Abstract
Atypical teratoid rhabdoid tumor (ATRT) is a rare, aggressive pediatric central nervous system (CNS) tumor that predominantly affects children under the age of 3. It is defined by the inactivation of the SMARCB1 gene, leading to the loss of INI1, a protein essential for cell lineage determination and cell differentiation. Current standard of care treatment requires aggressive multimodal therapy with maximal safe resection, high-dose chemotherapy with autologous stem cell rescue, and radiation, yet overall survival remains < 50%. These intensive regimens have improved overall survival but are associated with significant morbidity and long-term effects. Molecular profiling has significantly advanced the understanding of ATRTs, revealing four molecular subgroups, ATRT-TYR, ATRT-MYC, ATRT-SHH, and ATRT-SMARCA4, each with distinct clinical presentations, oncogenic pathways, and prognoses. Molecular characterization enables better prognostic stratification, guiding treatment decisions and allowing for more personalized therapeutic approaches. Targeted therapies based on these molecular insights remain experimental, and continued exploration of molecular mechanisms and how they differ amongst subgroups is pivotal for the development of less toxic, more effective targeted treatments.
1. Introduction
Atypical teratoid rhabdoid tumor (ATRT) is a rare, highly malignant central nervous system (CNS) tumor that primarily affects very young children. ATRTs account for 1–2% of pediatric CNS tumors but represent up to 20% and 40–50% of CNS tumors in children under the age of 3 and 1, respectively. The median age of diagnosis is 16–30 months, with 70–80% of diagnoses occurring before age 3 [1,2,3,4]. ATRTs are classified by the World Health Organization (WHO) as Grade IV embryonal tumors and exhibit aggressive phenotypes [1,5,6]. ATRTs demonstrate a tendency for rapid growth, are frequently metastatic at diagnosis, and often exhibit chemotherapy resistance, making them challenging to effectively treat [2,3,7]. These characteristics, in conjunction with its relatively recent discovery and characterization, have led to a historically dismal prognosis. The median survival time remains around 17 months [1,4,6,8]. Once universally fatal, this disease still carries a 5-year event-free survival (EFS) and overall survival (OS) of less than 50% with the current standard of care therapy [7,9]. Progress in OS comes at a significant cost to patients with intensive treatment regimens and numerous long-term side effects.
1.1. Discovery of ATRT
ATRTs were first reported in the literature by Biggs et al. in 1987 when describing a CNS tumor reminiscent of a renal malignant rhabdoid tumor [10]. The term “teratoid/rhabdoid” was later coined by Dr. Rorke in 1996, as she identified a biologically unique subset of embryonal tumors with a perplexingly heterogeneous histologic appearance. This terminology originated as a description of the poorly differentiated, multi-lineage morphology characteristically seen under the microscope when examining ATRTs [11]. The hallmark feature, as suggested by its name, is the presence of rhabdoid cells. These large round/oval cells feature abundant eosinophilic cytoplasm with large, eccentric nuclei and striking nucleoli that bear resemblance to rhabdomyoblasts. Dispersed amongst these cells are undifferentiated embryonal small round blue cells, epithelial morphology, and mesenchymal morphology (Figure 1). Further complicating the histopathology, these cell lineages are not all present in each tumor [2,12]. The degree of morphologic heterogeneity combined with the varied immunohistochemical staining seen amongst these tumors created a diagnostic conundrum, as evidenced by the relatively recent recognition of this tumor as a unique entity. The ATRT was not added to the WHO CNS classification system until the 3rd edition, published in 2000 [13].
