
The Current Landscape of Molecular Pathology for the Diagnosis and Treatment of Pediatric Medulloblastoma



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Histopathology and Molecular Pathology in the Diagnosis of Medulloblastoma
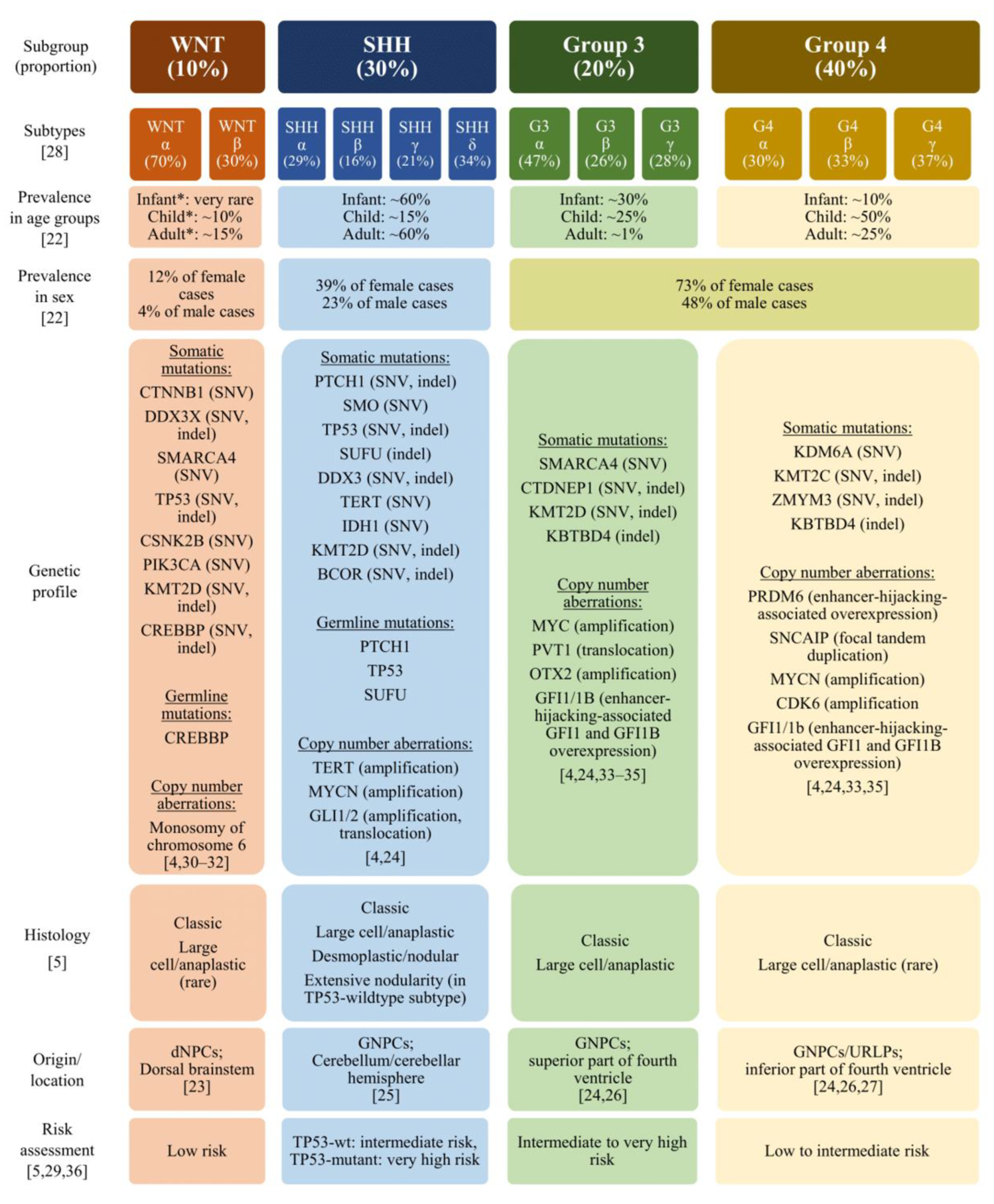
3. Considerations for Molecular Targets of Therapy
4. Discussion
Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MB | medulloblastoma |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| WNT | wingless |
| SHH | sonic hedgehog |
| SMO | smoothened |
| HDAC | histone deacetylases |
| CSI | standard craniospinal radiation |
| PFRT | posterior fossa radiation therapy |
| IFRT | involved field radiation therapy |
References
- Kline, N.E.; Sevier, N. Solid tumors in children. J. Pediatr. Nurs. 2003, 18, 96–102. [Google Scholar] [CrossRef]
- Rossi, A.; Caracciolo, V.; Russo, G.; Reiss, K.; Giordano, A. Medulloblastoma: From molecular pathology to therapy. Clin. Cancer Res. 2008, 14, 971–976. [Google Scholar] [CrossRef]
- Orr, B.A. Pathology, diagnostics, and classification of medulloblastoma. Brain Pathol. 2020, 30, 664–678. [Google Scholar] [CrossRef]
- Mahapatra, S.; Amsbaugh, M.J. Medulloblastoma. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar] [PubMed]
- Kijima, N.; Kanemura, Y. Molecular Classification of Medulloblastoma. Neurol. Med. Chir. 2016, 56, 687–697. [Google Scholar] [CrossRef]
- Choi, J.Y. Medulloblastoma: Current Perspectives and Recent Advances. Brain Tumor Res. Treat. 2023, 11, 28–38. [Google Scholar] [CrossRef]
- Cotter, J.A.; Hawkins, C. Medulloblastoma: WHO 2021 and Beyond. Pediatr. Dev. Pathol. 2022, 25, 23–33. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Archer, T.C.; Mahoney, E.L.; Pomeroy, S.L. Medulloblastoma: Molecular Classification-Based Personal Therapeutics. Neurotherapeutics 2017, 14, 265–273. [Google Scholar] [CrossRef]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef]
- Mani, S.; Chatterjee, A.; Dasgupta, A.; Shirsat, N.; Pawar, A.; Epari, S.; Sahay, A.; Sahu, A.; Moiyadi, A.; Prasad, M.; et al. Clinico-Radiological Outcomes in WNT-Subgroup Medulloblastoma. Diagnostics 2024, 14, 358. [Google Scholar] [CrossRef]
- Gajjar, A.; Robinson, G.W.; Smith, K.S.; Lin, T.; Merchant, T.E.; Chintagumpala, M.; Mahajan, A.; Su, J.; Bouffet, E.; Bartels, U.; et al. Outcomes by Clinical and Molecular Features in Children with Medulloblastoma Treated with Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J. Clin. Oncol. 2021, 39, 822–835. [Google Scholar] [CrossRef]
- Ellison, D.W.; Kocak, M.; Dalton, J.; Megahed, H.; Lusher, M.E.; Ryan, S.L.; Zhao, W.; Nicholson, S.L.; Taylor, R.E.; Bailey, S.; et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J. Clin. Oncol. 2011, 29, 1400–1407. [Google Scholar] [CrossRef]
- Northcott, P.A.; Hielscher, T.; Dubuc, A.; Mack, S.; Shih, D.; Remke, M.; Al-Halabi, H.; Albrecht, S.; Jabado, N.; Eberhart, C.G.; et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011, 122, 231–240. [Google Scholar] [CrossRef]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic prognostication within medulloblastoma subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef]
