

The Potential Role of Histone Modifications in Glioblastoma Therapy: Review Article

Abstract
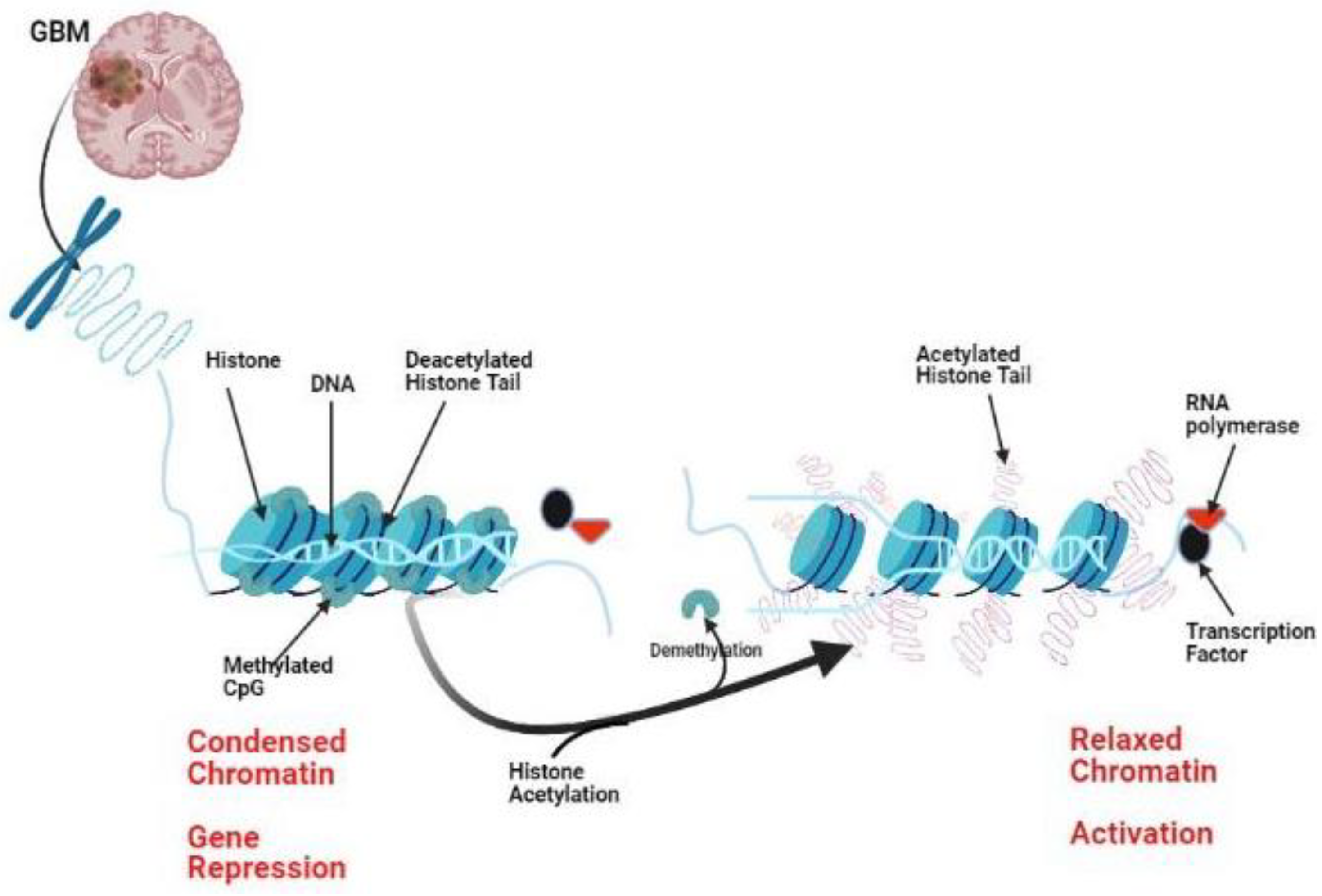
:1. Introduction
2. Histone Modifications in Glioma
2.1. Histone Deacetylation in Glioma
2.2. Histone Acetylation in Glioma
2.3. Histone Methylation in Glioma
2.4. Histone Demethylation in Glioma
2.5. Histone Ubiquitination in Glioma
2.6. Histone Sumoylation and Glioma
2.7. Histone Phosphorylation and Glioma
2.8. Targeting Histone-Modifying Enzymes in Glioma
3. HDAC Inhibitors (HDACi)
3.1. Vorinostat
3.2. Valproic Acid
3.3. Romidepsin (FK228)
3.4. Panobinostat
3.5. Targeting the Ubiquitin–Proteasome System in GBM
3.5.1. Controlling Proteasome
3.5.2. Controlling Ubiquitination Enzymes
3.6. Targeting SUMOylation in GBM
3.7. Targeting Epigenetic Modifiers of H3K27me3
3.8. Limitations of HDACi in Clinical Practice
4. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABTC | The Adult Brain Tumor Consortium |
| EIAEDs | Enzyme-inducing antiepileptic drugs |
| EORTC | European Organization for Research and Treatment of Cancer |
| GBM | Glioblastoma |
| DNA | Deoxynucleic acid |
| EGF | Epidermal Growth Factor |
| HDAC | Histone Deacetylase |
| HDACi | Histone Deacetylase Inhibitors |
| HATs | Histone Acetyltransferases |
| NCIC | National Cancer Institute of Canada |
| USP | Ubiquitin Specific Protease |
| TMZ | Temozolomide |
References
- Sherrod, B.A.; Gamboa, N.T.; Wilkerson, C.; Wilde, H.; Azab, M.A.; Karsy, M.; Jensen, R.L.; Menacho, S.T. Effect of patient age on glioblastoma perioperative treatment costs: A value driven outcome database analysis. J. Neuro-Oncol. 2019, 143, 465–473. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. deacetylase Epigenetic therapy of cancer with histone inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Bi, G.; Jiang, G. The molecular mechanism of HDAC inhibitors in anticancer effects. Cell. Mol. Immunol. 2006, 3, 285–289. [Google Scholar]
- Secrist, J.P.; Zhou, X.; Richon, V.M. HDAC inhibitors for the treatment of cancer. Curr. Opin. Investig. Drugs 2003, 4, 1422–1427. [Google Scholar]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACS): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Bezecny, P. Histone deacetylase inhibitors in glioblastoma: Preclinical and clinical experience. Med. Oncol. 2014, 31, 985. [Google Scholar] [CrossRef]
- Adamopoulou, E.; Naumann, U. HDAC inhibitors and their potential applications to glioblastoma therapy. Oncoimmunology 2013, 2, e25219. [Google Scholar] [CrossRef]
- Sturm, D.; Bender, S.; Jones, D.T.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Hamid, A.; Hussain, A.; Majeed, R.; Qurishi, Y.; Bhat, J.A.; Najar, R.A.; Qazi, A.K.; Zargar, M.A.; Singh, S.K.; et al. Understanding histone deacetylases in the cancer development and treatment: An epigenetic perspective of cancer chemotherapy. DNA Cell Biol. 2012, 31 (Suppl. 1), S62–S71. [Google Scholar] [CrossRef] [PubMed]
- Lucio-Eterovic, A.K.; Cortez, M.A.; Valera, E.T.; Motta, F.J.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: Class II and IV are hypoexpressed in glioblastomas. BMC Cancer 2008, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Chen, J.; Tan, Q.; Xie, C.; Li, C.; Zhan, W.; Wang, M. Silencing of histone deacetylase 2 suppresses malignancy for proliferation, migration, and invasion of glioblastoma cells and enhances temozolomide sensitivity. Cancer Chemother. Pharmacol. 2016, 78, 1289–1296. [Google Scholar] [CrossRef]