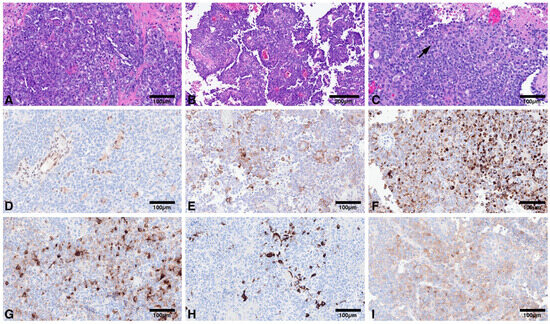
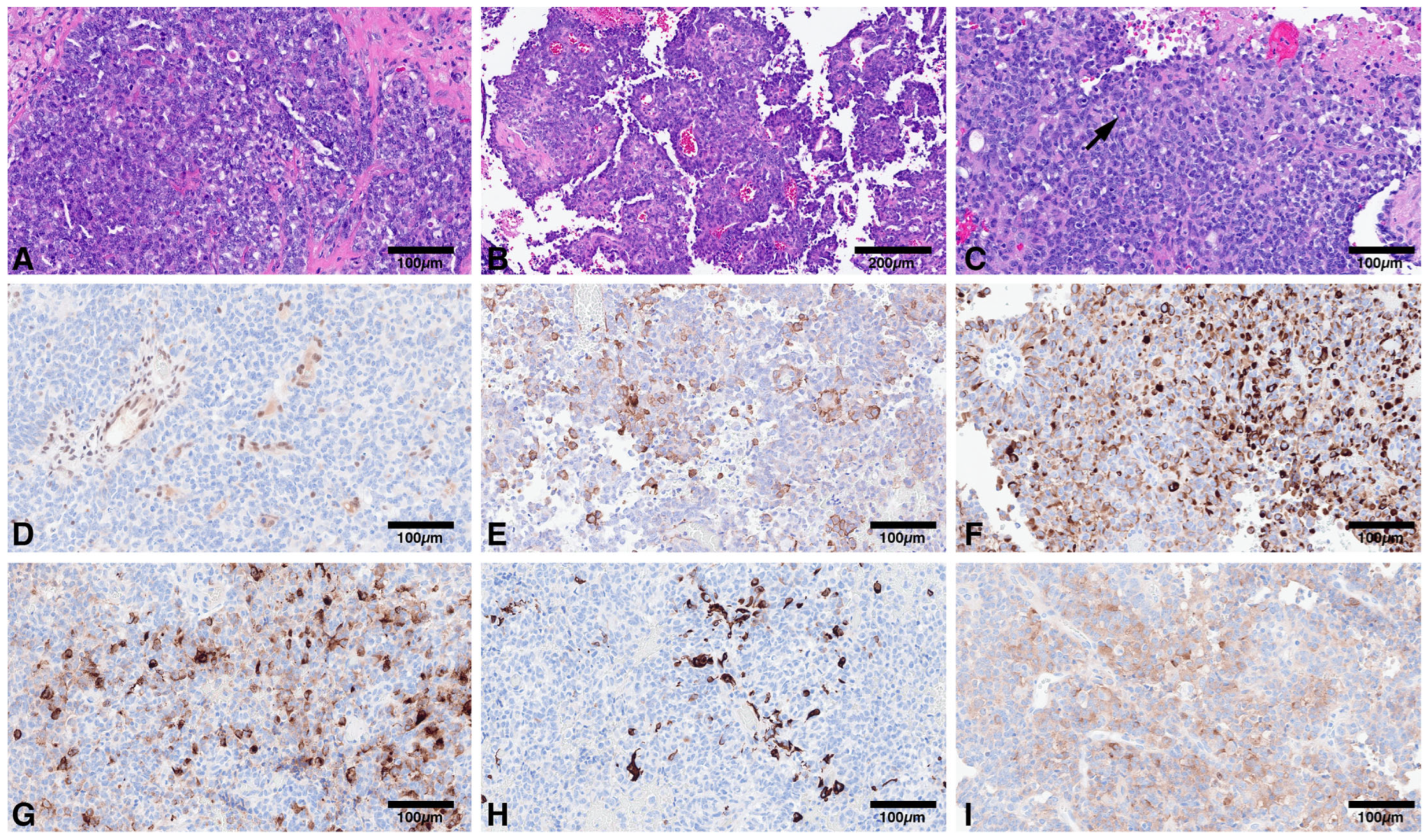
Figure 1.
(A): Compact population of round to oval embryonal cells arranged in sheets; (B): Pseudopapillary pattern may also be encountered; (C): Variable proportion of rhabdoid cells may be present (arrow); (D): Loss of nuclear immunoreactivity for INI1 in tumor cells, with retained expression in endothelial cells. Tumors are polyimmunophenotypic with immunoreactivity for smooth muscle actin (E); CAM 5.2 (F); EMA (G); GFAP (H); Synaptophysin (I).
Identification of the hallmark mutation in SWItch/sucrose nonfermentable (SWI/SNF)-related, matrix-associated, actin-dependent regulator of chromatin family B (SMARCB1), a tumor suppressor gene involved in the production of INI1 (otherwise known as BAF47 or SNF5), solidified ATRTs as a unique tumor entity despite its varied histopathologic appearance. INI1 is a necessary subunit of the SWI/SNF complex responsible for repositioning and restructuring nucleosomes through destabilization of histone-DNA interactions [1,3,6]. Additionally, a small subset of ATRTs is caused by the inactivation of SMARCA4, a gene responsible for producing an ATPase subunit of the SWI/SNF complex [1,2,3]. The actions of the SWI/SNF complex regulate chromatin structure and thus gene expression, a process essential for cell lineage determination and cell differentiation [3].
1.2. Current Diagnosis of ATRT
The diagnostic process for ATRTs is multifaceted, requiring the integration of several modalities to provide a thorough, accurate diagnosis. Suspicion for ATRTs begins with consistent imaging findings in the classic age demographic. On MRI, ATRTs appear as heterogeneous masses with irregular but well-defined borders, often featuring calcifications, hemorrhage, and central cystic necrosis. They can present in a variety of supra- and infratentorial locations and typically feature peritumoral edema, contrast enhancement, and strong diffusion restriction [4]. After resection, histologic evaluation identifies small round blue cells suggestive of an embryonal tumor, and in some specimens, the pathognomonic finding of rhabdoid cells. On immunohistochemical staining, INI1 or BRG1 expression is diagnostic for SMARCB1 and SMARCA4 mutations, respectively. The staining pattern of ATRTs is otherwise incredibly diverse, with frequent positivity for vimentin and epithelial membrane antigen and variable positivity for GFAP, actin, cytokeratin, and neuronal markers [12].
While immunohistochemistry can identify the diagnostic mutations of ATRTs, modern CNS tumor evaluations also include gene sequencing to confirm the presence of SMARCB1 or SMARCA4 mutations [2]. Next-generation sequencing (NGS) has prevailed as the superior technology for identifying diagnostically and therapeutically important mutations, as it is more time- and cost-efficient and requires smaller tissue samples compared to whole-genome sequencing. Many institutions have created targeted CNS neoplasm NGS panels that cover a broad array of pertinent mutations, which are routinely updated with new molecular discoveries [14]. Currently, the impact of NGS on ATRT diagnoses is less significant compared to many other pediatric CNS tumors, as most rhabdoid tumors exhibit fewer than 10 exomic alterations, most of which are not shared among tumors. Given the variability in co-occurring mutations, the data on their involvement in tumorigenesis and prognosis is limited [2].
