
A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates †


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
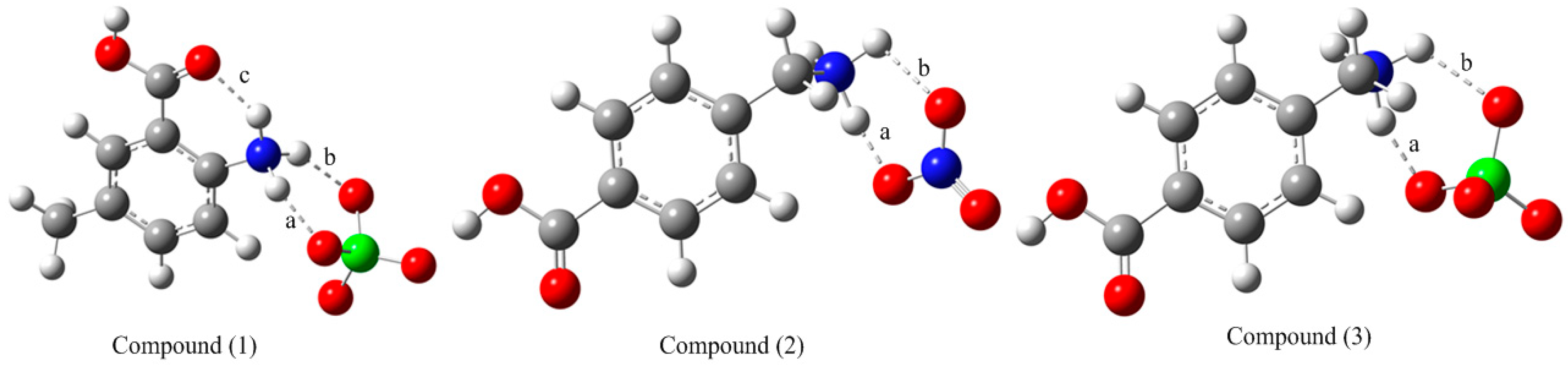
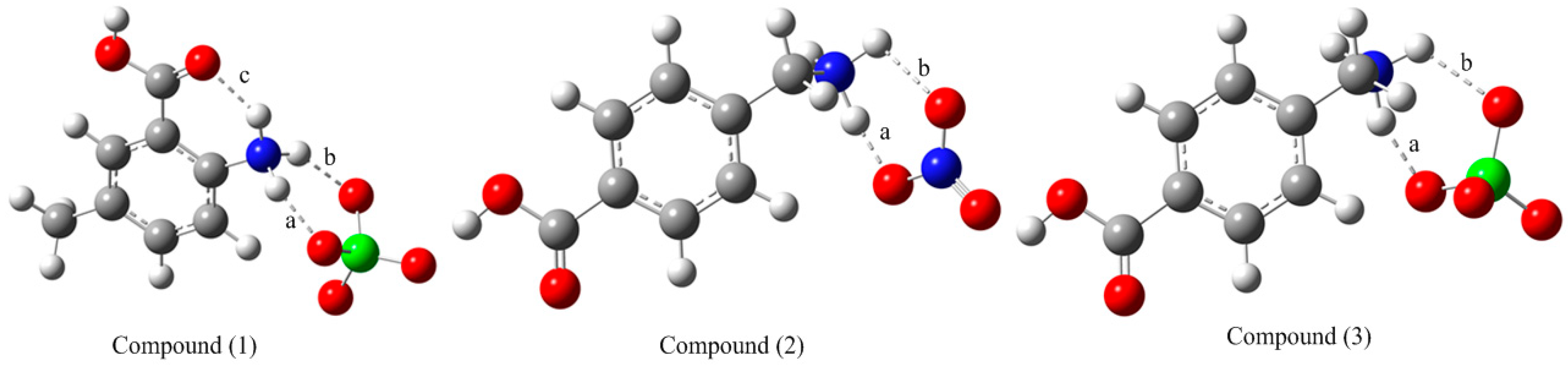
3.1. Comparative Theoretical and Experimental Structural Study