- Zhong, S.; Fan, Y.; Wu, B.; Wang, Y.; Jiang, S.; Ge, J.; Hua, C.; Zhao, G.; Chen, Y.; Xu, H. HDAC3 expression correlates with the prognosis and grade of patients with Glioma: A diversification analysis based on Transcriptome and clinical evidence. World Neurosurg. 2018, 119, e145–e158. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; He, X.; Liu, W.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Feng, J.; Yan, P.F.; Zhao, H.Y.; Zhang, F.C.; Zhao, W.H.; Feng, M. SIRT6 suppresses glioma cell growth via induction of apoptosis, inhibition of oxidative stress and suppression of JAK2/STAT3 signaling pathway activation. Oncol. Rep. 2016, 35, 1395–1402. [Google Scholar] [CrossRef]
- Chen, H.; Lin, R.; Zhang, Z.; Wei, Q.; Zhong, Z.; Huang, J.; Xu, Y. Sirtuin 1 knockdown inhibits glioma cell proliferation and potentiates temozolomide toxicity via facilitation of reactive oxygen species generation. Oncol. Lett. 2019, 17, 5343–5350. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Sealy, L.; Chalkley, R. DNA associated with hyperacetylated histone is preferentially digested by DNase I. Nucleic Acids Res. 1978, 5, 1863–1876. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L.; Liang, J.; Shi, L.; Yang, J.; Yi, X.; Zhang, D.; Han, X.; Yu, N.; Shang, Y. Histone acetyltransferase 1 promotes homologous recombination in DNA repair by facilitating histone turnover. J. Biol. Chem. 2013, 288, 18271–18278. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.; Auston, D.; Grant, P.; John, S.; Cook, R.G.; Workman, J.L.; Pillus, L. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 2001, 15, 3144–3154. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef]
- Diao, P.Y.; Li, S.X.; Peng, J.; Yang, J.H.; Pan, Y.C.; Xu, X.P.; Tang, H.; Hu, J.X.; Zhao, H.F.; Huang, G.D. Overexpression of EP300-interacting inhibitor of differentiation 3 predicts poor prognosis in patients with glioblastoma multiforme. Int. J. Clin. Exp. Pathol. 2020, 13, 979–9880. [Google Scholar]
- Lv, D.; Jia, F.; Hou, Y.; Sang, Y.; Alvarez, A.A.; Zhang, W.; Feng, H. Histone acetyltransferase KAT6A upregulates PI3K/AKT signaling through TRIM24 binding. Cancer Res. 2017, 77, 6190–6201. [Google Scholar] [CrossRef]
- Saidi, D.; Cheray, M.; Osman, A.M.; Stratoulias, V.; Lindberg, O.R.; Shen, X.; Blomgren, K.; Joseph, B. Glioma-induced SIRT1-dependent activation of hMOF histone H4 lysine 16 acetyltransferase in microglia promotes a tumor supporting phenotype. OncoImmunology 2018, 7, e1382790. [Google Scholar] [CrossRef]
- Bedford, M.T.; Richard, S. Arginine methylation: An emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Chiang, K.; Zielinska, A.E.; Shaaban, A.M.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Davies, C.C. PRMT5 is a critical regulator of breast Cancer stem cell function via histone methylation and FOXP1 expression. Cell Rep. 2017, 21, 3498–3513. [Google Scholar] [CrossRef] [PubMed]
- Xu, K. DNA and histone methylation in prostate Cancer. In Cancer Drug Discovery and Development; Humana Press Inc.: Cham, Switzerland, 2017; pp. 489–529. [Google Scholar] [CrossRef]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional role of G9a histone methyltransferase in cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulou, A.; Gargalionis, A.; Dalagiorgou, G.; Adamopoulos, C.; Papavassiliou, K.A.; Lea, R.W.; Piperi, C.; Papavassiliou, A.G. role of histone lysine methyltransferases SUV39H1 and SETDB1 in gliomagenesis: Modulation of cell proliferation, migration, and colony formation. NeuroMol. Med. 2014, 16, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Li, Q.; Yang, C.; Huo, D.; Wang, X.; Ai, C.; Kong, Y.; Sun, X.; Wang, W.; Zhou, Y.; et al. PRMT2 links histone H3R8 asymmetric dimethylation to oncogenic activation and tumorigenesis of glioblastoma. Nat. Commun. 2018, 9, 4552. [Google Scholar] [CrossRef]
- Loh, Y.H.; Zhang, W.; Chen, X.; George, J.; Ng, H.H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007, 21, 2545–2557. [Google Scholar] [CrossRef]