DNA methylation analysis is another advanced diagnostic tool used to subclassify ATRTs into their distinct subgroups of ATRT-TYR, ATRT-MYC, ATRT-SHH, and ATRT-SMARCA4 [8,15]. Although not yet considered standard of care, it is increasingly recognized as a valuable tool, given the evolving understanding of how subgroups influence clinical course, treatment efficacy, and prognosis, as will be explored in this review. As with DNA sequencing, targeted methylation arrays have emerged as the superior tool for CNS tumor analysis for similar reasons discussed above. The most commonly utilized technologies for evaluating these arrays are the DKFZ/Heidelberg and NCI/Bethesda classifiers, though many institutions are actively creating or using internally derived classifiers [16].
1.3. Rhabdoid Tumor Predisposition Syndrome and Germline Mutations
Rhabdoid tumor predisposition syndrome (RTPS) is an autosomal dominant hereditary cancer syndrome that leads to a markedly increased risk of developing malignant rhabdoid tumors (MRT) [17]. RTPS is caused by germline mutations in SMARCB1 (RTPS1) or SMARCA4 (RTPS2). Approximately one-third of SMARCB1-mutated MRTs harbor a germline mutation, and this number is estimated to be higher in SMARCA4-mutated MRTs, though the exact prevalence is difficult to quantify [18]. RTPS should be suspected in patients who present at an early age (<1 yr old), with synchronous MRTs, or with a family history of rhabdoid tumor(s), though testing for a germline mutation is recommended in all patients with a newly diagnosed MRT. Most germline SMARCB1 mutations are de novo, given their strong penetrance, whereas SMARCA4 mutations are often inherited from asymptomatic carriers, reflecting an incomplete penetrance pattern. Patients with RTPS typically present with an aggressive clinical course and carry an inferior prognosis compared to sporadic MRTs [5,17]. MRTs can present intracranially as ATRTs or extracranially in various anatomic locations, including the kidney, liver, lung, heart, and connective tissues. These patients are also at risk for a host of other non-rhabdoid tumors with SMARCB1 deficiency or alterations, including, but not limited to, epithelioid sarcoma, synovial sarcoma, renal medullary carcinoma, myoepithelial carcinomas, sinonasal carcinomas, schwannomas, and peripheral nerve sheath tumors [19,20]. Intensive surveillance with frequent whole-body imaging and routine clinical examinations is recommended for patients with RTPS, as early detection of MRTs and other non-rhabdoid tumors in these patients is critical [17].
2. Molecular Subtypes
ATRT was previously considered a single entity among the group of embryonal tumors. In 2016, Johann et al. first described 3 distinct molecular subgroups of ATRTs: ATRT-tyrosinase (TYR), ATRT-myelocytomatosis oncogene (MYC), and ATRT-sonic hedgehog (SHH) (Figure 2). With the use of gene expression profiles and DNA methylation profiles, previously histologically diagnosed ATRTs were able to be further subcategorized into one of these groups [8]. Many studies have corroborated these findings with various strategies. These molecular subtypes have notably different epidemiology, clinical presentations, tumorigenesis pathways, and prognoses, making accurate diagnosis pivotal. The heterogeneity between these entities highlights the importance of a thorough diagnostic process that utilizes methylomic and molecular profiling and an individualized treatment plan.
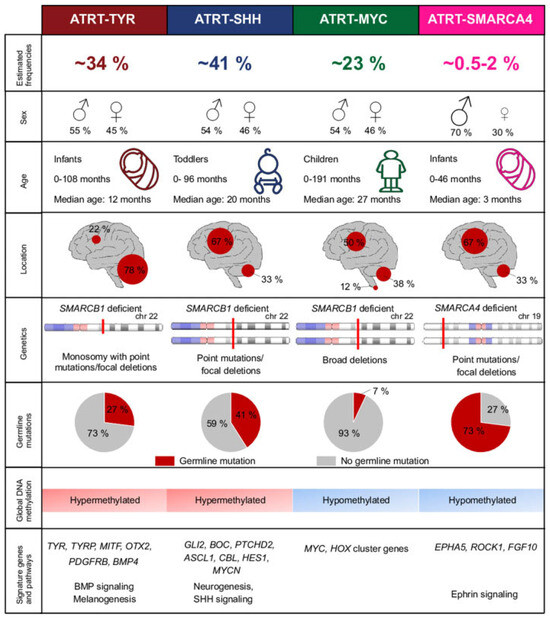
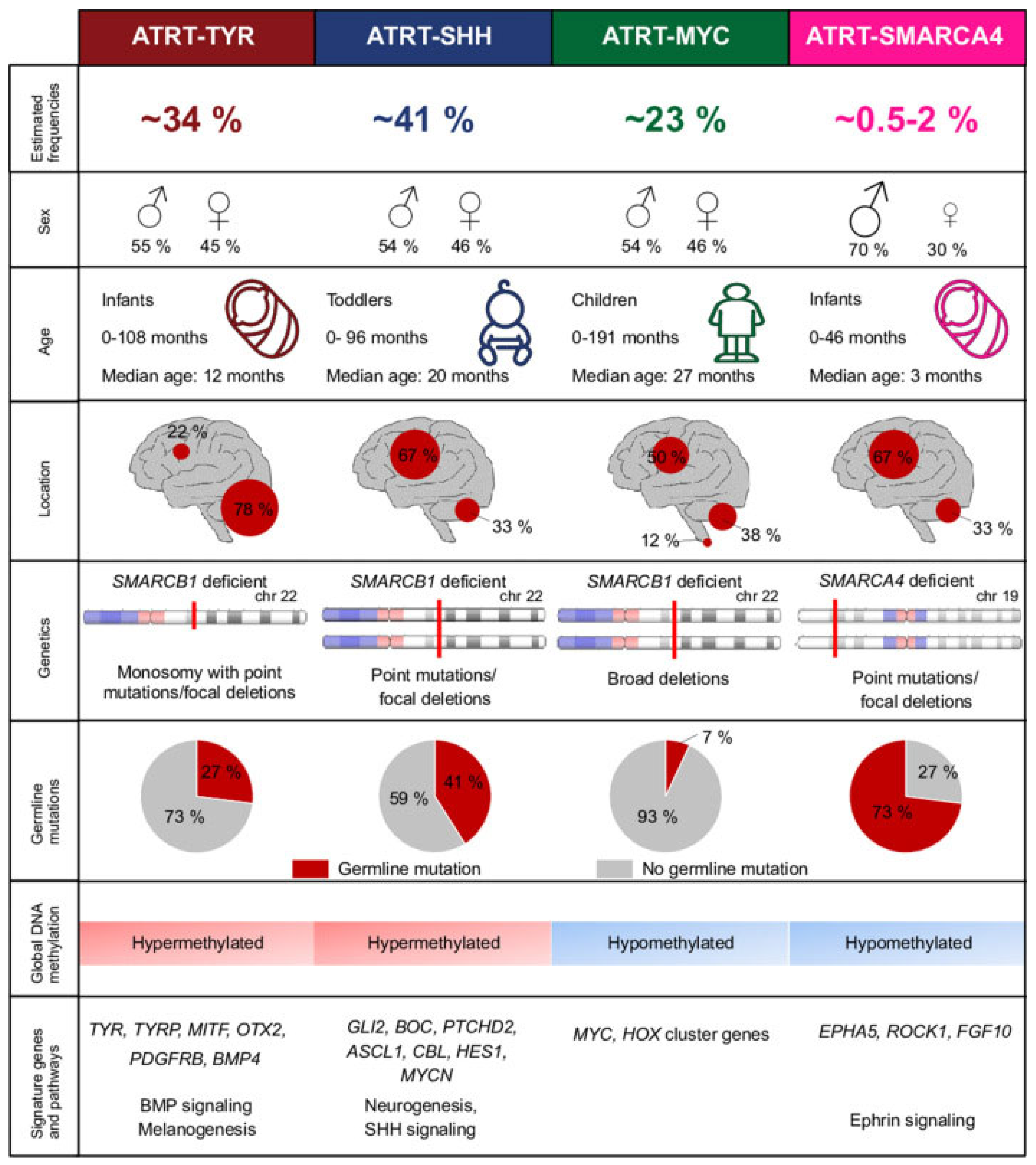
Figure 2.
Molecular subgroups of ATRT as seen in Holdhof, D. et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathologica, 2021 [18].
