1. Introduction
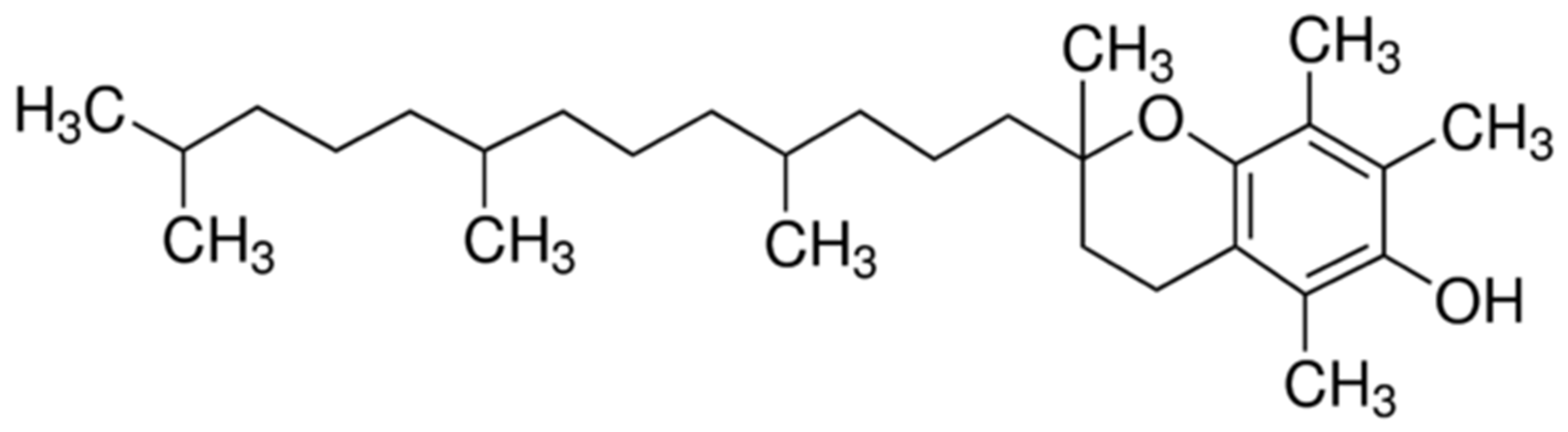
α-Tocopherol is the active form of vitamin E,
Figure 1. It functions as a hydroperoxyl radical scavenger and protects the organism from oxidative damage and plays a crucial role in cell signaling and regulating immune responses [
1].
Caseins are the major phosphoproteins of mammalian milk and exist as micelles made of polypeptides and consist of four types, i.e., αs1- (38%), αs2- (10%), β- (34%) and κ-casein (15%). Casein are almost similar in molecular weight: αS1-casein 23.62, αS2-casein 25.50, β-casein 24.09 and κ-casein 19.00 kD, and net negative charge [
2]. Bovine casein is a family of milk proteins with hydrophilic and hydrophobic regions that show block distribution within the protein chain. These amphiphilic properties offer great potential as a material for use as a matrix for transporting active molecules as vitamin.
Molecular docking techniques are used to predict how a protein interacts with small molecules, such as vitamins. This ability governs a significant part of the protein’s dynamics which may enhance/inhibit its interaction function as to which molecules are targeted [
3]. Two factors are of paramount importance in molecular docking studies: optimizing the ligand for the correct native conformation in the presence of which it can achieve a best fit orientation to bind with a protein of interest, and the conformational flexibility of ligand and protein [
4]. Thus, the accurate prediction of the binding modes between the ligand and protein is of fundamental importance in modern structure-based small molecules transport design.
The objective of the current work was to evaluate the interaction of α1-casein, the main fraction of the casein, with vitamin E by molecular docking calculations, using SwissDock and DockThor servers. Using specific scoring functions based on energy terms, the best protein–ligand binding models were obtained, and therefore the binding affinity between tocopherol and α1-casein.
2. Computational Methodology
2.1. Tocopherol Structure Preparation
The ligand in this study was α-tocopherol. The canonical SMILES of α-tocopherol was obtained from PubChem [
5]. Then the structure was minimized with the USCF Chimera program [
6]. Finally, the α-tocopherol structure obtained by Chimera was optimized employing Gaussian09 program suit [
7] with the hybrid density functional B3LYP and 6-31+G(d,p) basis sets.