- Chammas, P.; Mocavini, I.; Di Croce, L. Engaging chromatin: PRC2 structure meets function. Br. J. Cancer 2020, 122, 315–328. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Wu, Q.; Rivera, M.; Minhas, S.; Lathia, J.D.; Sloan, A.E.; Rich, J.N. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012, 19, 428–439. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Schwartzentruber, J.; Khuong-Quang, D.A.; Liu, X.Y.; Sturm, D.; Korshunov, A.; Majewski, J. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013, 125, 659–669. [Google Scholar] [CrossRef]
- Bozek, D.; Wang, A.; Hao, X.; Johnston, M.; Luchman, H.A.; Weiss, S. STEM-28.DOT1L epigenetically regulates GBM brain tumor stem cells. Neuro-Oncology 2017, 19 (Suppl. 6), 231–232. [Google Scholar] [CrossRef]
- Cheng, T.; Xu, Y. Effects of enhancer of zeste homolog 2 (EZH2) expression on brain glioma cell proliferation and tumorigenesis. Med. Sci. Monit. 2018, 24, 7249–7255. [Google Scholar] [CrossRef]
- Wang, S.; Tan, X.; Yang, B.; Yin, B.; Yuan, J.; Qiang, B.; Peng, X. The role of protein arginine-methyltransferase 1 in gliomagenesis. BMB Rep. 2012, 45, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Alinari, L.; Lustberg, M.E.; Martin, L.K.; Cordero-Nieves, H.M.; Banasavadi-Siddegowda, Y.; Baiocchi, R.A. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014, 74, 1752–1765. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Banelli, B.; Carra, E.; Barbieri, F.; Wurth, R.; Parodi, F.; Pattarozzi, A.; Carosio, R.; Forlani, A.; Allemanni, G.; Marubbi, D.; et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle 2015, 14, 3418–3429. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Nair, B.C.; Krishnan, S.K.; Gonugunta, V.K.; Zhang, Q.G.; Suzuki, T.; Vadlamudi, R.K. KDM1 is a novel therapeutic target for the treatment of gliomas. Oncotarget 2013, 4, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Shou, T.; Yang, H.; Lv, J.; Liu, D.; Sun, X. MicroRNA-3666 suppresses the growth and migration of glioblastoma cells by targeting KDM2A. Mol. Med. Rep. 2019, 19, 1049–1055. [Google Scholar] [CrossRef]
- Staberg, M.; Rasmussen, R.D.; Michaelsen, S.R.; Pedersen, H.; Jensen, K.E.; Villingshøj, M.; Hamerlik, P. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol. Oncol. 2018, 12, 406–420. [Google Scholar] [CrossRef]
- Wang, B.; Fan, X.; Ma, C.; Lei, H.; Long, Q.; Chai, Y. Downregulation of KDM4A suppresses the survival of Glioma cells by promoting autophagy. J. Mol. Neurosci. 2016, 60, 137–144. [Google Scholar] [CrossRef]
- Romani, M.; Daga, A.; Banelli, B.; Forlani, A.; Pistillo, M.P. Targeting of histone demethylases KDM5A and KDM6B inhibits the proliferation of temozolomide resistant glioblastoma cells. Cancers 2019, 11, 878. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Jeusset, L.M.-P.; McManus, K.J. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells 2019, 8, 165. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zhou, W.; Wu, Q.; Huang, Z.; Shi, Y.; Yang, K.; Chen, C.; Xie, Q.; Mack, S.C.; Wang, X.; et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J. Exp. Med. 2017, 214, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Oikonomaki, M.; Bady, P.; Hegi, M.E. Ubiquitin specific peptidase 15 (USP15) suppresses glioblastoma cell growth via stabilization of HECTD1 E3 ligase attenuating WNT pathway activity. Oncotarget 2017, 8, 110490–110502. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, P.; Ji, W.; Yu, Z.; Chen, H.; Jiang, L. Ubiquitin-specific protease 4 promotes glioblastoma multiforme via activating ERK pathway. Oncotargets Ther. 2019, 12, 1825–1839. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Ai, C.; Dong, F.; Xia, X.; Zhao, X.; Yang, C.; Kang, C.; Zhou, Y.; Zhao, Q.; Sun, X.; et al. Targeting of BMI-1 with PTC-209 inhibits glioblastoma development. Cell Cycle 2018, 17, 1199–1211. [Google Scholar] [CrossRef]