2.1. ATRT-TYR
ATRT-TYR represents approximately 30–40% of ATRT cases [7,8,21]. Clinically, ATRT-TYR tumors classically present in infants as an infratentorial mass. The median age of patients at diagnosis is 12 months, with 90% of patients presenting before age 3 [15]. 75% of these tumors can be found in the posterior fossa, often presenting as an exophytic growth at the cerebellopontine angle. Studies have shown an association with band-like contrast enhancement of these tumors on imaging [4]. Histopathologically, there is a correlation with epithelial predominant morphology, with a sensitivity of 49% and a specificity of 97% [12].
Upon genetic analysis, the mutations responsible for biallelic SMARCB1 inactivation in ATRT-TYR often involve copy number aberrations with a partial or complete loss of one chromosome 22 copy, as well as broad mutations [8,15]. ATRT-TYR displays a hypermethylated epigenetic environment [3,8,15,18]. As described in the name, ATRT-TYR tumors are associated with the overexpression of the enzyme tyrosinase, which is involved in the production of melanin in melanocytes, an important substrate for neural tube development [4]. Other implicated upregulated genes and pathways in oncogenesis include bone morphogenic protein (BMP), microphthalmia-associated transcription factor (MITF), orthodenticle homeobox 2 (OTX2), fibroblast growth factor receptor 2 (FGFR2), and janus kinase 1 (JAK1) [1,3,15].
2.2. ATRT-SHH
ATRT-SHH accounts for approximately 40–50% of ATRT diagnoses [7,8,21]. This subgroup of ATRT is characterized by an intermediate age group, with a median age of presentation of 20 months [15]. These tumors are known to present both supratentorially and infratentorially at equal rates. The SHH subtype 1 is known to favor the supratentorial region, often in the basal ganglia, whereas subtype 2 commonly presents infratentorially in the posterior fossa at the quadrigeminal cistern [4]. Notably, the SHH subtype is the only group known to involve tumors that span both the supra- and infratentorial regions [15]. On imaging, ATRT-SHH tumors often demonstrate lower rates of contrast enhancement compared to the other subgroups [4]. On review, this subtype exhibits a tendency towards small round blue cell tumor morphology, with a sensitivity of 57% and specificity of 83% [12].
Genetically, the mutations responsible for ATRT-SHH are commonly compound heterozygous point mutations [8,15]. Epigenetically, the SHH subtype is known to have a hypermethylated genome, similar to ATRT-TYR [3,8,15,18]. These tumors are associated with the activation of sonic hedgehog signaling through MYCN, GLI2, and BOC, as well as NOTCH signaling through ASCL1, HES5/6, and DTX [1,4]. SHH tumors, in comparison to their counterparts, are characterized by a neuronally differentiated phenotype [15].
2.3. ATRT-MYC
Approximately 10–20% of ATRT cases are classified into the MYC subgroup [7,8,21]. The MYC subgroup represents the oldest demographic, with a median age of 27 months at diagnosis [15]. These tumors are seen in both the supratentorial and infratentorial regions, with a slightly higher likelihood of supratentorial presentation [3]. In the supratentorial brain, they are classically found in cortical regions. In the infratentorial brain, they tend to present in extra-axial locations, notably the cranial nerves or internal auditory canal [4]. On imaging, MYC tumors have a higher propensity for peritumoral edema [15]. ATRT-MYC tumors are associated with a mesenchymal predominant morphology on histologic analysis, with a sensitivity of 46% and a specificity of 81% [12].