2.2. αs1-Casein Structure Preparation
The amino acid sequence of αs1-casein was obtained from the GenBank of NCBI [
8] and it corresponded to the NCBI Reference Sequence: NP_851372.1 of Bos taurus with 214 residues of amino acid. The three-dimensional structure was generated by I-TASSER server [
9] from the corresponding FASTA file. The protonation state of the ionizable residues at pH 7 was evaluated with the PROPKA program [
10]. The final structure was minimized with the USCF Chimera program according to the MM calculation method and was validated by the Mol-Probity server [
11].
2.3. Molecular Docking Studies
After the preparation of the protein and ligand structures, molecular docking calculations were performed by SwissDock [
12] and DockThor servers [
13]. These docking studies corresponded to a system with flexible ligand and rigid protein. Using specific scoring functions based on energy terms the best protein–ligand binding models were obtained. Interaction types and distances were evaluated with the USCF Chimera program and Discovery Studio Visualizer [
14].
3. Results and Discussion
The minimized structures of α-tocopherol and αs1-casein are shown in
Figure 2. Docking studies of α-tocopherol with αs1-casein were carried out by SwissDock and DockThor servers, and the results of the binding modes between these structures are given below.
Molecular Docking Calculation Conducted with Swissdock Server
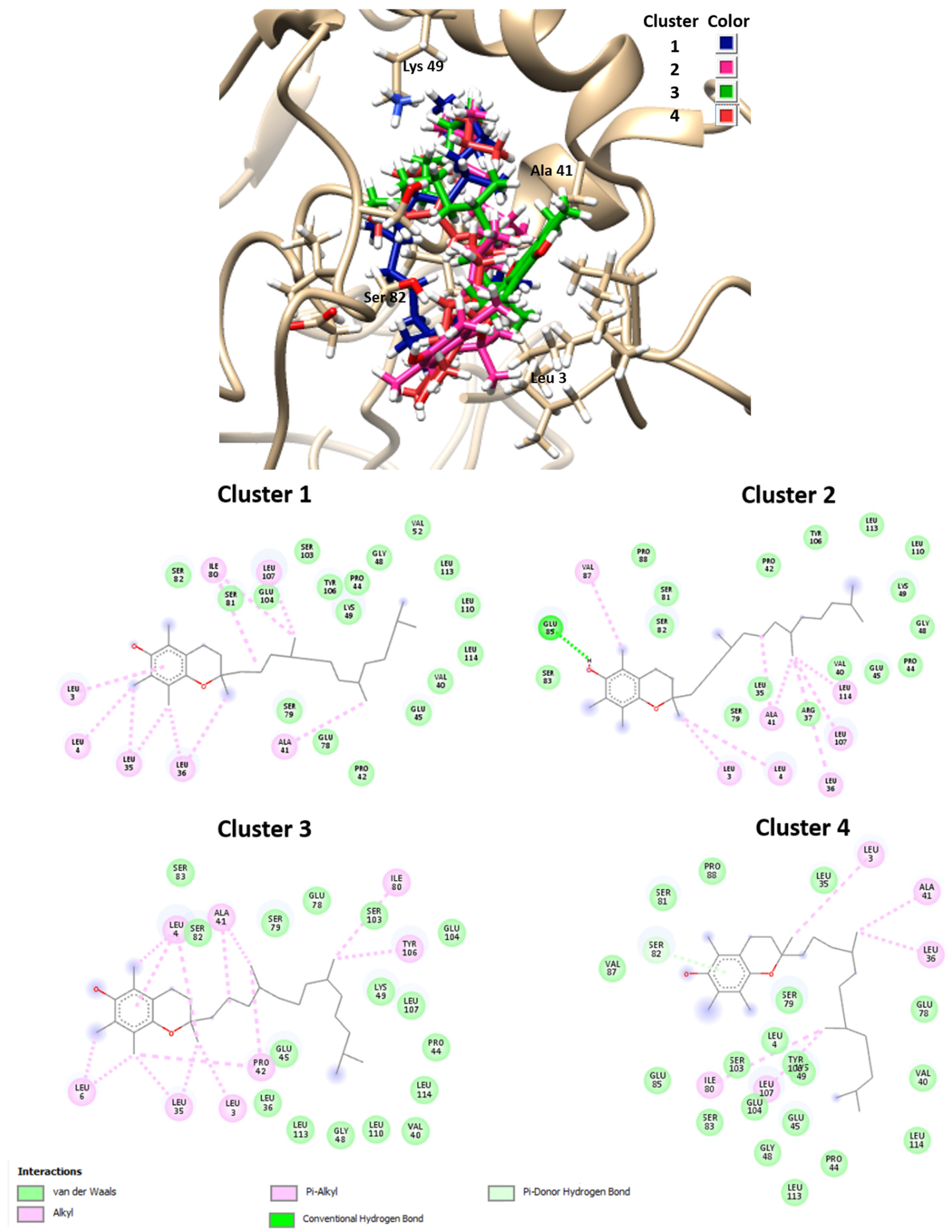
The results obtained by molecular docking protocols with SwissDock for the first cluster are shown in
Table 1 and
Figure 2. Predicted binding sites were clustered in 57 clusters with populations of 4–8 members. The cluster rank was predicted by the full fitness energy of the members. The best full fitness corresponded to the first member of each cluster.
