
An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2 †


Abstract
:1. Introduction
2. Materials and Methods
2.1. Receptor Preparation
2.2. Ligand Preparation
2.3. Molecular Docking Study
2.4. ADMET Analysis
3. Results and Discussion
3.1. Analyzing Molecular Docking Results and Binding Interactions
3.2. ADMET Analysis
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Wang, Q.; Su, L.; Liu, N.; Zhang, L.; Xu, W.; Fang, H. Cyclin Dependent Kinase 1 Inhibitors: A Review of Recent Progress. Curr. Med. Chem. 2011, 18, 2025–2043. [Google Scholar] [CrossRef]
- Martinsson-Ahlzén, H.-S.; Liberal, V.; Grünenfelder, B.; Chaves, S.R.; Spruck, C.H.; Reed, S.I. Cyclin-Dependent Kinase-Associated Proteins Cks1 and Cks2 Are Essential during Early Embryogenesis and for Cell Cycle Progression in Somatic Cells. Mol. Cell. Biol. 2008, 28, 5698–5709. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Lin, H.; Zhang, Z. CKS2 in human cancers: Clinical roles and current perspectives (Review). Mol. Clin. Oncol. 2015, 3, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Giordano, A.; Tommonaro, G. Curcumin and cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yu, J.; Cui, R.; Lin, J.; Ding, X. Curcumin in Treating Breast Cancer: A Review. J. Lab. Autom. 2016, 21, 723–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adiwidjaja, J.; McLachlan, A.J.; Boddy, A.V. Curcumin as a clinically-promising anticancer agent: Pharmacokinetics and drug interactions. Expert Opin. Drug Metab. Toxicol. 2017, 13, 953–972. [Google Scholar] [CrossRef] [PubMed]
