, 237–250. [Google Scholar] [CrossRef]
- Bi, D.; Wen, L.; Wu, Z.; Shen, Y. GABAergic dysfunction in excitatory and inhibitory (E/I) imbalance drives the pathogenesis of Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 1312–1329. [Google Scholar] [CrossRef]
- Ben-Azu, B.; Aderibigbe, A.O.; Ajayi, A.M.; Eneni, A.O.; Umukoro, S.; Iwalewa, E.O. Involvement of GABAergic, BDNF and Nox-2 mechanisms in the prevention and reversal of ketamine-induced schizophrenia-like behavior by morin in mice. Brain Res. Bull. 2018, 139, 292–306. [Google Scholar] [CrossRef]
- Robb, J.L.; Hammad, N.A.; Weightman Potter, P.G.; Chilton, J.K.; Beall, C.; Ellacott, K.L. The metabolic response to inflammation in astrocytes is regulated by nuclear factor-kappa B signaling. Glia 2020, 68, 2246–2263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Guo, Y.; Zhang, Y.; Gao, Y.; Ning, B. Metabolic reprogramming of astrocytes: Emerging roles of lactate. Neural. Regen. Res. 2026, 21, 421–432. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Kielian, T. Neuron–astrocyte interactions in neurodegenerative diseases: Role of neuroinflammation. Clin. Exp. Neuroimmunol. 2015, 6, 245–263. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Chai, Y.; Wang, Y.; Zhang, J.; Chen, X. Astrocyte-mediated inflammatory responses in traumatic brain injury: Mechanisms and potential interventions. Front. Immunol. 2025, 16, 1584577. [Google Scholar] [CrossRef] [PubMed]
- Wetherington, J.; Serrano, G.; Dingledine, R. Astrocytes in the epileptic brain. Neuron. 2008, 58, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Edison, P. Astroglial activation: Current concepts and future directions. Alzheimer’s Dement. 2024, 20, 3034–3053. [Google Scholar] [CrossRef]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. Roles of neuropathology-associated reactive astrocytes: A systematic review. Acta Neuropathol. Commun. 2023, 11, 42. [Google Scholar] [CrossRef]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and inflammatory adaptation of reactive astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, Q.; Guo, T.; Zhang, S.; Zheng, S.; Wang, R.; Deng, Q.; Yang, G.; Zhang, S.; Tang, L.; et al. Metabolic reprogramming in astrocytes results in neuronal dysfunction in intellectual disability. Mol. Psychiatry 2024, 29, 1569–1582. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Redondo-Flórez, L.; López-Mora, C.; Yáñez-Sepúlveda, R.; Tornero-Aguilera, J.F. New insights and potential therapeutic interventions in metabolic diseases. Int. J. Mol. Sci. 2023, 24, 10672. [Google Scholar] [CrossRef]
- Descalzi, G.; Gao, V.; Steinman, M.Q.; Suzuki, A.; Alberini, C.M. Lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Commun. Biol. 2019, 2, 247. [Google Scholar] [CrossRef]
- Jang, J.; Kim, S.R.; Lee, J.E.; Lee, S.; Son, H.J.; Choe, W.; Yoon, K.S.; Kim, S.S.; Yeo, E.J.; Kang, I. Molecular mechanisms of neuroprotection by ketone bodies and ketogenic diet in cerebral ischemia and neurodegenerative diseases. Int. J. Mol. Sci. 2023, 25, 124. [Google Scholar] [CrossRef]
- Chen, Z.; Yuan, Z.; Yang, S.; Zhu, Y.; Xue, M.; Zhang, J.; Leng, L. Brain energy metabolism: Astrocytes in neurodegenerative diseases. CNS Neurosci. Ther. 2023, 29, 24–36. [Google Scholar] [CrossRef]
- González-García, I.; Gruber, T.; García-Cáceres, C. Insulin action on astrocytes: From energy homeostasis to behaviour. J. Neuroendocrinol. 2021, 33, e12953. [Google Scholar] [CrossRef] [PubMed]