The genetics of ATRT-MYC are often characterized by focal homozygous deletions of SMARCB1 [1,8,15]. Epigenetically, the methylation patterns of ATRT-MYC more closely resemble most pediatric CNS tumors with a hypomethylated environment [3,8,15,18]. These genetic changes lead to the elevated expression of MYC, homeobox (HOX) cluster, and HOX transcript antisense intergenic RNA (HOTAIR) genes [1,15]. ATRT-MYC tumors bear the strongest resemblance to extracranial rhabdoid tumors [15].
2.4. ATRT-SMARCA4
ATRT-SMARCA4 constitutes 0.5–2% of ATRT cases [18]. Given that it is the rarest of the subgroups, much less is known and understood about this tumor type. Clinically, data demonstrates that these tumors are often seen in a younger infantile age and have a higher likelihood of associated germline mutation [3,18]. Less is known about radiographic and histopathologic associations with this specific tumor type. The mutations responsible for the SMARCA4 subgroup are often point mutations or focal deletions, and their genome, similar to ATRT-MYC, demonstrates hypomethylation [18]. These tumors are associated with overexpression of ephrin type-A receptor (EPHA5), rho-associated coiled-coil containing protein kinase 1 (ROCK1), and fibroblast growth factor 10 (FGF10) [3].
2.5. Prognostic Factors
Considering the intensity of the therapy currently employed to treat ATRT, there has been a strong effort to better understand the prognostic factors for this disease. Frühwald et al. identified age, metastatic status, and pathologic subgroup as independent prognostic factors in a study of 134 patients. Age was recognized as the strongest factor, with a 5-year OS of 45.3% for those >1 year old compared to 16.7% for patients < 1 year old. Metastasis at presentation was also associated with significantly worse outcomes, with a 5-year OS of only 16.9% in contrast to 43% in non-metastatic disease [7]. Of the subgroups, ATRT-TYR had the best prognosis, likely due to the higher likelihood of localized disease at presentation [22]. The 5-year OS of TYR and non-TYR groups were 48% and 24%, respectively. The OS between the SHH and MYC groups was not statistically significant [7].
3. Current Standard of Care Therapy
3.1. Surgery
The current standard of care for ATRTs begins with maximal safe resection of the tumor. As discussed, these tumors are incredibly aggressive and often present as a large mass with impressive vascularity, frequently inhabiting eloquent areas of the brain. Additionally, 20–40% of cases present with metastatic disease [2,3]. Given the difficulty of resection, the rate of gross total resection (GTR) is limited to approximately 30–68% [2]. Various studies have shown conflicting data regarding the prognostic implications of GTR. Early studies demonstrated a significant survival benefit in patients who underwent GTR, but later studies were unable to support this claim [2,23]. The protocols initiated after surgery have varied significantly, making comparisons between various studies challenging. GTR, however, remains the standard of care at this time, given that the therapeutic options for ATRTs remain limited.
3.2. Radiation
The use and necessity of radiation in the treatment of ATRTs has been a widely debated topic for many years. Many treatment regimens have demonstrated radiosensitivity in ATRTs, and radiation has been shown to be a positive prognostic factor. However, the adverse outcomes on neurocognition and neuroendocrine function, as well as the risk of serious complications, including radiation necrosis, have been the catalyst for many studies that have explored the ability to delay or defer radiation [2,3,4]. As with surgery, the data on the timing, technique, and necessity of radiation administration have shown inconsistent results. There have been several studies that have demonstrated a few long-term survivors whose treatment protocol avoided radiation [2,24]. There have also been studies that have demonstrated that the use of surgery with chemotherapy only, even high-dose chemotherapy (HDCT) with autologous stem cell rescue (ASCR), shows significantly inferior outcomes [23]. In terms of timing, early radiation following surgery seems to be advantageous, as many local recurrences are known to occur quickly during induction chemotherapy. However, numerous studies have also demonstrated that delayed RT does not have a detrimental impact on OS [2,3,4,25]. With the large percentage of patients who present with metastatic disease, the need for craniospinal irradiation (CSI), as opposed to focal irradiation, is another prominent question. The prevailing consensus for CSI is that outcomes are superior for those with metastatic disease. There has been conflicting data, however, about the benefit of CSI as a preventative treatment for focal disease. Currently, most guidelines recommend CSI only for metastatic disease in children > 3 years old [4,23].
3.3. Chemotherapy
With the relative rarity of ATRTs, there was a paucity of trials dedicated to ATRT patients following its discovery as a unique tumor entity. Thus, the majority of early treatment data on ATRTs arose from retrospective review of general treatment protocols for a variety of pediatric CNS tumors [1]. With the goal of avoiding radiation in infants and young children, many early trials focused on single-modality standard chemotherapy treatments [2]. When patients with ATRTs were isolated, data showed little to no survival benefit in patients who underwent chemotherapy-only regimens. A notable trial by Strother et al. in 2013 described a cohort of 36 ATRT patients who received one of two prolonged standard chemotherapy regimens that demonstrated a 0% survival rate [26]. It quickly became clear that multimodal therapy was necessary to improve survival. The first ATRT-dedicated trial was led by Dana-Farber Cancer Institute based on a protocol designed for parameningeal rhabdomyosarcoma. The study began in 2004 and combined established chemotherapy regimens with intrathecal chemotherapy and radiation, demonstrating a 2-year EFS and OS of 53% and 70% [27].