- Devassy, J.G.; Nwachukwu, I.D.; Jones, P.J.H. Curcumin and cancer: Barriers to obtaining a health claim. Nutr. Rev. 2015, 73, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Wei, Y.; Lee, R.J.; Zhao, L. Liposomal curcumin and its application in cancer Physical property. Int. J. Nanomed. 2017, 12, 6027–6044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Farias, M.; Carrasco-Pozo, C. The anti-cancer effect of quercetin: Molecular implications in cancer metabolism. Int. J. Mol. Sci. 2019, 20, 3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Huang, S.; Yin, X.; Zan, Y.; Guo, Y.; Han, L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 2018, 208, 123–130. [Google Scholar] [CrossRef]
- Tang, S.M.; Deng, X.T.; Zhou, J.; Li, Q.P.; Ge, X.X.; Miao, L. Pharmacological basis and new insights of quercetin action in respect to its anticancer effects. Biomed. Pharmacother. 2020, 121, 109604. [Google Scholar] [CrossRef]
- Maurya, A.K.; Vinayak, M. Quercetin regresses Dalton’s lymphoma growth via suppression of PI3K/AKT signaling leading to upregulation of p53 and decrease in energy metabolism. Nutr. Cancer 2015, 67, 354–363. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Alsahli, M.A.; Almatroudi, A.; Verma, A.K.; Aloliqi, A.; Allemailem, K.S.; Khan, A.A.; Rahmani, A.H. Potential therapeutic targets of quercetin, a plant flavonol, and its role in the therapy of various types of cancer through the modulation of various cell signaling pathways. Molecules 2021, 26, 1315. [Google Scholar] [CrossRef]
- Jayaprakasam, B.; Zhang, Y.; Seeram, N.P.; Nair, M.G. Growth inhibition of human tumor cell lines by withanolides from Withania somnifera leaves. Life Sci. 2003, 74, 125–132. [Google Scholar] [CrossRef]
- Wang, H.C.; Tsai, Y.L.; Wu, Y.C.; Chang, F.R.; Liu, M.H.; Chen, W.Y.; Wu, C.C. Withanolides-induced breast cancer cell death is correlated with their ability to inhibit heat protein 90. PLoS ONE 2012, 7, e37764. [Google Scholar] [CrossRef] [PubMed]
