
Theoretical Assessment of PMMA’s Potential to Remove Beta Blockers from the Aquatic Environment Using Atomistic Calculations †

 ,
,  ,
, 
 and
and 
Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Obaideen, K.; Shehata, N.; Sayed, E.T.; Abdelkareem, M.A.; Mahmoud, M.S.; Olabi, A.G. The Role of Wastewater Treatment in Achieving Sustainable Development Goals (SDGs) and Sustainability Guideline. Energy Nexus 2022, 7, 100112. [Google Scholar] [CrossRef]
- EL-Ghoul, Y.; Alminderej, F.M.; Alsubaie, F.M.; Alrasheed, R.; Almousa, N.H. Recent Advances in Functional Polymer Materials for Energy, Water, and Biomedical Applications: A Review. Polymers 2021, 13, 4327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gitungo, S.; Dyksen, J.E.; Raczko, R.F.; Axe, L. Indicator Compounds Representative of Contaminants of Emerging Concern (CECs) Found in the Water Cycle in the United States. Int. J. Environ. Res. Public Health 2021, 18, 1288. [Google Scholar] [CrossRef]
- Love, D.; Slovisky, M.; Costa, K.A.; Megarani, D.; Mehdi, Q.; Colombo, V.; Ivantsova, E.; Subramaniam, K.; Bowden, J.A.; Bisesi, J.H., Jr.; et al. Toxicity Risks Associated with the Beta-Blocker Metoprolol in Marine and Freshwater Organisms: A Review: Toxicity of Metoprolol. Environ. Toxicol. Chem. 2024, 43, 2530–2544. [Google Scholar] [CrossRef]
- Zhou, L.; Sleiman, M.; Ferronato, C.; Chovelon, J.-M.; de Sainte-Claire, P.; Richard, C. Sulfate Radical Induced Degradation of Β2-Adrenoceptor Agonists Salbutamol and Terbutaline: Phenoxyl Radical Dependent Mechanisms. Water Res. 2017, 123, 715–723. [Google Scholar] [CrossRef]
- Quaresma, A.V.; Sousa, B.A.; Silva, K.T.S.; Silva, S.Q.; Werle, A.A.; Afonso, R.J.C.F. Oxidative Treatments for Atenolol Removal in Water: Elucidation by Mass Spectrometry and Toxicity Evaluation of Degradation Products. Rapid Commun. Mass Spectrom. 2019, 33, 303–313. [Google Scholar] [CrossRef]
- Krunić, D.; Armaković, S.; Bilić, A.; Armaković, S.J.; Gavanski, L. Understanding the Interactions between PMMA Polymer and Common Industrial Gasses: A Computational xTB, DFT, SAPT and MD Study. Mol. Phys. 2025, e2456108. [Google Scholar] [CrossRef]
- Singh, V.; Patra, S.; Arul Murugan, N.; Toncu, D.-C.; Tiwari, A. Recent Trends in Computational Tools and Data-Driven Modeling for Advanced Materials. Mater. Adv. 2022, 3, 4069–4087. [Google Scholar] [CrossRef]