- Perez-Catalan, N.A.; Doe, C.Q.; Ackerman, S.D. The role of astrocyte-mediated plasticity in neural circuit development and function. Neural Dev. 2021, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.S.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Park, M.K.; Lee, S.H.; Lee, C.J.; Yang, H.W.; Woo, S.Y.; et al. Effects of Pyruvate Kinase M2 (PKM2) Gene Deletion on Astrocyte-Specific Glycolysis and Global Cerebral Ischemia-Induced Neuronal Death. Antioxidants 2023, 12, 491. [Google Scholar] [CrossRef]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Butt, A.; Li, B.; Illes, P.; Zorec, R.; Semyanov, A.; Tang, Y.; Sofroniew, M.V. Astrocytes in human central nervous system diseases: A frontier for new therapies. Signal Transduct. Target. Ther. 2023, 8, 396. [Google Scholar] [CrossRef]
- Matés, J.M.; Campos-Sandoval, J.A.; de Los Santos-Jiménez, J.; Márquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef]
- Raj Rai, S.; Bhattacharyya, C.; Sarkar, A.; Chakraborty, S.; Sircar, E.; Dutta, S.; Sengupta, R. Glutathione: Role in oxidative/nitrosative stress, antioxidant defense, and treatments. ChemistrySelect 2021, 6, 4566–4590. [Google Scholar] [CrossRef]
- Maszka, P.; Kwasniak-Butowska, M.; Cysewski, D.; Slawek, J.; Smolenski, R.T.; Tomczyk, M. Metabolomic footprint of disrupted energetics and amino acid metabolism in neurodegenerative diseases: Perspectives for early diagnosis and monitoring of therapy. Metabolites 2023, 13, 369. [Google Scholar] [CrossRef]
- Fontana, A.C. Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 2015, 134, 982–1007. [Google Scholar] [CrossRef]
- Meneghini, V.; Peviani, M.; Luciani, M.; Zambonini, G.; Gritti, A. Delivery platforms for CRISPR/Cas9 genome editing of glial cells in the central nervous system. Front. Genome Ed. 2021, 3, 644319. [Google Scholar] [CrossRef]
- Seifert, G.; Schilling, K.; Steinhäuser, C. Astrocyte dysfunction in neurological disorders: A molecular perspective. Nat. Rev. Neurosci. 2006, 7, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood–brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Ghosh, S.; Majhi, S.; Hoque, A.N.; Chakrabarti, A. Systems biology approaches as a tool in understanding neurodevelopmental disorders: Some case studies. In Systems Biology Approaches: Prevention, Diagnosis, and Understanding Mechanisms of Complex Diseases; Springer: Singapore, 2024; pp. 511–536. [Google Scholar]
- Ben-Azu, B.; Adebayo, O.G.; Fokoua, A.R.; Onuelu, J.E.; Asiwe, J.N.; Moke, E.G.; Omogbiya, I.A.; Okpara, O.L.; Okoro, J.E.; Oghenevwerutevwe, O.M.; et al. Containment of neuroimmune challenge by diosgenin confers amelioration of neurochemical and neurotrophic dysfunctions in ketamine-induced schizophrenia in mice. Brain Disorders. 2024, 13, 100122. [Google Scholar] [CrossRef]
- Héja, L.; Barabás, P.; Nyitrai, G.; Kékesi, K.A.; Lasztóczi, B.; Tőke, O.; Tárkányi, G.; Madsen, K.; Schousboe, A.; Dobolyi, Á.; et al. Glutamate uptake triggers transporter-mediated GABA release from astrocytes. PLoS ONE 2009, 4, e7153. [Google Scholar] [CrossRef] [PubMed]
- Kabała, K.; Janicka, M. Relationship between the GABA pathway and signaling of other regulatory molecules. Int. J. Mol. Sci. 2024, 25, 10749. [Google Scholar] [CrossRef]