- Palliyaguru, D.L.; Singh, S.V.; Kensler, T.W. Withania somnifera: From prevention to treatment of cancer. Mol. Nutr. Food Res. 2016, 60, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, R.; Khalil, R.; Green, R.; Mohapatra, S.S.; Mohapatra, S. Withania somnifera (Ashwagandha) and withaferin a: Potential in integrative oncology. Int. J. Mol. Sci. 2019, 20, 5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukund, V.; Mukund, D.; Sharma, V.; Mannarapu, M.; Alam, A. Genistein: Its role in metabolic diseases and cancer. Crit. Rev. Oncol. Hematol. 2017, 119, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; Nabavi, S.M. Genistein and Cancer: Current Status, Challenges, and future directions. Adv. Nutr. 2015, 6, 408–419. [Google Scholar] [CrossRef]
- Ji, Z.; Huo, C.; Yang, P. Genistein inhibited the proliferation of kidney cancer cells via CDKN2a hypomethylation: Role of abnormal apoptosis. Int. Urol. Nephrol. 2020, 52, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Pavese, J.M.; Farmer, R.L.; Bergan, R.C. Inhibition of cancer cell invasion and metastasis by genistein. Cancer Metastasis Rev. 2010, 29, 465–482. [Google Scholar] [CrossRef] [Green Version]
- Mukund, V. Genistein: Its Role in Breast Cancer Growth and Metastasis. Curr. Drug Metab. 2020, 21, 6–10. [Google Scholar] [CrossRef]
- Wood, D.J.; Korolchuk, S.; Tatum, N.J.; Wang, L.Z.; Endicott, J.A.; Noble, M.E.M.; Martin, M.P. Differences in the Conformational Energy Landscape of CDK1 and CDK2 Suggest a Mechanism for Achieving Selective CDK Inhibition. Cell Chem. Biol. 2019, 26, 121–130.e5. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Walker, M. In Silico Methods for Predicting Drug Toxicity. Methods Mol. Biol. 2016, 1425, 63–83. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, L.G.; Santos, R.N.D.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Kaur, T.; Madgulkar, A.; Bhalekar, M.; Asgaonkar, K. Molecular Docking in Formulation and Development. Curr. Drug Discov. Technol. 2019, 16, 30–39. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in Docking. Adv. Docking 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- Norinder, U.; Bergström, C.A.S. Prediction of ADMET properties. ChemMedChem 2006, 1, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/ Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, S.; Leszczynski, J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin. Drug Discov. 2020, 15, 1473–1487. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Psimadas, D.; Georgoulias, P.; Valotassiou, V.; Loudos, G. Molecular Nanomedicine Towards Cancer. J. Pharm. Sci. 2012, 101, 2271–2280. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Moriguchi, I.; Shuichi, H.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1994, 17, 1460–1462. [Google Scholar] [CrossRef] [Green Version]
- Penzotti, J.E.; Landrum, G.A.; Putta, S. Building predictive ADMET models for early decisions in drug discovery. Curr. Opin. Drug Discov. Dev. 2004, 7, 7–61. [Google Scholar]
- Ritchie, T.J.; MacDonald, S.J.F.; Peace, S.; Pickett, S.D.; Luscombe, C.N. Increasing small molecule drug developability in sub-optimal chemical space. MedChemComm 2013, 4, 673–680. [Google Scholar] [CrossRef]
- Ottaviani, G.; Gosling, D.J.; Patissier, C.; Rodde, S.; Zhou, L.; Faller, B. What is modulating solubility in simulated intestinal fluids? Eur. J. Pharm. Sci. 2010, 41, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Waller, D.G.; Sampson, A.P. Pharmacokinetics. In Medical Pharmacology and Therapeutics, 5th ed.; Waller, D.G., Sampson, A.P., Eds.; Elsevier B.V.: Southampton, UK, 2018; pp. 33–62. [Google Scholar] [CrossRef]