From geometries of predicted binding sites corresponding to clusters 1–4 ranged from −1030.70 and −1024.66 Kcal/mol of full fitness values, the interaction types present in between α-tocopherol and the αs1-casein chain were identified. The observed interactions between vitamin E and the amino acid residues consisted of several hydrophobic interactions, especially with the residues corresponding to the sequence from 3–119 residues. The cavity of the binding site contains aliphatic hydrophobic residues such as Leu3, Leu4, Leu6, Leu35, Leu36, Leu107, Le114, Ala41, Val87, Pro42, Ile80, with a distance of interaction between tocopherol and residues of 3.8–5.4 Å. Only a hydrogen bond was observed between the phenyl group of vitamin E and the carboxylate group of Glu85 residue with an interaction distance of 2.13 Å. Similar interactions were observed with binding sites of other clusters not shown in
Figure 3. These results indicate that the α-tocopherol interacts with the hydrophobic sites of the αs1-casein, i.e., with amino acid chain segments that present essentially a random coil structure.
Docking calculations previously studied with SwissDock were compared with new calculations carried out with the DockThor server.
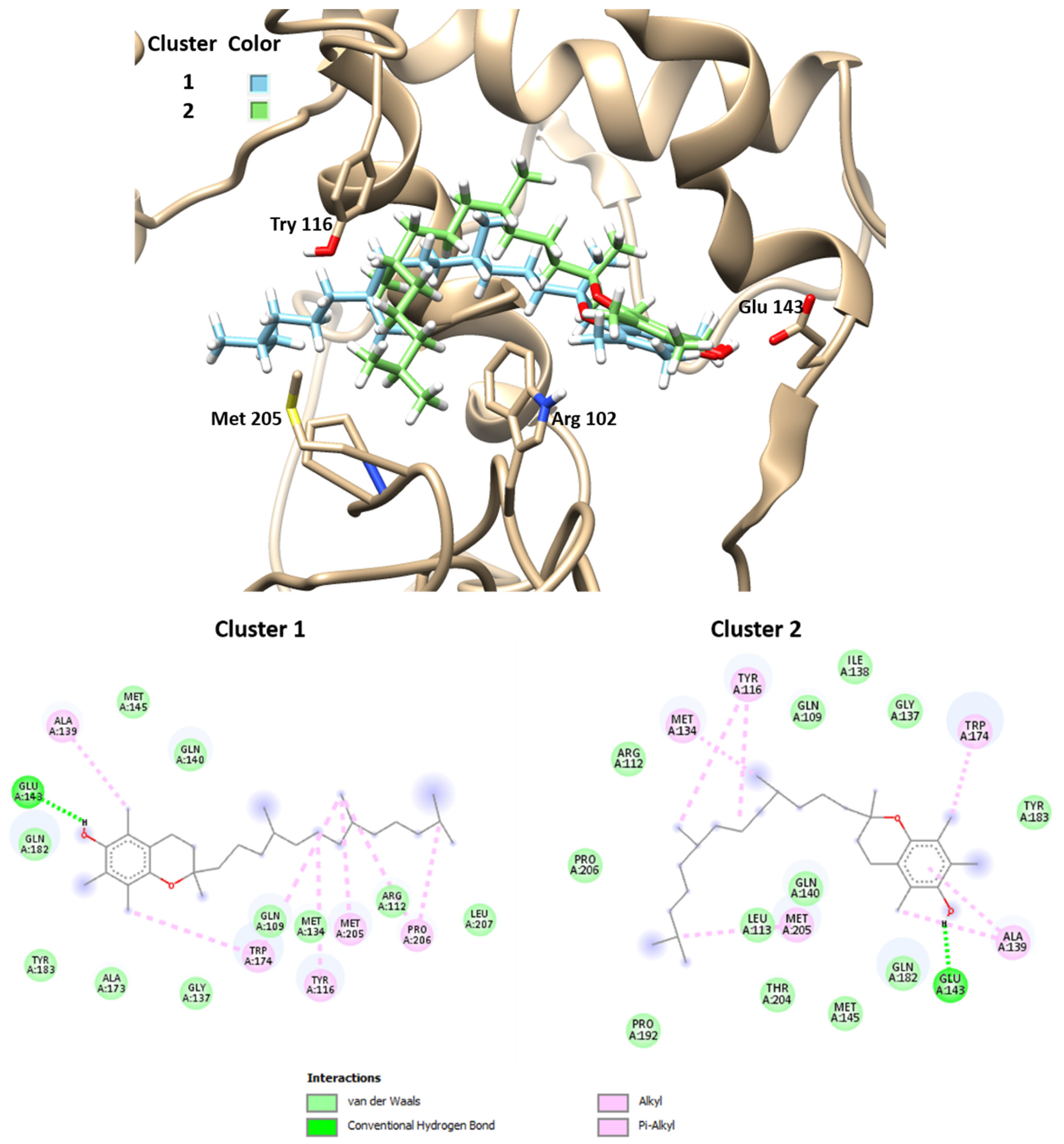
Figure 4 and
Table 2 present the best results obtained by molecular docking protocols with DockThor. Only two results were selected, i.e., the best two predicted binding sites, with an affinity score of −8.6 Kcal/mol of the members. Cluster 2 presents more total energy than cluster 1, with a difference of root-mean-square deviation (RMSD) between both of 3.66 Å. These news modes of interactions of ligand–protein are different from that obtained with SwissDock.
The observed interactions between vitamin E and amino acid residues in these modes consisted of several hydrophobic interactions, especially with the residues corresponding to the sequence from 109–206 residues. The cavity of the binding site contains aliphatic and aromatic hydrophobic residues such as Gln109, Arg112, Tyr116, Met134, Ala139. Trp174, Met205, with a distance of interaction between tocopherol and residues of 4.2–5.5 Å. Hydrogen bonds were observed between the phenyl group of vitamin E and the carboxylate group of Glu143 with distances of 1.75–1.98 Å. These results indicate that the α-tocopherol interacts with hydrophobic sites of the αs1-casein, such as the binding site modes predicted by the docking calculation with SwissDock.
4. Conclusions