3.4. High Dose Chemotherapy and Autologous Stem Cell Rescue
The Head Start protocol was the first attempt to further intensify chemotherapy with the goal of avoiding radiation. It introduced the idea of HDCT with ASCR as a treatment for ATRTs. Results produced a 3-year EFS of 21% and OS of 26% [23,28]. The Children’s Oncology Group study ACNS0333 (NCT00653068) further expanded on the idea of HSCT with ASCR paired with RT, the timing and technique of which were determined by patient age and disease extent at the time of presentation. ACNS0333 began with maximal safe resection followed by two cycles of induction chemotherapy based on a prior successful multi-agent regimen paired with high dose methotrexate [23,25]. For patients with localized disease who were >6 months old with infratentorial disease or >12 months old with supratentorial disease, focal RT occurred prior to consolidation HDCT with ASCR. For those with non-metastatic disease below the defined age cutoffs, focal RT occurred following consolidation. For those with metastatic disease, age-adjusted CSI was completed following consolidation. The 4-year EFS and OS for patients were 37% and 43%, respectively [25]. This protocol remains the mainstay of treatment for ATRT at most referral centers. Currently, because data on prognostic factors is still relatively new and developing, most patients receive this regimen as described in the study, irrespective of their tumor characteristics.
4. Molecular Targeted Therapies
The innumerable acute toxicities paired with the significant long-term effects on developmental and functional outcomes from current therapies highlight the pressing need for effective targeted therapies. Not only do they hold the potential to limit the adverse effects of therapy, they also offer the potential to serve as an additional line of treatment for those patients who are refractory to current standard of care, improving both the rate and quality of survival [1,3].
4.1. Tumorigenesis and Tumor Microenvironment of ATRT
The genetic profile, immunogenicity, and tumorigenesis of ATRTs are quite unique when compared to many pediatric CNS tumor counterparts. As discussed, ATRTs are well known for their marked genetic simplicity and lack of identified recurrent mutations outside of SMARCB1. In stark contrast, however, their epigenetic environments are markedly heterogeneous, demonstrating that ATRT is a primarily epigenetically driven cancer. Whole-genome bisulfite sequencing reveals vastly different methylation patterns between subgroups, leading to variable silencing of tumor suppressor genes and activation of oncogenes amongst subgroups [8]. Another unique feature of ATRTs is their highly immunologically active tumor microenvironment. Similarly to the epigenetics of the ATRT subgroups, there is heterogeneity seen between the microenvironments of the subgroups [1]. Epigenetic modifiers, which work to restore typical gene expression patterns, and immunotherapies, which work to turn “cold” tumor microenvironments into “hot” immunologically active environments, are some of the most promising targeted treatments at the forefront of ATRT research.
4.2. Current Targeted Therapy Research
The goal of epigenetic modifiers is to decrease excess methylation and increase deficient acetylation that leads to silencing of tumor suppressor and neuronal differentiation genes through closed chromatin. Deletion of SMARCB1 prevents SWI/SNF inhibition of EZH2, a histone methyltransferase subunit of PRC2. This allows PCR2 to silence genes via histone methylation, notably through trimethylation of histone H3 at lysine 27 (H3K27me3) [1,23]. EZH2 inhibition has been shown to prevent tumor formation in orthotopic models; thus, the EZH2 inhibitor tazemetostat has been studied in several phase 1 trials. One study of 21 patients showed 1 complete response and 5 objective responses [23]. Another study of 8 patients showed no objective response in patients, though 2 exhibited stable disease for several months [5,23]. The drug decitabine, a cytosine analog, works through a similar mechanism at a DNA level by disrupting DNA methylation through DNA methyltransferase inhibition. Decitabine was trialed in a study of relapsed/refractory rhabdoid tumors and demonstrated radiologic response in 6 of 22 patients, 4 of which were ATRT patients [5]. The other class of epigenetic modifiers actively being studied is histone deacetylase (HDAC) inhibitors, including panobinostat and vorinostat. These drugs work by a different approach of increasing acetyl groups on DNA to prevent gene silencing. These medications were also effective in slowing tumor growth and prolonging survival in xenograft models [23]. Studies have demonstrated that these medications can improve outcomes on their own but hold even more promise when combined in multimodal regimens.
Immunotherapy is another rapidly developing field of targeted treatments for pediatric CNS tumors. Some of the most notable immunotherapies currently being studied for the treatment of ATRTs include checkpoint inhibitors (notably PD-1 inhibitors), dendritic cell vaccines, oncolytic virus vaccines, and chimeric-antigen receptor (CAR) T cells. They each have shown some level of efficacy in the treatment of ATRTs, though essentially all studies have been limited to orthotopic models or a broad population of variable CNS tumors in which ATRT patients make up only a small minority. Many trials are ongoing to further investigate these therapies [1]. There is still much to learn about the oncogenic mechanisms that drive ATRTs that could be the impetus for additional unique therapeutics specific to ATRTs.
5. Discussion
The introduction and integration of molecular data into the diagnostic criteria for CNS tumors revolutionized the field of Neuro-Oncology. The revised edition of WHO CNS 4, published in 2016, was the first iteration to formally introduce the concept of molecular data integration, but WHO CNS 5, published in 2021, greatly elaborated upon this concept. WHO CNS 5 integrated data from next generation sequencing and DNA methylation profiling with histology and immunohistochemistry that had long been established in the field. It further expanded upon the concept of sub-classification, allowing for a more precise diagnostic and prognostication process [29,30]. Most importantly, the molecular and epigenetic data described in WHO CNS 5 opens the door to further exploration and development of effective targeted therapies for patients with CNS neoplasms. WHO CNS 5 was the first iteration to formally list the ATRT subgroups that had been established in the field since their discovery in 2016. Over the last decade since their discovery, the understanding of these subgroups, their molecular changes, and their associated prognoses has vastly advanced.