- Saghir, S.A.; Ansari, R.A. Pharmacokinetics. Ref. Modul. Biomed. Sci. 2018, 1–9. [Google Scholar] [CrossRef]
- Turfus, S.C.; Delgoda, R.; Picking, D.; Gurley, B.J. Pharmacokinetics. In Pharmacognosy. Fundamentals, Applications and Strategies; Badal, S., Delgoda, R., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 495–512. [Google Scholar] [CrossRef]
- Paixão, P.; Gouveia, L.F.; Morais, J.A. Prediction of the human oral bioavailability by using in vitro and in silico drug related parameters in a physiologically based absorption model. Int. J. Pharm. 2012, 429, 84–98. [Google Scholar] [CrossRef]
- Montanari, F.; Ecker, G.F. Prediction of drug-ABC-transporter interaction—Recent advances and future challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Szakács, G.; Váradi, A.; Özvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef]
- Sharom, F.J. ABC Multidrug Transporters: Structure, Function and Role in Chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Edwards, D.A.; Langer, R. A linear theory of transdermal transport phenomena. J. Pharm. Sci. 1994, 83, 1315–1334. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Y.; Li, J.Y.; Hao, G.F.; Yang, G.F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2020, 25, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Pedrys, A.; Duszkiewicz, R.; Kucia, U. Ligand Potency, Efficiency and Drug-likeness: A Story of Intuition, Misinterpretation and Serendipity. Curr. Protein Pept. Sci. 2019, 20, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Kurosawa, T.; Mikami, Y.; Nakamura, H. Prediction of synthetic accessibility based on commercially available compound databases. J. Chem. Inf. Model. 