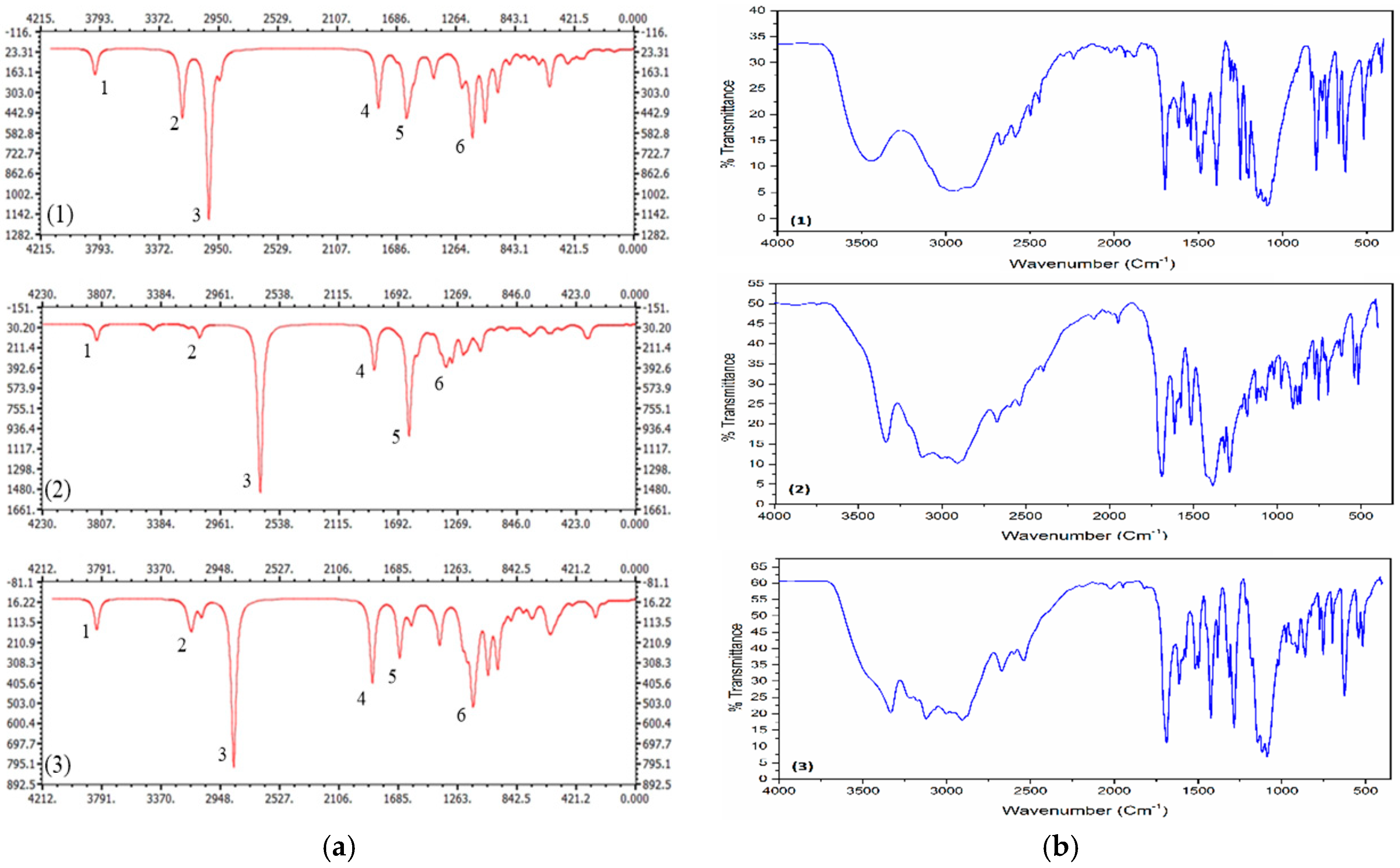
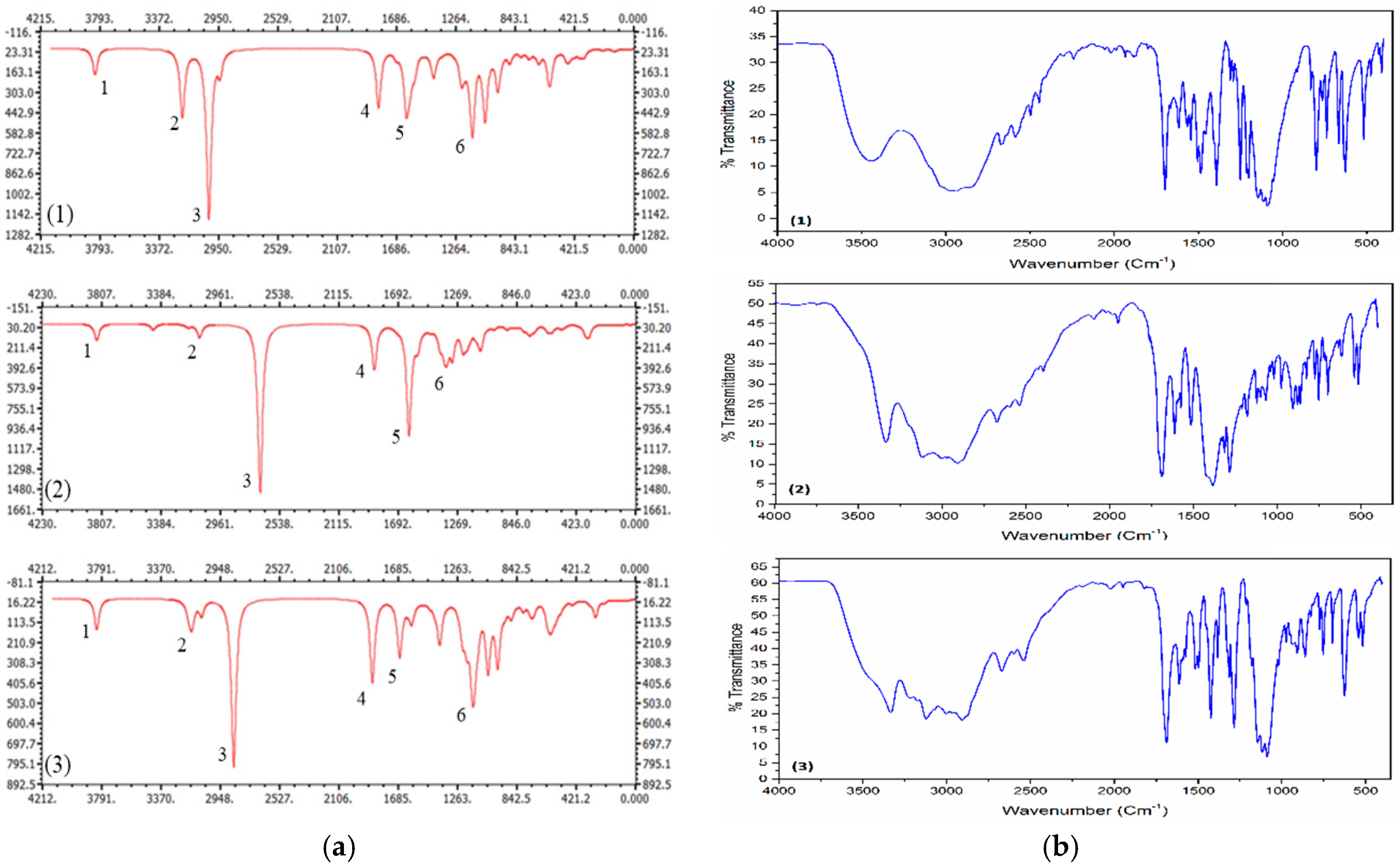
3.2. Spectral Analyses
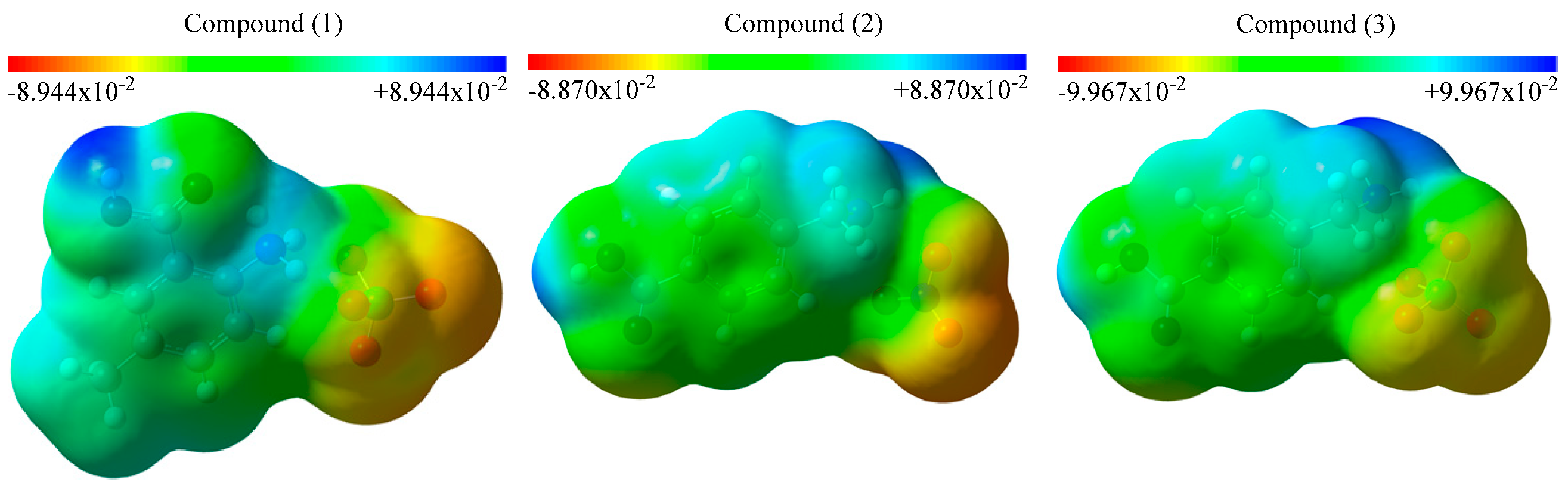