This improved understanding has led to many studies aimed at translating molecular information into effective therapeutics, as discussed above. In the interim, this knowledge is crucial for implementing individualized therapeutic plans with current standard of care therapies for patients based on their tumor characteristics. Given the intensity of the current multimodal protocols, this prognostic information allows for productive conversations about attempting to delay or avoid aggressive therapies in those with a favorable prognosis or, conversely, providing them upfront for those with a poor prognosis.
6. Future Directions
These numerous advances are certainly worthy of recognition and have undoubtedly progressed the field, but the molecular data gathered about ATRTs has yet to translate into the widespread implementation of evidence-based targeted treatments for this aggressive disease. Many trials are currently underway, working towards actualizing this goal, but targeted treatments for ATRTs remain experimental at this time. As part of this ongoing work, there is still much to discover about molecular changes in each subgroup, notably the epigenetic alterations seen in ATRTs with their otherwise relatively stable genome. After identifying effective targeted treatments, there is also much to learn about which treatments are most effective for each subgroup, given that most current targeted treatment studies analyze efficacy for ATRTs without taking subgroup data into account. While there is still much to explore, a promising future presents itself in the management of ATRTs by integrating molecular knowledge into the diagnostic process and treatment plans.
Author Contributions
Concept and design: A.C. and S.R.; Drafting of the manuscript and Critical revision: A.C., A.K., A.S., E.V. and S.R. All authors have read and agreed to the published version of the manuscript.
Funding
No funding was required for this publication. Kirti Gupta provided histopathological images for Figure 1.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript.
| ATRT | Atypical teratoid rhabdoid tumor |
| CNS | central nervous system |
| WHO | World Health Organization |
| EFS | event-free survival |
| OS | overall survival |
| MRT | malignant rhabdoid tumor |
| PNET | primitive neuroectodermal tumors |
| ETMR | embryonal tumors with multilayered rosettes |
| SWI/SNF | SWitch/sucrose nonfermentable |
| SMARCB1 | actin-dependent regulator of chromatin B |
| SMARCA4 | chromatin remodeling complex subunit ATPase 4 |
| NGS | next genome sequencing |
| RTPS | rhabdoid tumor predisposition syndrome |
| ATRT-TYR | tyrosinase |
| ATRT-MYC | myelocytomatosis oncogene |
| ATRT-SHH | sonic hedgehog |
| BMP | bone morphogenic protein |
| MITF | microphthalmia-associated transcription factor |
| OTX2 | orthodenticle homeobox 2 |
| FGFR2 | fibroblast growth factor receptor 2 |
| JAK1 | janus kinase 1 |
| HOX | homeobox cluster |
| HOTAIR | HOX transcript antisense intergenic RNA |
| EPHA5 | ephrin type-A receptor |
| ROCK1 | rho-associated coiled-coil containing protein kinase 1 |
| FGF10 | fibroblast growth factor 10 |
| GTR | gross total resection |
| HDCT | high dose chemotherapy |
| ASCR | autologous stem cell rescue |
| CSI | craniospinal irradiation |
| H3K27me3 | histone H3 at lysine 27 |
| HDAC | histone deacetylase |
| CAR | chimeric-antigen receptor |
References
- Tran, S.; Plant-Fox, A.S.; Chi, S.N.; Narendran, A. Current advances in immunotherapy for atypical teratoid rhabdoid tumor (ATRT). Neuro-Oncol. Pract. 2023, 10, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Nesvick, C.L.; Lafay-Cousin, L.; Raghunathan, A.; Bouffet, E.; Huang, A.A.; Daniels, D.J. Atypical teratoid rhabdoid tumor: Molecular insights and translation to novel therapeutics. J. Neuro-Oncol. 2020, 150, 47–56. [Google Scholar] [CrossRef]
- Rechberger, J.S.; Nesvick, C.L.; Daniels, D.J. Atypical teratoid rhabdoid tumor (ATRT): Disease mechanisms and potential drug targets. Expert. Opin. Ther. Targets 2022, 26, 187–192. [Google Scholar] [CrossRef]
- Timmermann, B.; Alapetite, C.; Dieckmann, K.; Kortmann, R.D.; Lassen-Ramshad, Y.; Maduro, J.H.; Ramos Albiac, M.; Ricardi, U.; Weber, D.C. ESTRO-SIOPE guideline: Clinical management of radiotherapy in atypical teratoid/rhabdoid tumors (AT/RTs). Radiother. Oncol. 2024, 196, 110227. [Google Scholar] [CrossRef] [PubMed]
- Gastberger, K.; Fincke, V.E.; Mucha, M.; Siebert, R.; Hasselblatt, M.; Frühwald, M.C. Current Molecular and Clinical Landscape of ATRT-The Link to Future Therapies. Cancer Manag. Res. 2023, 15, 1369–1393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, H. Molecular targeted therapies for pediatric atypical teratoid/rhabdoid tumors. Pediatr. Investig. 2022, 6, 111–122. [Google Scholar] [CrossRef]
- Frühwald, M.C.; Hasselblatt, M.; Nemes, K.; Bens, S.; Steinbügl, M.; Johann, P.D.; Kerl, K.; Hauser, P.; Quiroga, E.; Solano-Paez, P.; et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol. 2020, 22, 1006–1017. [Google Scholar] [CrossRef]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef]