2014, 54, 3259–3267. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Zeiger, E. The test that changed the world: The Ames test and the regulation of chemicals. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 841, 43–48. [Google Scholar] [CrossRef]
- Föllmann, W.; Degen, G.; Oesch, F.; Hengstler, J.G. Ames Test, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Jain, A.K.; Singh, D.; Dubey, K.; Maurya, R.; Mittal, S.; Pandey, A.K. Models and Methods for In Vitro Toxicity, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Gad, S.C. Maximum Tolerated Dose, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Stampfer, H.G.; Gabb, G.M.; Dimmitt, S.B. Why maximum tolerated dose? Br. J. Clin. Pharmacol. 2019, 85, 2213–2217. [Google Scholar] [CrossRef]
- Gad, S.C. LD50/LC50 (Lethal Dosage 50/Lethal Concentration 50), 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
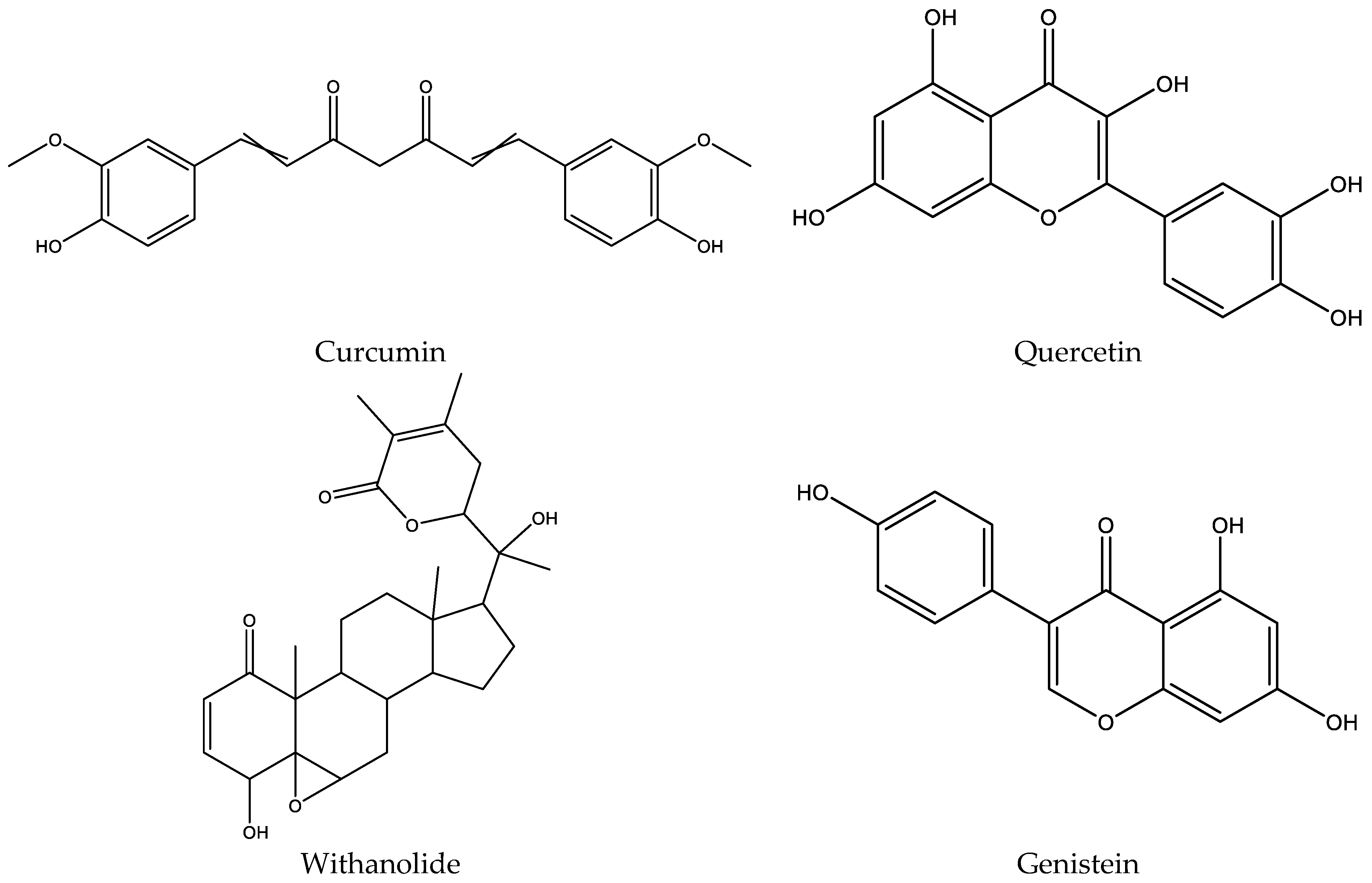

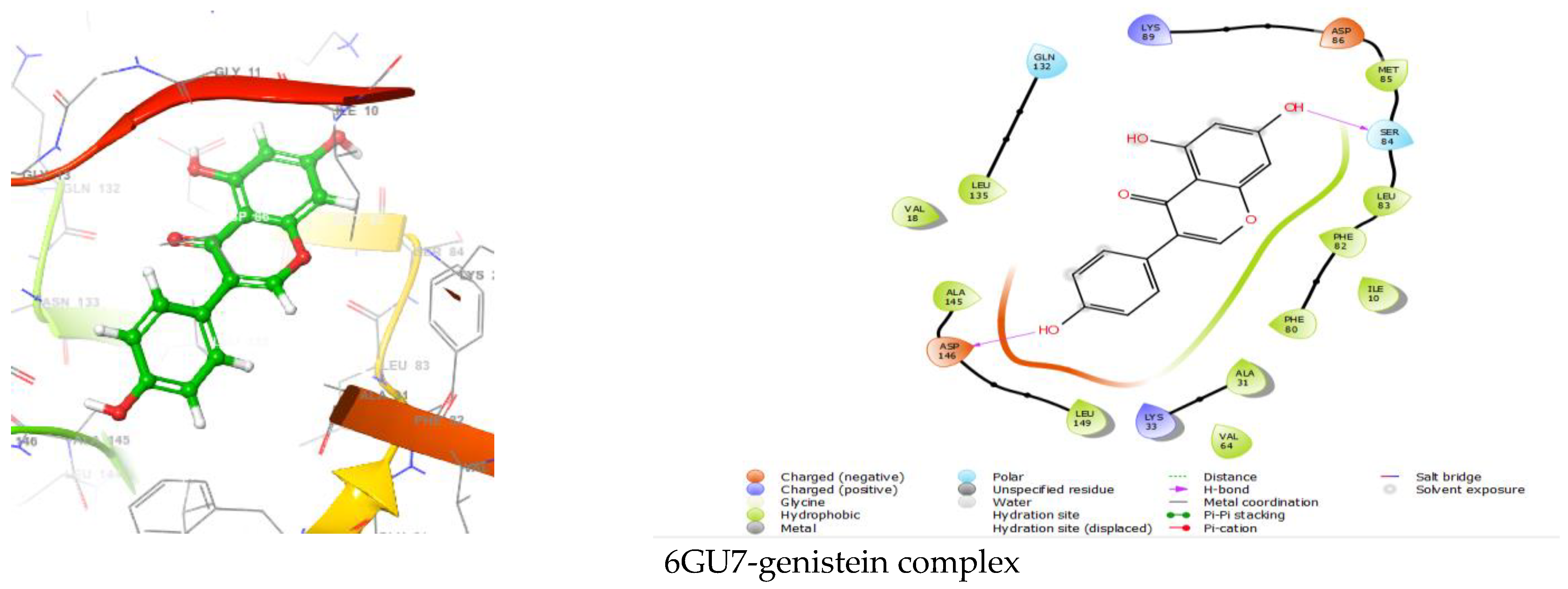
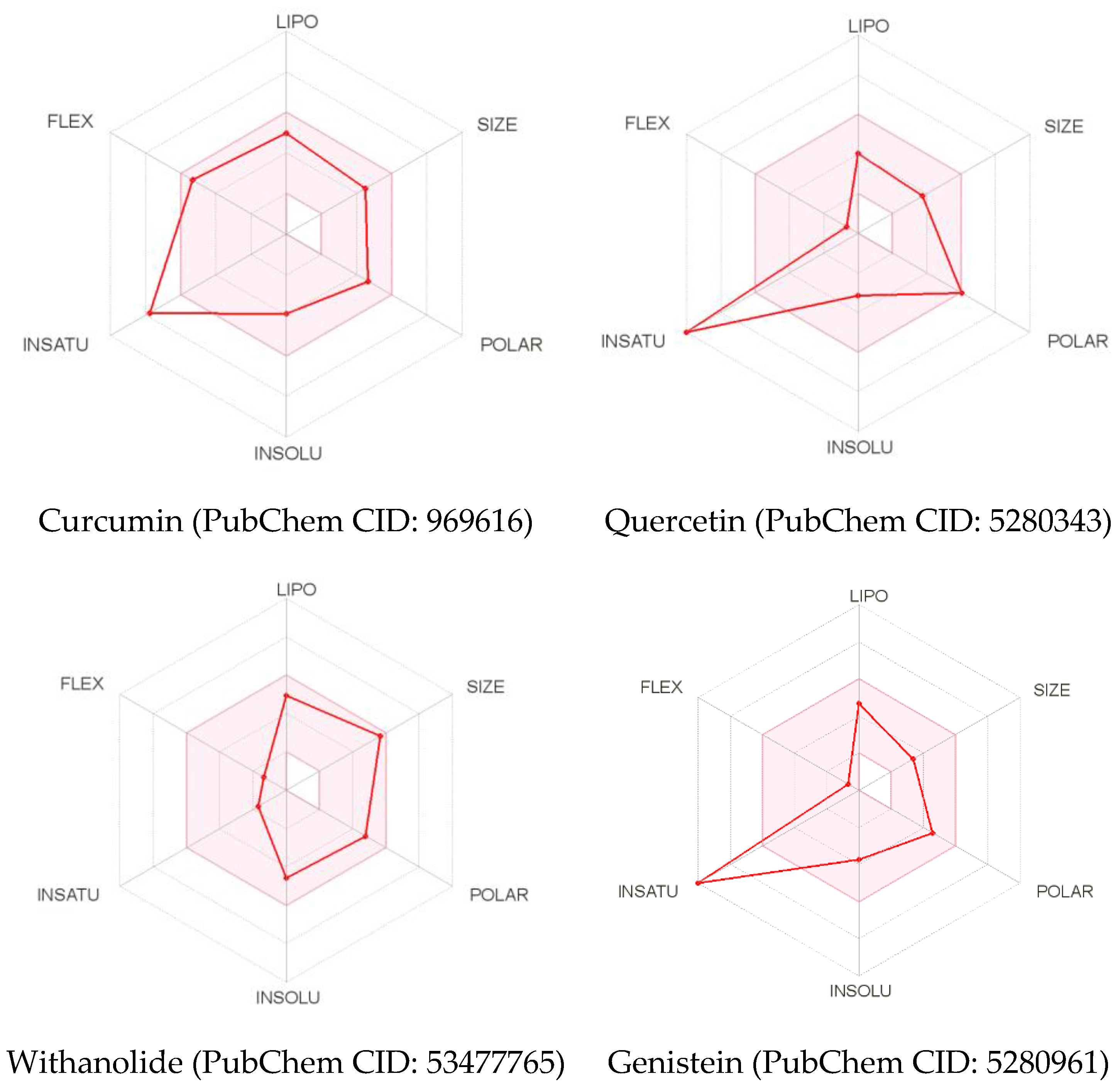

{kind=link}
{kind=link}
{kind=link}
{kind=link}
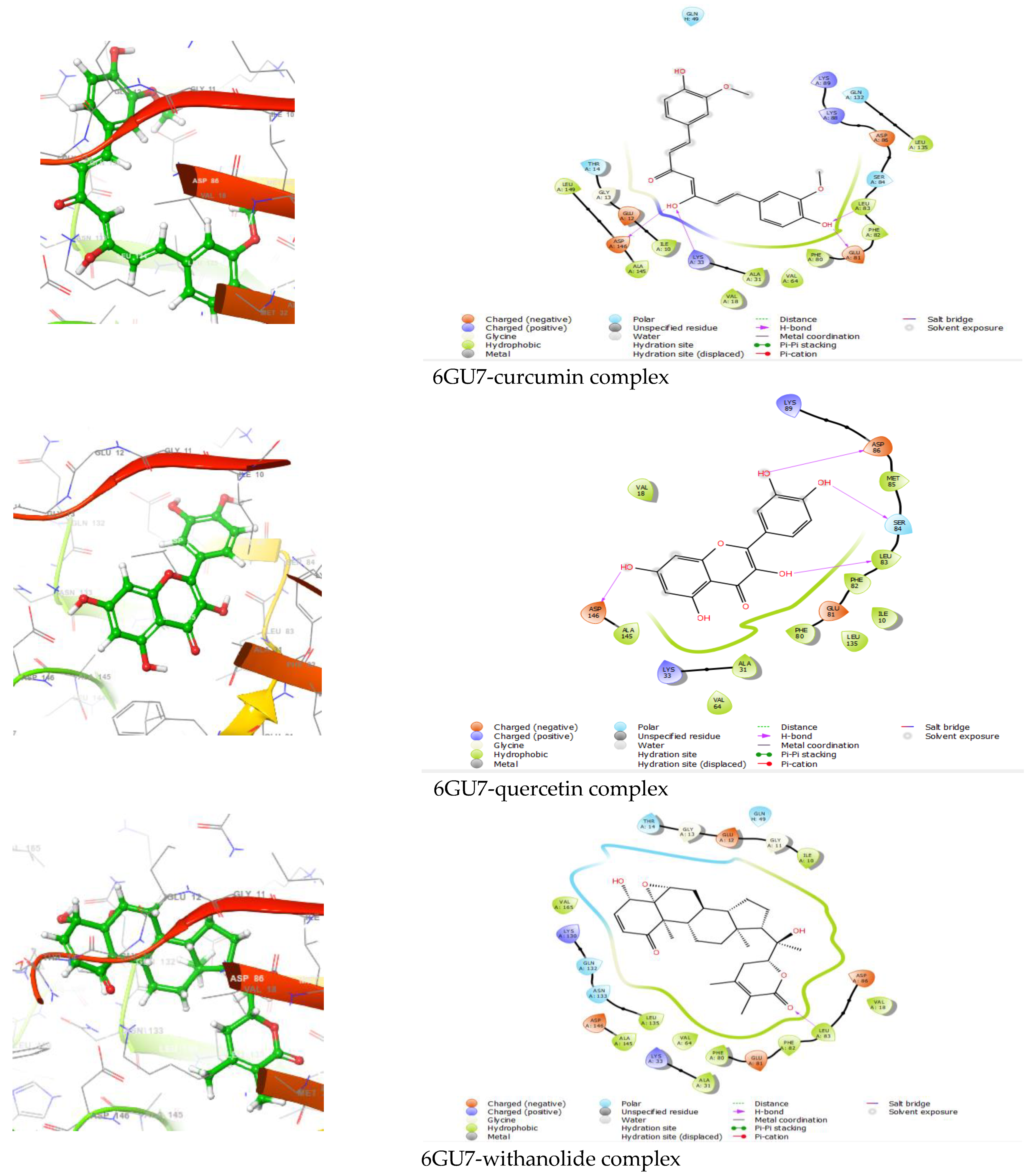
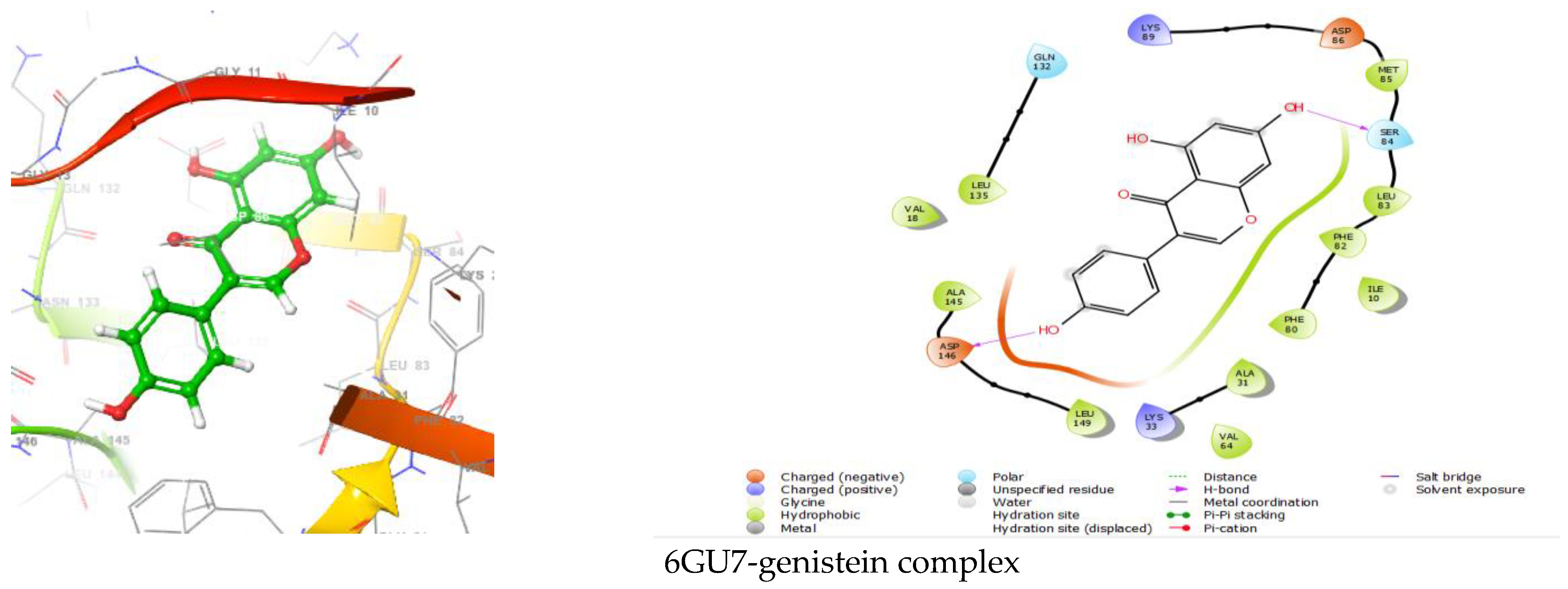
| Protein–Ligand Complex | Docking Score (kcal/mol) | H-Bond | Non-Bonding Interactions |
|---|---|---|---|
| 6GU7-curcumin | −9.419 | ASP146, LYS33, GLU81, LEU83 | Polar THR14, SER84, GLN132, GLN49 Hydrophobic LEU149, ILE10, ALA145, VAL18, ALA31, VAL64, PHE80, PHE82, LEU83, LEU135 Charged (Negative) GLU12, ASP146, GLU81, ASP86 Charged (Positive) LYS33, LYS88, LYS89 |
| 6GU7-quercetin | −8.709 | ASP146, LEU83, SER84, ASP86 | Polar SER84 Hydrophobic Val18, ALA145, VAL64, ALA31, PHE 80, LEU135, ILE10, PHE82, LEU83, MET85 Charged (Negative) ASP146, GLU81, ASP86 Charged (Positive) LYS33, LYS89 |