- Onodera, S.; Nakamura, Y.; Azuma, T. Gorlin Syndrome: Recent Advances in Genetic Testing and Molecular and Cellular Biological Research. Int. J. Mol. Sci. 2020, 21, 7559. [Google Scholar] [CrossRef]
- Kolodziejczak, A.S.; Guerrini-Rousseau, L.; Planchon, J.M.; Ecker, J.; Selt, F.; Mynarek, M.; Obrecht, D.; Sill, M.; Autry, R.J.; Stutheit-Zhao, E.; et al. Clinical outcome of pediatric medulloblastoma patients with Li-Fraumeni syndrome. Neuro-Oncology 2023, 25, 2273–2286. [Google Scholar] [CrossRef]
- Hovestadt, V.; Ayrault, O.; Swartling, F.J.; Robinson, G.W.; Pfister, S.M.; Northcott, P.A. Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat. Rev. Cancer 2020, 20, 42–56. [Google Scholar] [CrossRef]
- Menyhárt, O.; Giangaspero, F.; Győrffy, B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) medulloblastomas. J. Hematol. Oncol. 2019, 12, 29. [Google Scholar] [CrossRef]
- Voskamp, M.J.; Li, S.; van Daalen, K.R.; Crnko, S.; Ten Broeke, T.; Bovenschen, N. Immunotherapy in Medulloblastoma: Current State of Research, Challenges, and Future Perspectives. Cancers 2021, 13, 5387. [Google Scholar] [CrossRef]
- Gatto, L.; Franceschi, E.; Tosoni, A.; Di Nunno, V.; Bartolini, S.; Brandes, A.A. Molecular Targeted Therapies: Time for a Paradigm Shift in Medulloblastoma Treatment? Cancers 2022, 14, 333. [Google Scholar] [CrossRef]
- Wen, J.; Hadden, M.K. Medulloblastoma drugs in development: Current leads, trials and drawbacks. Eur. J. Med. Chem. 2021, 215, 113268. [Google Scholar] [CrossRef]
- Mahajan, A.; Shih, H.; Penas-Prado, M.; Ligon, K.; Aldape, K.; Hu, L.S.; Loughan, A.R.; Basso, M.R.; Leeper, H.E.; Nahed, B.V.; et al. The Alliance AMBUSH Trial: Rationale and Design. Cancers 2022, 14, 414. [Google Scholar] [CrossRef]
- Gajjar, A.J.; Orr, B.; Ellison, D.; Lafay-Cousin, L.; Minturn, J.; Fisher, M.; Bendel, A.; Hwang, E.; Murray, J.; Landi, D.; et al. MDB-49. Outcomes of Sonic Hedgehog (Shh) Medulloblastoma (Mb) in Children and Adults Treated with Risk-Adapted Therapy on a Prospective International Protocol (SJMB12). Neuro-oncology 2024, 26. [Google Scholar] [CrossRef]
- Hau, P.; Frappaz, D.; Hovey, E.; McCabe, M.G.; Pajtler, K.W.; Wiestler, B.; Seidel, C.; Combs, S.E.; Dirven, L.; Klein, M.; et al. Development of Randomized Trials in Adults with Medulloblastoma-The Example of EORTC 1634-BTG/NOA-23. Cancers 2021, 13, 3451. [Google Scholar] [CrossRef] [PubMed]
- Michalski, J.M.; Janss, A.J.; Vezina, L.G.; Smith, K.S.; Billups, C.A.; Burger, P.C.; Embry, L.M.; Cullen, P.L.; Hardy, K.K.; Pomeroy, S.L.; et al. Children’s Oncology Group Phase III Trial of Reduced-Dose and Reduced-Volume Radiotherapy with Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J. Clin. Oncol. 2021, 39, 2685–2697. [Google Scholar] [CrossRef]