- Fan, L.; Chen, Z.; Wu, X.; Cai, X.; Feng, S.; Lu, J.; Wang, H.; Liu, N. Ubiquitin-specific protease 3 promotes glioblastoma cell invasion and epithelial-mesenchymal transition via stabilizing snail. Mol. Cancer Res. 2019, 17, 1975–1984. [Google Scholar] [CrossRef]
- Grunda, J.M.; Nabors, L.B.; Palmer, C.A.; Chhieng, D.C.; Steg, A.; Mikkelsen, T.; Diasio, R.B.; Zhang, K.; Allison, D.; Grizzle, W.E.; et al. Increased expression of thymidylate synthetase (TS), ubiquitin specific protease 10 (USP10) and survivin is associated with poor survival in glioblastoma multiforme (GBM). J. Neuro-Oncol. 2006, 80, 261–274. [Google Scholar] [CrossRef]
- Wang, Z.; Song, Q.; Xue, J.; Zhao, Y.; Qin, S. Ubiquitin-specific protease 28 is overexpressed in human glioblastomas and contributes to glioma tumorigenicity by regulating MYC expression. Exp. Biol. Med. 2016, 241, 255–264. [Google Scholar] [CrossRef]
- Park, H.J.; Yun, D.-J. New Insights into the Role of the Small Ubiquitin-like Modifier (SUMO) in Plants. Int. Rev. Cell Mol. Biol. 2013, 300, 161–209. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, Y.; Wang, X.; Liang, Z.; He, G.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Protein SUMOylation modification and its associations with disease. Open Biol. 2017, 7, 170167. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ji, S. Inhibition of Ubc9-induced CRMP2 SUMOylation disrupts Glioblastoma cell proliferation. J. Mol. Neurosci. 2019, 69, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Z.; Xia, Z.; Wang, X.; Ma, Y.; Sheng, Z.; Gu, Q.; Shen, G.; Zhou, L.; Zhu, H.; et al. SAE1 promotes human glioma progression through activating AKT SUMOylation-mediated signaling pathways. Cell Commun. Signal. 2019, 17, 82. [Google Scholar] [CrossRef]
- Rossetto, D.; Avvakumov, N.; Côté, J. Histone phosphorylation A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.T.; Lee, S.Y.; Xu, Y.M.; Zheng, D.; Cho, Y.Y.; Zhu, F.; Kim, H.G.; Li, S.Q.; Zhang, Z.; Bode, A.M.; et al. Phosphorylation of histone H2B serine 32 is linked to cell transformation. J. Biol. Chem. 2011, 286, 26628–26637. [Google Scholar] [CrossRef]
- Choi, H.S.; Choi, B.Y.; Cho, Y.Y.; Mizuno, H.; Kang, B.S.; Bode, A.M.; Dong, Z. Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation. Cancer Res. 2005, 65, 5818–5827. [Google Scholar] [CrossRef]
- Pacaud, R.; Cheray, M.; Nadaradjane, A.; François, M. Vallette, and Pierre-François Cartron. Histone H3 Phosphorylation in GBM: A New Rational to Guide the Use of Kinase Inhibitors in anti-GBM Therapy. Theranostics 2015, 5, 12–22. [Google Scholar] [CrossRef]
- Azab, M.A.; Alomari, A.; Azzam, A.Y. Featuring how calcium channels and calmodulin affect glioblastoma behavior. A review article. Cancer Treat. Res. Commun. 2020, 25, 100255. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol. Cancer Res. 2008, 6, 21–30. [Google Scholar] [CrossRef]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Herndon, J.E.; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II study of Bevacizumab and Vorinostat for patientswith recurrent World Health Organization grade 4 malignant Glioma. Oncologist 2018, 23, 157. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.B.; Lipp, E.S.; Miller, E.; Herndon, J.E., II; McSherry, F.; Desjardins, A.; Reardon, D.A. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J. Neuro-Oncol. 2018, 137, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, F.M.; Lamborn, K.R.; Kuhn, J.G.; Wen, P.Y.; Yung, W.K.A.; Gilbert, M.R.; Chang, S.M.; Lieberman, F.S.; Prados, M.D.; Fine, H.A. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American brain tumor consortium study 03–03. Neuro-Oncology 2011, 13, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L.; et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro-Oncology 2015, 17, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Ramesh, K.; Huang, V.; Gurbani, S.S.; Cordova, J.S.; Schreibmann, E.; Weinberg, B.D.; Sengupta, S.; Voloschin, A.D.; Holdhoff, M.; et al. Final Report on Clinical Outcomes and Tumor Recurrence Patterns of a Pilot Study Assessing Efficacy of Belinostat (PXD-101) with Chemoradiation for Newly Diagnosed Glioblastoma. Tomography 2022, 8, 688–700. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Pinheiro, R.; Braga, C.; Santos, G.; Bronze, M.R.; Perry, M.J.; Moreira, R.; Brites, D.; Falcão, A.S. Targeting Gliomas: Can a new alkylating hybrid compound make a difference? ACS Chem. Neurosci. 2017, 8, 50–59. [Google Scholar] [CrossRef]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS ONE 2012, 7, e30815. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef]
- Carew, J.S.; Giles, F.J.; Nawrocki, S.T. Histone deacetylase inhibitors: Mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008, 269, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.-S.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Everix, L.; Seane, E.N.; Ebenhan, T.; Goethals, I.; Bolcaen, J. Introducing HDAC-Targeting Radiopharmaceuticals for Glioblastoma Imaging and Therapy. Pharmaceuticals 2023, 16, 227. [Google Scholar] [CrossRef]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31. [Google Scholar] [CrossRef]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P.; Gynecologic Oncology Group. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A gynecologic oncology group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Galanis, E.; Jaeckle, K.A.; Maurer, M.J.; Reid, J.M.; Ames, M.M.; Reilly, J.S.H.F.; Loboda, A.; Nebozhyn, M.; Fantin, V.R.; Richon, V.M.; et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: A north central cancer treatment group study. J. Clin. Oncol. 2009, 27, 2052–2058. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central Cancer treatment group trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef]
- Lee, E.Q.; Puduvalli, V.K.; Reid, J.M.; Kuhn, J.G.; Lamborn, K.R.; Cloughesy, T.F.; Chang, S.M.; Drappatz, J.; Yung, W.K.A.; Mark, R.; et al. Gilbert Phase I study of vorinostat in combination with temozolomide in patients with malignant gliomas. Clin. Cancer Res. 2011, 18, 6032–6039. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Chowdhary, S.; Potthast, L.; Prabhu, A.; Tsai, Y.-Y.; Sarcar, B.; Kahali, S.; Brem, S.; Yu, H.M.; Rojiani, A.; et al. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro-Oncology 2012, 14, 93–100. [Google Scholar] [CrossRef]
- Qian, D.Z.; Wang, X.; Kachhap, S.K.; Kato, Y.; Wei, Y.; Zhang, L.; Atadja, P.; Pili, R. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004, 64, 6626–6634. [Google Scholar] [CrossRef] [PubMed]
- Sarcar, B.; Kahali, S.; Chinnaiyan, P. Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor SN38 in glioblastoma cell lines. J. Neuro-Oncol. 2010, 99, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: A north central cancer treatment group study. Neuro-Oncology 2012, 14, 215–221. [Google Scholar] [CrossRef]
- Rampazzo, E.; Manfreda, L.; Bresolin, S.; Cani, A.; Mariotto, E.; Bortolozzi, R.; Della Puppa, A.; Viola, G.; Persano, L. Histone Deacetylase Inhibitors Impair Glioblastoma Cell Motility and Proliferation. Cancers 2022, 14, 1897. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Gorlia, T.; Cairncross, J.G.; van den Bent, M.J.; Mason, W.; Belanger, K.; Brandes, A.A.; Bogdahn, U.; Macdonald, D.R.; Forsyth, P.; et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 2011, 77, 1156–1164. [Google Scholar] [CrossRef]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Wen, P.Y.; Schiff, D. Valproic acid as the AED of choice for patients with glioblastoma? The jury is out. Neurology 2011, 77, 1114–1115. [Google Scholar] [CrossRef]