- Ma, X.J.; Li, D.; Wang, L.; Hao, S.Y.; Zhang, L.W.; Zhang, J.T.; Wu, Z. Overall Survival of Primary Intracranial Atypical Teratoid Rhabdoid Tumor Following Multimodal Treatment: A Pooled Analysis of Individual Patient Data. Neurosurg. Rev. 2020, 43, 281–292. [Google Scholar] [CrossRef]
- Biggs, P.; Garen, P.; Powers, J.; Garvin, J. Malignant rhabdoid tumor of the central nervous system. Hum. Pathol. 1987, 18, 332–337. [Google Scholar] [CrossRef]
- Rorke, L.B.; Packer, R.; Biegel, J. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J. Neuro-Oncol. 1995, 24, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Zin, F.; Cotter, J.A.; Haberler, C.; Dottermusch, M.; Neumann, J.; Schüller, U.; Schweizer, L.; Thomas, C.; Nemes, K.; Johann, P.D.; et al. Histopathological patterns in atypical teratoid/rhabdoid tumors are related to molecular subgroup. Brain Pathol. 2021, 31, e12967. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Chen, Y.; M de Blank, P.; Ondracek, A.; Farah, P.; Gittleman, H.; Wolinsky, Y.; Kruchko, C.; Cohen, M.L.; Brat, D.J.; et al. The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001–2010. Neuro Oncol. 2014, 16, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, J.; Rothhammer-Hampl, T.; Zoubaa, S.; Bumes, E.; Pukrop, T.; Kölbl, O.; Corbacioglu, S.; Schmidt, N.O.; Proescholdt, M.; Hau, P.; et al. A comprehensive DNA panel next generation sequencing approach supporting diagnostics and therapy prediction in neurooncology. Acta Neuropathol. Commun. 2020, 8, 124. [Google Scholar] [CrossRef]
- Ho, B.; Johann, P.D.; Grabovska, Y.; De Dieu Andrianteranagna, M.J.; Yao, F.; Frühwald, M.; Hasselblatt, M.; Bourdeaut, F.; Williamson, D.; Huang, A.; et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors—A reinvestigation and current consensus. Neuro Oncol. 2020, 22, 613–624. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Nemes, K.; Bens, S.; Bourdeaut, F.; Johann, P.; Kordes, U.; Seibert, R.; Frühwald, M. Rhabdoid Tumor Predisposition Syndrome; University of Washington: Seattle, WA, USA, 2017; pp. 1–25. [Google Scholar]
- Holdhof, D.; Johann, P.D.; Spohn, M.; Bockmayr, M.; Safaei, S.; Joshi, P.; Masliah-Planchon, J.; Ho, B.; Andrianteranagna, M.; Bourdeaut, F.; et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathol. 2021, 141, 291–301. [Google Scholar] [CrossRef]
- Parker, N.A.; Al-Obaidi, A.; Deutsch, J.M. SMARCB1/INI1-deficient tumors of adulthood. F1000Research 2020, 9, 662. [Google Scholar] [CrossRef]
- Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. [Google Scholar] [CrossRef]
- Kram, D.E.; Henderson, J.J.; Baig, M.; Chakraborty, D.; Gardner, M.A.; Biswas, S.; Khatua, S. Embryonal Tumors of the Central Nervous System in Children: The Era of Targeted Therapeutics. Bioengineering 2018, 5, 78. [Google Scholar] [CrossRef]
- Upadhyaya, S.A.; Robinson, G.W.; Onar-Thomas, A.; Orr, B.A.; Johann, P.; Wu, G.; Billups, C.A.; Tatevossian, R.G.; Dhanda, S.K.; Srinivasan, A.; et al. Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional Trials. Clin. Cancer Res. 2021, 27, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Alva, E.; Rubens, J.; Chi, S.; Rosenberg, T.; Reddy, A.; Raabe, E.H.; Margol, A. Recent progress and novel approaches to treating atypical teratoid rhabdoid tumor. Neoplasia 2023, 37, 100880. [Google Scholar] [CrossRef] [PubMed]
- Lafay-Cousin, L.; Hawkins, C.; Carret, A.S.; Johnston, D.; Zelcer, S.; Wilson, B.; Jabado, N.; Scheinemann, K.; Eisenstat, D.; Fryer, C.; et al. Central nervous system atypical teratoid rhabdoid tumours: The Canadian Paediatric Brain Tumour Consortium experience. Eur. J. Cancer 2012, 48, 353–359. [Google Scholar] [CrossRef]
- Reddy, A.T.; Strother, D.R.; Judkins, A.R.; Burger, P.C.; Pollack, I.F.; Krailo, M.D.; Buxton, A.B.; Williams-Hughes, C.; Fouladi, M.; Mahajan, A.; et al. Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report from the Children’s Oncology Group Trial ACNS0333. J. Clin. Oncol. 2020, 38, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Strother, D.R.; Lafay-Cousin, L.; Boyett, J.M.; Burger, P.; Aronin, P.; Constine, L.; Duffner, P.; Kocak, M.; Kun, L.E.; Horowitz, M.E.; et al. Benefit from prolonged dose-intensive chemotherapy for infants with malignant brain tumors is restricted to patients with ependymoma: A report of the Pediatric Oncology Group randomized controlled trial 9233/34. Neuro Oncol. 2014, 16, 457–465. [Google Scholar] [CrossRef]
- Chi, S.N.; Zimmerman, M.A.; Yao, X.; Cohen, K.J.; Burger, P.; Biegel, J.A.; Rorke-Adams, L.B.; Fisher, M.J.; Janss, A.; Mazewski, C.; et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J. Clin. Oncol. 2009, 27, 385–389. [Google Scholar] [CrossRef]
- Zaky, W.; Dhall, G.; Ji, L.; Haley, K.; Allen, J.; Atlas, M.; Bertolone, S.; Cornelius, A.; Gardner, S.; Patel, R.; et al. Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: The Head Start III experience. Pediatr. Blood Cancer 2014, 61, 95–101. [Google Scholar] [CrossRef]
- Smith, H.L.; Wadhwani, N.; Horbinski, C. Major Features of the 2021 WHO Classification of CNS Tumors. Neurotherapeutics 2022, 19, 1691–1704. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).