- Leary, S.E.S.; Packer, R.J.; Li, Y.; Billups, C.A.; Smith, K.S.; Jaju, A.; Heier, L.; Burger, P.; Walsh, K.; Han, Y.; et al. Efficacy of Carboplatin and Isotretinoin in Children with High-risk Medulloblastoma: A Randomized Clinical Trial From the Children’s Oncology Group. JAMA Oncol. 2021, 7, 1313–1321. [Google Scholar] [CrossRef]
- Lazow, M.A.; Palmer, J.D.; Fouladi, M.; Salloum, R. Medulloblastoma in the Modern Era: Review of Contemporary Trials, Molecular Advances, and Updates in Management. Neurotherapeutics 2022, 19, 1733–1751. [Google Scholar] [CrossRef]
- Morfouace, M.; Shelat, A.; Jacus, M.; Freeman, B.B.; Turner, D.; Robinson, S.; Zindy, F.; Wang, Y.D.; Finkelstein, D.; Ayrault, O.; et al. Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell 2014, 25, 516–529. [Google Scholar] [CrossRef]
- Hill, R.M.; Plasschaert, S.L.A.; Timmermann, B.; Dufour, C.; Aquilina, K.; Avula, S.; Donovan, L.; Lequin, M.; Pietsch, T.; Thomale, U.; et al. Relapsed Medulloblastoma in Pre-Irradiated Patients: Current Practice for Diagnostics and Treatment. Cancers 2021, 14, 126. [Google Scholar] [CrossRef]
- Holmberg, K.O.; Borgenvik, A.; Zhao, M.; Giraud, G.; Swartling, F.J. Drivers Underlying Metastasis and Relapse in Medulloblastoma and Targeting Strategies. Cancers 2024, 16, 1752. [Google Scholar] [CrossRef]
- Levy, A.S.; Krailo, M.; Chi, S.; Villaluna, D.; Springer, L.; Williams-Hughes, C.; Fouladi, M.; Gajjar, A. Temozolomide with irinotecan versus temozolomide, irinotecan plus bevacizumab for recurrent medulloblastoma of childhood: Report of a COG randomized Phase II screening trial. Pediatr. Blood Cancer 2021, 68, e29031. [Google Scholar] [CrossRef] [PubMed]
- Peyrl, A.; Chocholous, M.; Sabel, M.; Lassaletta, A.; Sterba, J.; Leblond, P.; Nysom, K.; Torsvik, I.; Chi, S.N.; Perwein, T.; et al. Sustained Survival Benefit in Recurrent Medulloblastoma by a Metronomic Antiangiogenic Regimen: A Nonrandomized Controlled Trial. JAMA Oncol. 2023, 9, 1688–1695. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koch, A.; Childress, A.; Vallee, E.; Steller, A.; Raskin, S. The Current Landscape of Molecular Pathology for the Diagnosis and Treatment of Pediatric Medulloblastoma. J. Mol. Pathol. 2025, 6, 11. https://doi.org/10.3390/jmp6020011
Koch A, Childress A, Vallee E, Steller A, Raskin S. The Current Landscape of Molecular Pathology for the Diagnosis and Treatment of Pediatric Medulloblastoma. Journal of Molecular Pathology. 2025; 6(2):11. https://doi.org/10.3390/jmp6020011
Chicago/Turabian StyleKoch, Alayna, Ashley Childress, Emma Vallee, Alyssa Steller, and Scott Raskin. 2025. "The Current Landscape of Molecular Pathology for the Diagnosis and Treatment of Pediatric Medulloblastoma" Journal of Molecular Pathology 6, no. 2: 11. https://doi.org/10.3390/jmp6020011
APA StyleKoch, A., Childress, A., Vallee, E., Steller, A., & Raskin, S. (2025). The Current Landscape of Molecular Pathology for the Diagnosis and Treatment of Pediatric Medulloblastoma. Journal of Molecular Pathology, 6(2), 11. https://doi.org/10.3390/jmp6020011