- Valiyaveettil, D.; Malik, M.; Joseph, D.M.; Ahmed, S.F.; Kothwal, S.A.; Vijayasaradhi, M. Effect of valproic acid on survival in glioblastoma: A prospective single-arm study. South Asian J. Cancer 2018, 7, 159–162. [Google Scholar] [CrossRef]
- Wu, Y.; Dong, L.; Bao, S.; Wang, M.; Yun, Y.; Zhu, R. FK228 augmented temozolomide sensitivity in human glioma cells by blocking PI3K/AKT/mTOR signal pathways. Biomed. Pharmacother. 2016, 84, 462–469. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Quinzii, C.M.; Westhoff, M.-A.; Karpel-Massler, G.; Sieglin, M.D. Inhibition of HDAC1/2 Along with TRAP1 Causes Synthetic Lethality in Glioblastoma Model Systems. Cells 2020, 9, 1661. [Google Scholar] [CrossRef]
- Govindarajan, V.; Shah, A.H.; Di, L.; Rivas, S.; Suter, R.K.; Eichberg, D.G.; Luther, E.; Lu, V.; Morell, A.A.; Ivan, M.E.; et al. Systematic Review of Epigenetic Therapies for Treatment of IDH-mutant Glioma. World Neurosurg. 2022, 162, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Tcherpakov, M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 2010, 143, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Stinchcombe, T.E.; Mitchell, B.S.; Shea, T.C.; Baldwin, A.S.; Stahl, S.; Adams, J.; Esseltine, D.L.; Elliott, P.J.; Pien, C.S.; et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin. Oncol. 2002, 20, 4420–4427. [Google Scholar] [CrossRef]
- Aghajanian, C.; Soignet, S.; Dizon, D.S.; Pien, C.S.; Adams, J.; Elliott, P.J.; Sabbatini, P.; Miller, V.; Hensley, M.L.; Pezzulli, S.; et al. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin. Cancer Res. 2002, 8, 2505–2511. [Google Scholar]
- Wang, W.; Cho, H.Y.; Rosenstein-Sisson, R.; Marín Ramos, N.I.; Price, R.; Hurth, K.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C. Intratumoral delivery of bortezomib: Impact on survival in an intracranial glioma tumor model. J. Neurosurg. 2018, 128, 695–700. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Villalonga-Planells, R.; Coll-Mulet, L.; Martínez-Soler, F.; Castaño, E.; Acebes, J.J.; Giménez-Bonafé, P.; Gil, J.; Tortosa, A. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS ONE 2011, 6, e18588. [Google Scholar] [CrossRef]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Berberich, A.; Kessler, T.; Thomé, C.M.; Pusch, S.; Hielscher, T.; Sahm, F.; Oezen, I.; Schmitt, L.M.; Ciprut, S.; Hucke, N.; et al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res. 2019, 25, 253–265. [Google Scholar] [CrossRef]
- Her, N.G.; Oh, J.W.; Oh, Y.J.; Han, S.; Cho, H.J.; Lee, Y.; Ryu, G.H.; Nam, D.H. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 2018, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ma, D.J.; Calligaris, D.; Zhang, S.; Feathers, R.W.; Vaubel, R.A.; Meaux, I.; Mladek, A.C.; Parrish, K.E.; Jin, F.; et al. Efficacy of the MDM2 Inhibitor SAR405838 in Glioblastoma Is Limited by Poor Distribution Across the Blood-Brain Barrier. Mol. Cancer Ther. 2018, 17, 1893–1901. [Google Scholar] [CrossRef]
- Hao, X.; Bahia, R.K.; Cseh, O.; Bozek, D.A.; Blake, S.; Rinnenthal, J.; Weyer-Czernilofsky, U.; Rudolph, D.; Artee Luchman, H. BI-907828, a novel potent MDM2 inhibitor, inhibits glioblastoma brain tumor stem cells in vitro and prolongs survival in orthotopic xenograft mouse models. Neuro-Oncology 2023, 25, 913–926. [Google Scholar] [CrossRef]
- Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03107780 (accessed on 1 May 2023).
- NCT Neuro Master Match-N2M2 (NOA-20)—Full Text View. 2023. Available online: https://clinicaltrials.gov (accessed on 1 May 2023).
- A Study to Determine How BI 907828 is Taken up in the Tumor and to Determine the Highest Dose of BI 907828 that Could be Tolerated in Combination with Radiation Therapy in People with a Brain Tumor Called Glioblastoma—Full Text View. 2023. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05376800 (accessed on 1 May 2023).