- Balkhi, H.M.; Gul, T.; Banday, M.Z.; Haq, E. Glutamate excitotoxicity: An insight into the mechanism. Int. J. Adv. Res. 2014, 2, 361–373. [Google Scholar]
- Papa, M.; De Luca, C.; Petta, F.; Alberghina, L.; Cirillo, G. Astrocyte–neuron interplay in maladaptive plasticity. Neurosci. Biobehav. Rev. 2014, 42, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, X. Nano-drug delivery systems (NDDS) in metabolic dysfunction-associated steatotic liver disease (MASLD): Current status, prospects and challenges. Front. Pharmacol. 2024, 15, 1419384. [Google Scholar] [CrossRef]
- Surnar, B.; Shah, A.S.; Park, M.; Kalathil, A.A.; Kamran, M.Z.; Ramirez Jaime, R.; Toborek, M.; Nair, M.; Kolishetti, N.; Dhar, S. Brain-accumulating nanoparticles for assisting astrocytes to reduce human immunodeficiency virus and drug abuse-induced neuroinflammation and oxidative stress. ACS Nano 2021, 15, 15741–15753. [Google Scholar] [CrossRef]
- Seifert, G.; Carmignoto, G.; Steinhäuser, C. Astrocyte dysfunction in epilepsy. Brain Res. Rev. 2010, 63, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef]
- Holt, M.G. Astrocyte heterogeneity and interactions with local neural circuits. Essays Biochem. 2023, 67, 93–106. [Google Scholar]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef]
- Xin, W.; Bonci, A. Functional astrocyte heterogeneity and implications for their role in shaping neurotransmission. Front. Cell. Neurosci. 2018, 12, 141. [Google Scholar] [CrossRef]
- Furman, J.L.; Sama, D.M.; Gant, J.C.; Beckett, T.L.; Murphy, M.P.; Bachstetter, A.D.; Van Eldik, L.J.; Norris, C.M. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci. 2012, 32, 16129–16140. [Google Scholar] [CrossRef]
- Lozzi, B.; Huang, T.W.; Sardar, D.; Huang, A.Y.; Deneen, B. Regionally distinct astrocytes display unique transcription factor profiles in the adult brain. Front. Neurosci. 2020, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I.; Malkov, A.E.; Waseem, T.; Mukhtarov, M.; Buldakova, S.; Gubkina, O.; Zilberter, M.; Zilberter, Y. Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J. Cereb. Blood Flow Metab. 2014, 34, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Bachoo, R.M.; Kim, R.S.; Ligon, K.L.; Maher, E.A.; Brennan, C.; Billings, N.; Chan, S.; Li, C.; Rowitch, D.H.; Wong, W.H.; et al. Molecular diversity of astrocytes with implications for neurological disorders. Proc. Natl. Acad. Sci. USA 2004, 101, 8384–8389. [Google Scholar] [CrossRef]
- Montgomery, D.L. Astrocytes: Form, functions, and roles in disease. Vet. Pathol. 1994, 31, 145–167. [Google Scholar] [CrossRef]
- Reid, J.K.; Kuipers, H.F. She doesn’t even go here: The role of inflammatory astrocytes in CNS disorders. Front. Cell. Neurosci. 2021, 15, 704884. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Agulhon, C.; Schmidt, E.; Oheim, M.; Ropert, N. New tools for investigating astrocyte-to-neuron communication. Front. Cell. Neurosci. 2013, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Parkin, G.M.; Udawela, M.; Gibbons, A.; Dean, B. Glutamate transporters, EAAT1 and EAAT2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J. Psychiatry 2018, 8, 51–63. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Liu, J.; Gao, J.; Guo, L.; Ma, G.; Guan, M.; Xia, C.; He, J.; Yang, Y.; Wu, Y.; Xu, J.; et al. Astrocyte-specific secretome profiling reveals its correlation with neurological disorders. bioRxiv 2024. [Google Scholar] [CrossRef]