| 6GU7-withanolide | −7.174 | LEU83 | Polar GLN132, ASN133, THR14, GLN49 Hydrophobic VAL165, LEU135, ALA145, VAL64, PHE80, ALA31, PHE82, LEU83, VAL18, ILE10 Charged (Negative) ASP146, GLU81, ASP86, GLU12 Charged (Positive) LYS130, LYS33 |
| 6GU7-genistein | −6.301 | ASP146, SER 84 | Polar GLN132, SER84 Hydrophobic LEU135, VAL18, ALA145, LEU149, VAL64, VAL31, PHE80, ILE10, PHE82, LEU83, MET85 Charged (Negative) ASP146, ASP86 Charged (Positive) LYS33, LYS89 |
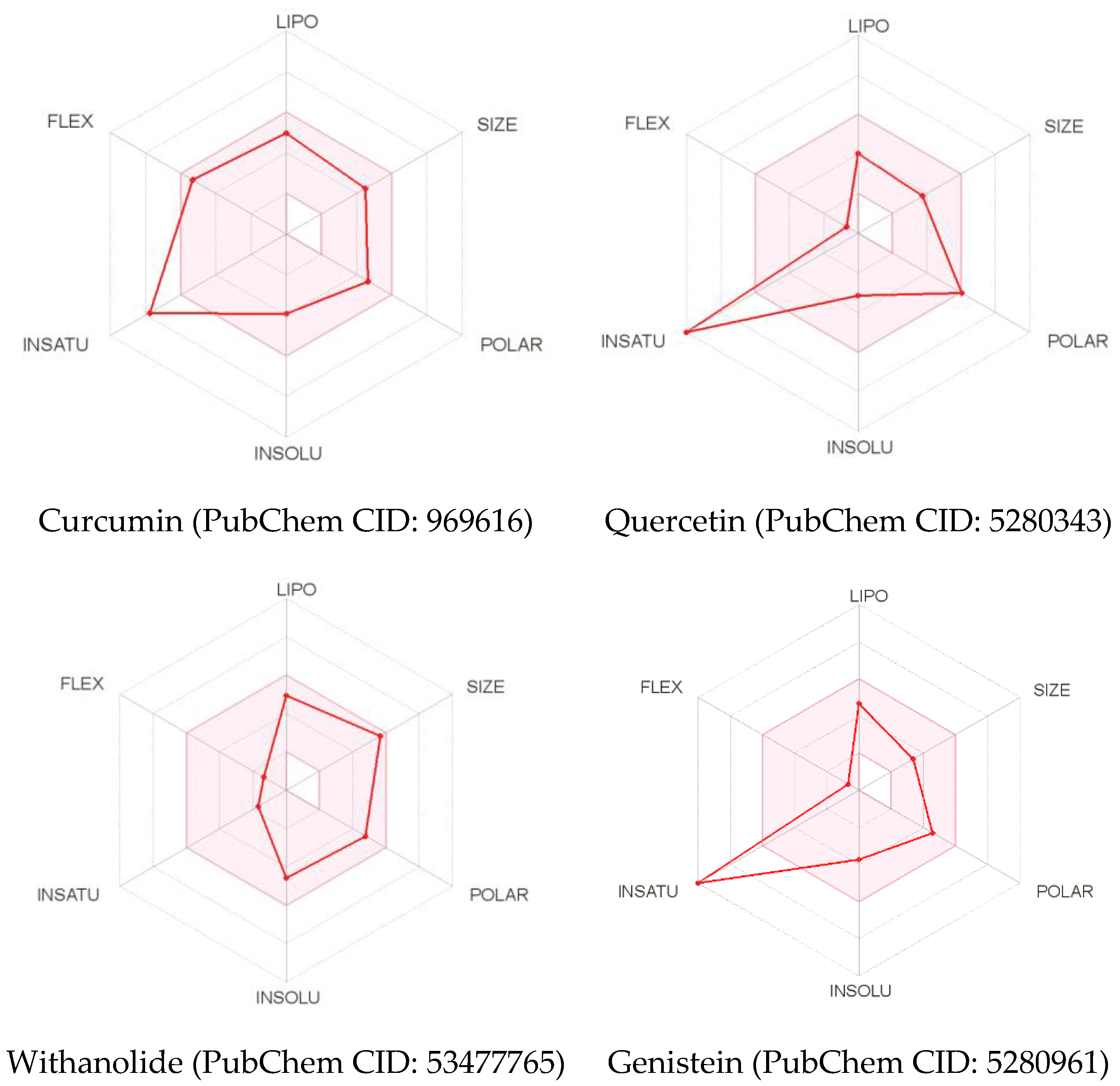
| Curcumin | Quercetin | Withanolide | Genistein | |
|---|---|---|---|---|
| Physicochemical Properties | ||||
| Molecular weight (g/mol) | 368.38 | 302.24 | 470.60 | 270.24 |
| Heavy atoms | 27 | 22 | 34 | 20 |
| Fraction Csp3 | 0.14 | 0.00 | 0.79 | 0.00 |
| Rotatable bonds | 8 | 1 | 2 | 1 |
| H-bond acceptors | 6 | 7 | 6 | 5 |
| H-bond donors | 2 | 5 | 2 | 3 |
| TPSA (Å2) | 93.06 | 131.36 | 96.36 | 90.90 |
| Lipophilicity | ||||
| Log Po/w (iLOGP) | 3.27 | 1.63 | 3.62 | 1.91 |
| Log Po/w (XLOGP3) | 3.20 | 1.54 | 3.12 | 2.67 |
| Log Po/w (WLOGP) | 3.15 | 1.99 | 3.50 | 2.58 |
| Log Po/w (MLOGP) | 1.47 | −0.56 | 2.75 | 0.52 |
| Log Po/w (SILICOS-IT) | 4.04 | 1.54 | 3.78 | 2.52 |
| Water Solubility | ||||
| Log S (ESOL) | −3.94 | −3.16 | −4.59 | −3.72 |
| Solubility (mg/mL; mol/L) | 4.22 × 10−2; 1.15 × 10−4 | 2.11 × 10−1; 6.98 × 10−4 | 1.21 × 10−2; 2.56 × 10−5 | 5.11 × 10−2; 1.89 × 10−4 |
| Class | Soluble | Soluble | Moderately soluble | Soluble |
| Pharmacokinetics | ||||
| GI absorption | High | High | High | High |
| BBB permeant | No | No | No | No |
| P-gp substrate | No | No | No | No |
| Log Kp (skin permeation) (cm/s) | −6.28 | −7.05 | −6.96 | −6.05 |
| Druglikeness | ||||
| Lipinski | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