- Yang, W.; Wang, L.; Roehn, G.; Pearlstein, R.D.; Ali-Osman, F.; Pan, H.; Goldbrunner, R.; Krantz, M.; Harms, C.; Paschen, W. Small ubiquitin-like modifier 1-3 conjugation [corrected] is activated in human astrocytic brain tumors and is required for glioblastoma cell survival. Cancer Sci. 2013, 104, 70–77. [Google Scholar] [CrossRef]
- Yang, Y.; Xia, Z.; Wang, X.; Zhao, X.; Sheng, Z.; Ye, Y.; He, G.; Zhou, L.; Zhu, H.; Xu, N.; et al. Small-Molecule Inhibitors Targeting Protein SUMOylation as Novel Anticancer Compounds. Mol. Pharmacol. 2018, 94, 885–894. [Google Scholar] [CrossRef]
- Huang, W.; He, T.; Chai, C.; Yang, Y.; Zheng, Y.; Zhou, P.; Qiao, X.; Zhang, B.; Liu, Z.; Wang, J.; et al. Triptolide inhibits the proliferation of prostate cancer cells and down-regulates SUMO-specific protease 1 expression. PLoS ONE 2012, 7, e37693. [Google Scholar] [CrossRef]
- Uno, M.; Koma, Y.; Ban, H.S.; Nakamura, H. Discovery of 1-[4-(N-benzylamino)phenyl]-3-phenylurea derivatives as non-peptidic selective SUMO-sentrin specific protease (SENP)1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5169–5173. [Google Scholar] [CrossRef]
- Brave, M.; Dagher, R.; Farrell, A.; Abraham, S.; Ramchandani, R.; Gobburu, J.; Booth, B.; Jiang, X.; Sridhara, R.; Justice, R. Topotecan in combination with cisplatin for the treatment of stage IVB, recurrent, or persistent cervical cancer. J. Oncol. 2006, 20, 1401–1404. [Google Scholar]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. J. Nat. Rev. Cancer. 2006, 6, 789. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Ye, D.; Gessler, F.A.; Lee, Y.J.; Peruzzotti-Jametti, L.; Baumgarten, P.; Johnson, K.R.; Maric, D.; Yang, W.; Kögel, D.; et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci Rep. 2017, 7, 7425. [Google Scholar] [CrossRef] [PubMed]
- Leszczynska, K.B.; Jayaprakash, C.; Kaminska, B.; Mieczkowski, J. Emerging Advances in Combinatorial Treatments of Epigenetically Altered Pediatric High-Grade H3K27M Gliomas. Front. Genet. 2021, 12, 742561. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Hirata, Y.; Sasaki, T.; Kanki, H.; Choong, C.-J.; Nishiyama, K.; Kubo, G.; Hotei, A.; Taniguchi, M.; Mochizuki, H. New 5-aryl-substituted 2-Aminobenzamide-type HDAC inhibitors with a Diketopiperazine group and their ameliorating effects on ischemia-induced neuronal cell death. Sci. Rep. 2018, 8, 1400. [Google Scholar] [CrossRef]
- Jordan, J.T.; Wen, P.Y. Novel chemotherapeutic approaches in adult high-grade Gliomas. Cancer Treat. Res. 2015, 163, 117–142. [Google Scholar] [CrossRef]
- Mayo, M.W.; Denlinger, C.E.; Broad, R.M.; Yeung, F.; Reilly, E.T.; Shi, Y.; Jones, D.R. Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-κB through the Akt pathway. J. Biol. Chem. 2003, 278, 18980–18989. [Google Scholar] [CrossRef]
- Seo, Y.J.; Kang, Y.; Muench, L.; Reid, A.; Caesar, S.; Jean, L.; Wagner, F.; Holson, E.; Haggarty, S.J.; Weiss, P.; et al. Image guided synthesis reveals potent blood-brain barrier permeable histone deacetylase inhibitors. ACS Chem. Neurosci. 2014, 5, 588–596. [Google Scholar] [CrossRef]
- Seo, Y.J.; Muench, L.; Reid, A.; Chen, J.; Kang, Y.; Hooker, J.M.; Volkow, N.D.; Fowler, J.S.; Kim, S.W. Radionuclide Labeling and Evaluation of Candidate Radioligands for Pet Imaging of Histone Deacetylase in the Brain. Bioorg. Med. Chem. Lett. 2013, 23, 6700–6705. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Kim, S.W.; Alexoff, D.; Xu, Y.W.; Shea, C.; Reid, A.; Volkow, N.; Fowler, J.S. Histone Deacetylase Inhibitor MS-275 Exhibits Poor Brain Penetration: Pharmacokinetic Studies of [11C]MS-275 Using Positron Emission Tomography. ACS Chem. Neurosci. 2010, 1, 65–73. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| HAT Involved | Mechanism Involved | Reference |
|---|---|---|
| KAT6A/MYST3 | Glioma cell-induced proliferation through H3K23ac/TRIM24-PI3K/AKT pathway. | [27] |
| KAT8 | Manipulation of the H4K16 acetylation level in microglia, using the intrinsic H4K16 acetyltransferase activities, adjusted the microglia’s tumor-supporting function. | [28] |
| KAT3B | An inhibitor for KAT3B acetyltransferase is highly expressed in GBM and correlates with a dismal prognosis. | [26] |
| Methyltransferases | ||
|---|---|---|
| KAMTS | PRMTS | |
| Type 1 | Type 2 | |
| SET1 | PRMT1 | PRMT5 |
| SET2 | PRMT2 | PRMT7 |
| SMYD | PRMT3 | PRMT9 |
| SUV4-20 | PRMT6 | |
| SET7/9 | PRMT8 | |
| SUV39 | PRMT4 | |
| Methyltransferases | Cell Line Used | Effect | Reference |
|---|---|---|---|
| KMT1A | Glioma cell lines (GOS-3, 1321N1, T98G, U87MG) | Positive correlation with aggressive tumors | [34] |