- Carter, S.F.; Chiotis, K.; Nordberg, A.; Rodriguez-Vieitez, E. Longitudinal association between astrocyte function and glucose metabolism in autosomal dominant Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 348–356. [Google Scholar] [CrossRef]
- Sarkar, S.; Guha, S.; Biswas, S.C. Role of Reactive Astrocytes in Alzheimer’s Disease. In The Biology of Glial Cells: Recent Advances; Springer: Singapore, 2022; pp. 199–242. [Google Scholar]
- Serrano-Pozo, A.; Li, H.; Li, Z.; Muñoz-Castro, C.; Jaisa-Aad, M.; Healey, M.A.; Welikovitch, L.A.; Jayakumar, R.; Bryant, A.G.; Noori, A.; et al. Astrocyte transcriptomic changes along the spatiotemporal progression of Alzheimer’s disease. Nat. Neurosci. 2024, 27, 2384–2400. [Google Scholar] [CrossRef] [PubMed]
- Valori, C.F.; Possenti, A.; Brambilla, L.; Rossi, D. Challenges and Opportunities of Targeting Astrocytes to Halt Neurodegenerative Disorders. Cells 2021, 10, 2019. [Google Scholar] [CrossRef]
- Afridi, R.; Rahman, M.H.; Suk, K. Implications of glial metabolic dysregulation in the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2022, 174, 105874. [Google Scholar] [CrossRef]
- Hamby, M.E.; Sofroniew, M.V. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 2010, 7, 494–506. [Google Scholar] [CrossRef]
- Tan, R.; Hong, R.; Sui, C.; Yang, D.; Tian, H.; Zhu, T.; Yang, Y. The role and potential therapeutic targets of astrocytes in central nervous system demyelinating diseases. Front. Cell. Neurosci. 2023, 17, 1233762. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Hoi, M.P.M. Emerging roles of astrocytes in blood-brain barrier disruption upon amyloid-beta insults in Alzheimer’s disease. Neural. Regen. Res. 2023, 18, 1890–1902. [Google Scholar] [PubMed]
- Finsterwald, C.; Magistretti, P.J.; Lengacher, S. Astrocytes: New targets for the treatment of neurodegenerative diseases. Curr. Pharm. Des. 2015, 21, 3570–3581. [Google Scholar] [CrossRef]
- Pereira, M.J.; Ayana, R.; Holt, M.G.; Arckens, L. Chemogenetic manipulation of astrocyte activity at the synapse—A gateway to manage brain disease. Front. Cell Dev. Biol. 2023, 11, 1193130. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, U.G.; Oyovwi, M.O.; Jeroh, E.; Esuku, D.T.; Ben-Azu, B. Dysfunctional Astrocyte Metabolism: A Driver of Imbalanced Excitatory/Inhibitory Tone and Support for Therapeutic Intervention Targets. J. Mol. Pathol. 2025, 6, 12. https://doi.org/10.3390/jmp6020012
Joseph UG, Oyovwi MO, Jeroh E, Esuku DT, Ben-Azu B. Dysfunctional Astrocyte Metabolism: A Driver of Imbalanced Excitatory/Inhibitory Tone and Support for Therapeutic Intervention Targets. Journal of Molecular Pathology. 2025; 6(2):12. https://doi.org/10.3390/jmp6020012
Chicago/Turabian StyleJoseph, Uchechukwu G., Mega O. Oyovwi, Ejayeta Jeroh, Daniel T. Esuku, and Benneth Ben-Azu. 2025. "Dysfunctional Astrocyte Metabolism: A Driver of Imbalanced Excitatory/Inhibitory Tone and Support for Therapeutic Intervention Targets" Journal of Molecular Pathology 6, no. 2: 12. https://doi.org/10.3390/jmp6020012
APA StyleJoseph, U. G., Oyovwi, M. O., Jeroh, E., Esuku, D. T., & Ben-Azu, B. (2025). Dysfunctional Astrocyte Metabolism: A Driver of Imbalanced Excitatory/Inhibitory Tone and Support for Therapeutic Intervention Targets. Journal of Molecular Pathology, 6(2), 12. https://doi.org/10.3390/jmp6020012