| Ghose | Yes | Yes | No; 1 violation: #atoms > 70 | Yes |
| Veber | Yes | Yes | Yes | Yes |
| Egan | Yes | Yes | Yes | Yes |
| Muegge | Yes | Yes | Yes | Yes |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 |
| Medicinal Chemistry | ||||
| PAINS | 0 alert | 1 alert: catechol_A | 0 alert | 0 alert |
| Brenk | 2 alerts: beta_keto_anhydride, michael_acceptor_1 | 1 alert: catechol | 1 alert: Three-membered_heterocycle | 0 alert |
| Leadlikeness | No; 2 violations: MW > 350, Rotors > 7 | Yes | No; 1 violation: MW > 350 | Yes |
| Synthetic accessibility | 2.97 | 3.23 | 6.85 | 2.87 |
| Toxicological Properties | ||||
| AMES toxicity | No | No | No | No |
| Max. tolerated dose (human) (log mg/kg/day) | 0.081 | 0.499 | 0.867 | 0.478 |
| hERG I inhibitor | No | No | No | No |
| hERG II inhibitor | No | No | No | No |
| Oral Rat Acute Toxicity (LD50) (mol/kg) | 1.833 | 2.471 | 2.831 | 2.268 |
| Oral Rat Chronic Toxicity (LOAEL) (log mg/kg_bw/day) | 2.228 | 2.612 | 1.776 | 2.189 |
| Hepatotoxicity | No | No | No | No |
| Skin Sensitization | No | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saikat, A.S.M. An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chem. Proc. 2022, 8, 5. https://doi.org/10.3390/ecsoc-25-11721
Saikat ASM. An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chemistry Proceedings. 2022; 8(1):5. https://doi.org/10.3390/ecsoc-25-11721
Chicago/Turabian StyleSaikat, Abu Saim Mohammad. 2022. "An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2" Chemistry Proceedings 8, no. 1: 5. https://doi.org/10.3390/ecsoc-25-11721
APA StyleSaikat, A. S. M. (2022). An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chemistry Proceedings, 8(1), 5. https://doi.org/10.3390/ecsoc-25-11721