- Ehlert, S.; Stahn, M.; Spicher, S.; Grimme, S. Robust and Efficient Implicit Solvation Model for Fast Semiempirical Methods. J. Chem. Theory Comput. 2021, 17, 4250–4261. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended Tight-Binding Quantum Chemistry Methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Pracht, P.; Caldeweyher, E.; Ehlert, S.; Grimme, S. A Robust Non-Self-Consistent Tight-Binding Quantum Chemistry Method for large Molecules. ChmRxiv, 2019. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All Spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Valero, R.; Costa, R.; de P., R. Moreira, I.; Truhlar, D.G.; Illas, F. Performance of the M06 Family of Exchange-Correlation Functionals for Predicting Magnetic Coupling in Organic and Inorganic Molecules. J. Chem. Phys. 2008, 128, 114103. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Ciofini, I.; Adamo, C.; Valero, R.; Zhao, Y.; Truhlar, D.G. On the Performances of the M06 Family of Density Functionals for Electronic Excitation Energies. J. Chem. Theory Comput. 2010, 6, 2071–2085. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Armaković, S.; Armaković, S.J. Atomistica.Online—Web Application for Generating Input Files for ORCA Molecular Modelling Package Made with the Anvil Platform. Mol. Simul. 2023, 49, 117–123. [Google Scholar] [CrossRef]
- Armaković, S.; Armaković, S.J. Online and Desktop Graphical User Interfaces for Xtb Programme from Atomistica.Online Platform. Mol. Simul. 2024, 50, 560–570. [Google Scholar] [CrossRef]
- Helmich-Paris, B.; de Souza, B.; Neese, F.; Izsák, R. An Improved Chain of Spheres for Exchange Algorithm. J. Chem. Phys. 2021, 155, 104109. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, L224108. [Google Scholar] [CrossRef]
- Neese, F. The SHARK Integral Generation and Digestion System. J. Comp. Chem. 2022, 44, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIRES Comput. Molec. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. Approximate Second-Order SCF Convergence for Spin Unrestricted Wavefunctions. Chem. Phys. Lett. 2000, 325, 93–98. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 5.0. WIRES Comput. Molec. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. WIRES Comput. Molec. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Lu, T. A Comprehensive Electron Wavefunction Analysis Toolbox for Chemists, Multiwfn. J. Chem. Phys. 2024, 161, 082503. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Van Der Waals Potential: An Important Complement to Molecular Electrostatic Potential in Studying Intermolecular Interactions. J. Mol. Model. 2020, 26, 315. [Google Scholar] [CrossRef]