We used an in silico calculation to study the binding modes of α-tocopherol with αs1-casein employing SwissDock and DockThor servers. Specific scoring functions based on energy terms were obtained from different protein–ligand binding models. The observed interactions between α-tocopherol and amino acid residues consisted of hydrophobic interactions of an electrostatic nature. A few hydrogen bonds were observed between the phenyl group of α-tocopherol and the carboxylate group of the glutamic acid residue. These results suggest that there exists a major interaction of α-tocopherol with random coil structure and interaction with segments formed by α-helix and β-sheet. This implies that in random coil segments there is a predominance of hydrophobic domains.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the reported data are presented in this document.
Acknowledgments
Authors thank the Capital Semilla of Argentina (2019), Universidad Nacional del Litoral (UNL), Santa Fe, Argentina, and Universidad Tecnológica Nacional (UTN), San Francisco, Argentina (grant PID 2018 UTN No. 5489) for financial support of this work.
References
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Glab, T.K.; Boratynski, J. Potential of casein as a carrier for biologically active agents. Top. Curr. Chem. 2017, 375, 71. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.K.; Mishra, N. A review on molecular docking: Novel tool for drug discovery. JSM Chem. 2016, 4, 1029. [Google Scholar]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov (accessed on October 2020).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A. Gaussian 09; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2008. [Google Scholar]
- GenBank. Available online: https://www.ncbi.nlm.nih.gov/genbank/ (accessed on October 2020).
- Tasser Server. Available online: https://zhanglab.ccmb.med.umich.edu/I-TASSER/ (accessed on October 2020).
- Propka. Available online: http://server.poissonboltzmann.org/pdb2pqr (accessed on October 2020).
- Mol Probity. Available online: http://molprobity.biochem.duke.edu/index.php (accessed on October 2020).
- SwissDock Server. Available online: http://www.swissdock.ch/ (accessed on October 2020).
- DockThor Server. Available online: https://dockthor.lncc.br/v2/ (accessed on October 2020).
- Discovery Studio Visualizer, v20.1.0.19259; 2009. Available online: https://discover.3ds.com/discovery-studio-visualizer/ (accessed on October 2020).
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).







{kind=link}
{kind=link}
{kind=link}
{kind=link}