| KMT2A | Cell lines isolated from primary human GBM | Glioma stem cells were blunted following silencing of KMT2A | [38] |
| KMT3A | Patient-derived tumor cells | Expressed in high-grade pediatric glioma | [39] |
| KMT4 | Xenograft models | Inhibition of KMT4 reduced stem cell expression of stemness markers | [40] |
| KMT6 | Patient-derived GBM cultures | Reduced expression levels of KMT6 are associated with low expression of oncogenes as c-myc. | [41] |
| PRMT1 | T98G, U87MG, and A172 cell lines and mouse xenografts | Highly expressed in glioma cell lines | [42] |
| PRMT2 | U87 and T98G cell lines | Expressed in high-grade gliomas and associated with poor prognosis. | [35] |
| PRMT5 | U373MG and LN229 cell lines | The expression is high in the high-grade glioma | [43] |
| Ubiquitin Specific Enzymes | Preclinical Study | Reference |
|---|---|---|
| USP1 | USP1 is overexpressed in glioma stem cells. Inhibition of USP1 increased radiosensitivity of GBM cells. | [56] |
| USP3 | USP3 is highly expressed in GBM and correlates with poor prognosis. | [57] |
| USP4 | USP4 is highly expressed in GBM cells. | [55] |
| USP10 | USP 10 is overexpressed and linked to poor survival in GBM patients. | [58] |
| USP13 | USP13 is highly expressed in GBM and is required by glioma stem cells to maintain its stemness features. | [53] |
| USP15 | USP15 attenuates the WNT pathway mediated by stabilization of HECTD1, supporting a tumor-suppressing role of USP15 in GBM cells. | [54] |
| USP28 | USP 28 is overexpressed in GBM cell lines and is associated with a high grade of glioma. | [59] |
| HDAC Inhibitor | Combination Therapy | Tumor Type | BBB Cross Rate | Result | Sponsor | Reference |
|---|---|---|---|---|---|---|
| Vorinostat | Temozolomide + Isotretinoin, bortezomib Bevacizumab Temozolomide + Bevacizumab Bevacizumab + Irinotecan, Temsirolimus Radiotherapy | Recurrent GBM Recurrent GBM Recurrent GBM Diffuse intrinsic pontine glioma High-grade glioma and anaplastic astrocytoma | Low | Still active No change in overall survival or progression-free survival compared to bevacizumab therapy. Progression-free survival for six months was not affected. Active Active | Duke University Durham Duke University Durham National Cancer Institute | [73] [74] |
| Valproic acid | VPA, temozolomide, and radiotherapy | Newly diagnosed GBM in adults | Good | Active | National Cancer Institute | |
| Romidepsin | Recurrent GBM | Low | Completed and showed that romidepsin is ineffective in the treatment of recurrent GBM. | NM | [75] | |
| Panobinostat | Bevacizumab Convection-enhanced delivery (CED) | Pediatric intrinsic pontine glioma Recurrent GBM Diffuse pontine glioma | Good | Active Adding this agent to bevacizumab did not improve the outcome compared to bevacizumab alone. Active | [76] | |
| Belinostat (PXD101) | Belinostat | Adults with newly diagnosed GBM | Good | Phase I | NM | [77] |
| Belinostat (PXD101) | TMZ and Radiation | Adults with newly diagnosed GBM | Good | Phase II | Emory University | NCT02137759 |
| Title | MDM2 Inhibitor | Status | Type of Cancer | NCT Number | Reference |
|---|---|---|---|---|---|
| Testing the Ability of AMG 232 (KRT 232) to Reach Into the Tumor in Patients With Brain Cancer | Navtemadlin | Suspended | Recurrent GBM Newly Diagnosed Glioblastoma MGMT-unmethylated Glioblastoma Gliosarcoma | NCT03107780 | [119] |
| NCT Neuro Master Match—N2M2 (NOA-20) (N2M2) | Idasanutlin | Active recruiting | GBM | NCT03158389 | [120] |
| A Study to Determine How BI 907828 is Taken up in the Tumor and to Determine the Highest Dose of BI 907828 That Could be Tolerated in Combination With Radiation Therapy in People With a Brain Tumor Called Glioblastoma | BI 907828 | Active recruiting | GBM | NCT05376800 | [121] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azab, M.A. The Potential Role of Histone Modifications in Glioblastoma Therapy: Review Article. J. Mol. Pathol. 2023, 4, 196-212. https://doi.org/10.3390/jmp4040018
Azab MA. The Potential Role of Histone Modifications in Glioblastoma Therapy: Review Article. Journal of Molecular Pathology. 2023; 4(4):196-212. https://doi.org/10.3390/jmp4040018
Chicago/Turabian StyleAzab, Mohammed A. 2023. "The Potential Role of Histone Modifications in Glioblastoma Therapy: Review Article" Journal of Molecular Pathology 4, no. 4: 196-212. https://doi.org/10.3390/jmp4040018
APA StyleAzab, M. A. (2023). The Potential Role of Histone Modifications in Glioblastoma Therapy: Review Article. Journal of Molecular Pathology, 4(4), 196-212. https://doi.org/10.3390/jmp4040018