- Lu, T.; Manzetti, S. Wavefunction and Reactivity Study of Benzo[a]Pyrene Diol Epoxide and Its Enantiomeric Forms. Struct. Chem. 2014, 25, 1521–1533. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Eargle, J.; Wright, D.; Luthey-Schulten, Z. Multiple Alignment of Protein Structures and Sequences for VMD. Bioinformatics 2006, 22, 504–506. [Google Scholar] [CrossRef]
- Stone, J.; Gullingsrud, J.; Grayson, P.; Schulten, K. A System for Interactive Molecular Dynamics Simulation. In Proceedings of the 2001 ACM Symposium on Interactive 3D Graphics; Hughes, J.F., Séquin, C.H., Eds.; ACM SIGGRAPH: New York, NY, USA, 2001; pp. 191–194. [Google Scholar]

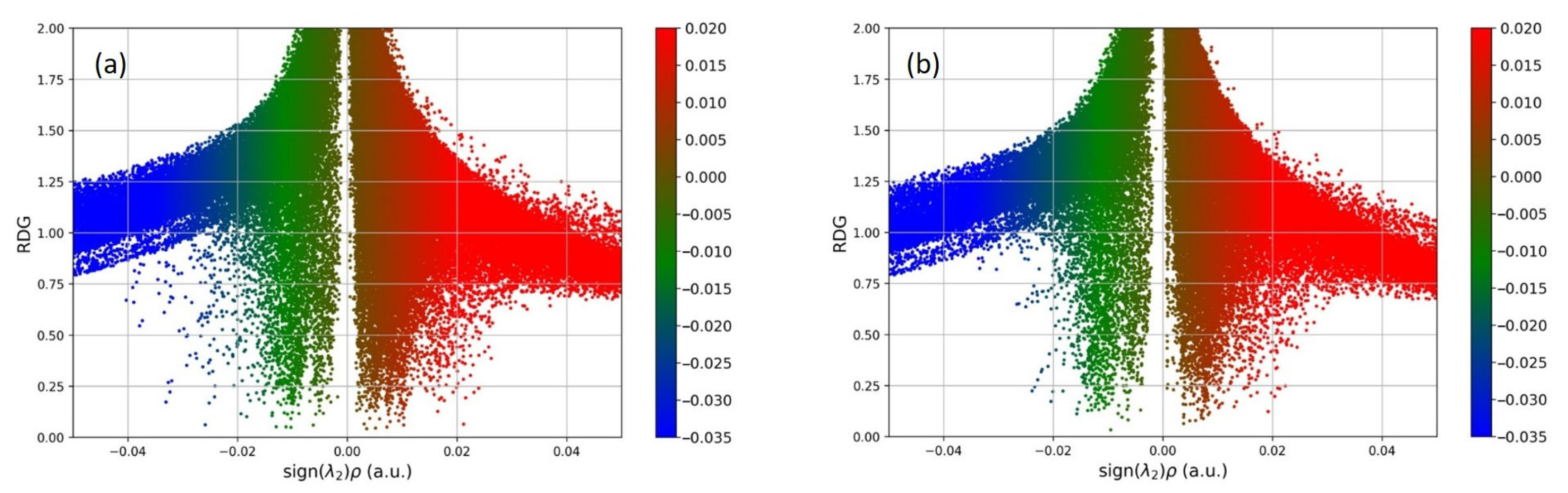
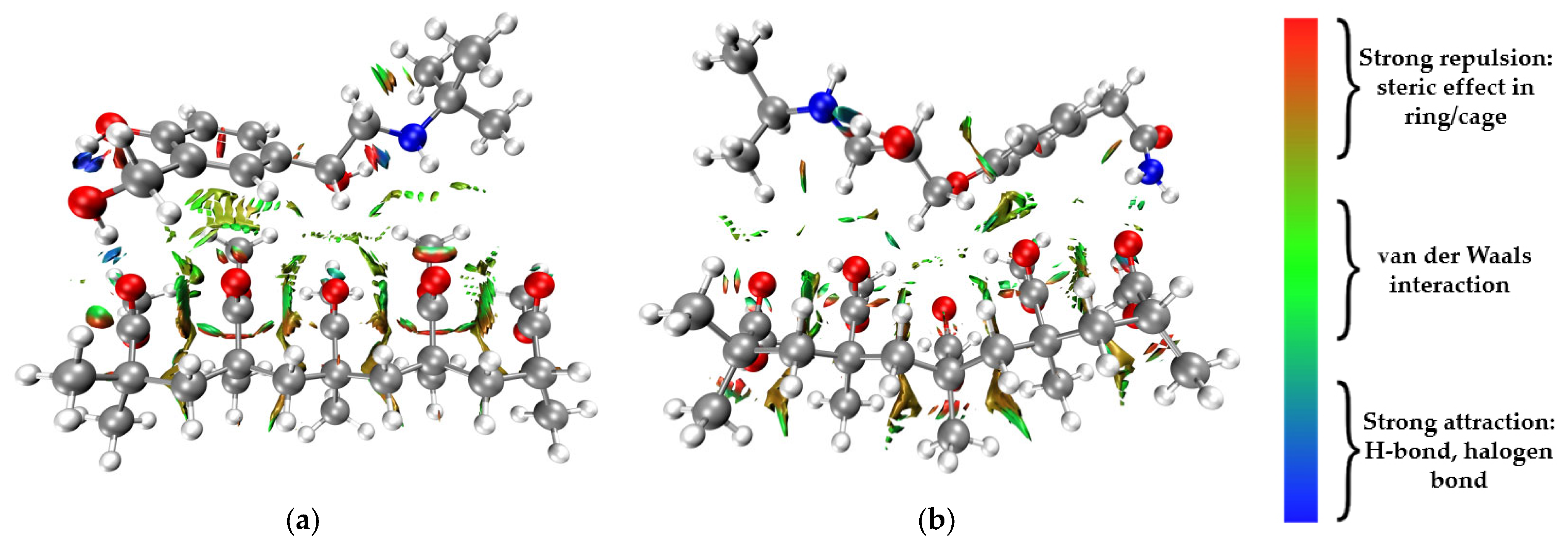

{kind=link}
{kind=link}
{kind=link}
| System | E [kcal/mol] |
|---|---|
| PMMA/salbutamol | −18.56 |
| PMMA/atenolol | −15.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelemiš, S.; Bilić, A.; Krunić, D.; Armaković, S.J.; Armaković, S. Theoretical Assessment of PMMA’s Potential to Remove Beta Blockers from the Aquatic Environment Using Atomistic Calculations. Eng. Proc. 2025, 99, 13. https://doi.org/10.3390/engproc2025099013
Pelemiš S, Bilić A, Krunić D, Armaković SJ, Armaković S. Theoretical Assessment of PMMA’s Potential to Remove Beta Blockers from the Aquatic Environment Using Atomistic Calculations. Engineering Proceedings. 2025; 99(1):13. https://doi.org/10.3390/engproc2025099013
Chicago/Turabian StylePelemiš, Svetlana, Andrijana Bilić, Dušica Krunić, Sanja J. Armaković, and Stevan Armaković. 2025. "Theoretical Assessment of PMMA’s Potential to Remove Beta Blockers from the Aquatic Environment Using Atomistic Calculations" Engineering Proceedings 99, no. 1: 13. https://doi.org/10.3390/engproc2025099013
APA StylePelemiš, S., Bilić, A., Krunić, D., Armaković, S. J., & Armaković, S. (2025). Theoretical Assessment of PMMA’s Potential to Remove Beta Blockers from the Aquatic Environment Using Atomistic Calculations. Engineering Proceedings, 99(1), 13. https://doi.org/10.3390/engproc2025099013