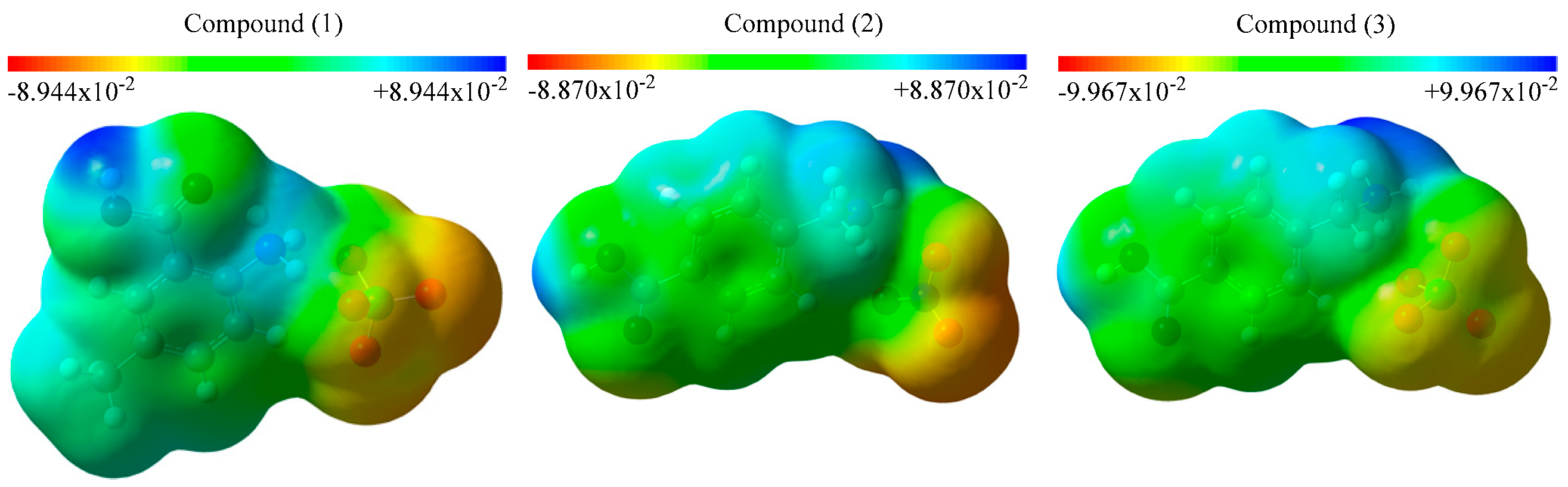
3.3. Molecular Electrostatic Potential (MEP) Maps and Contour Diagrams
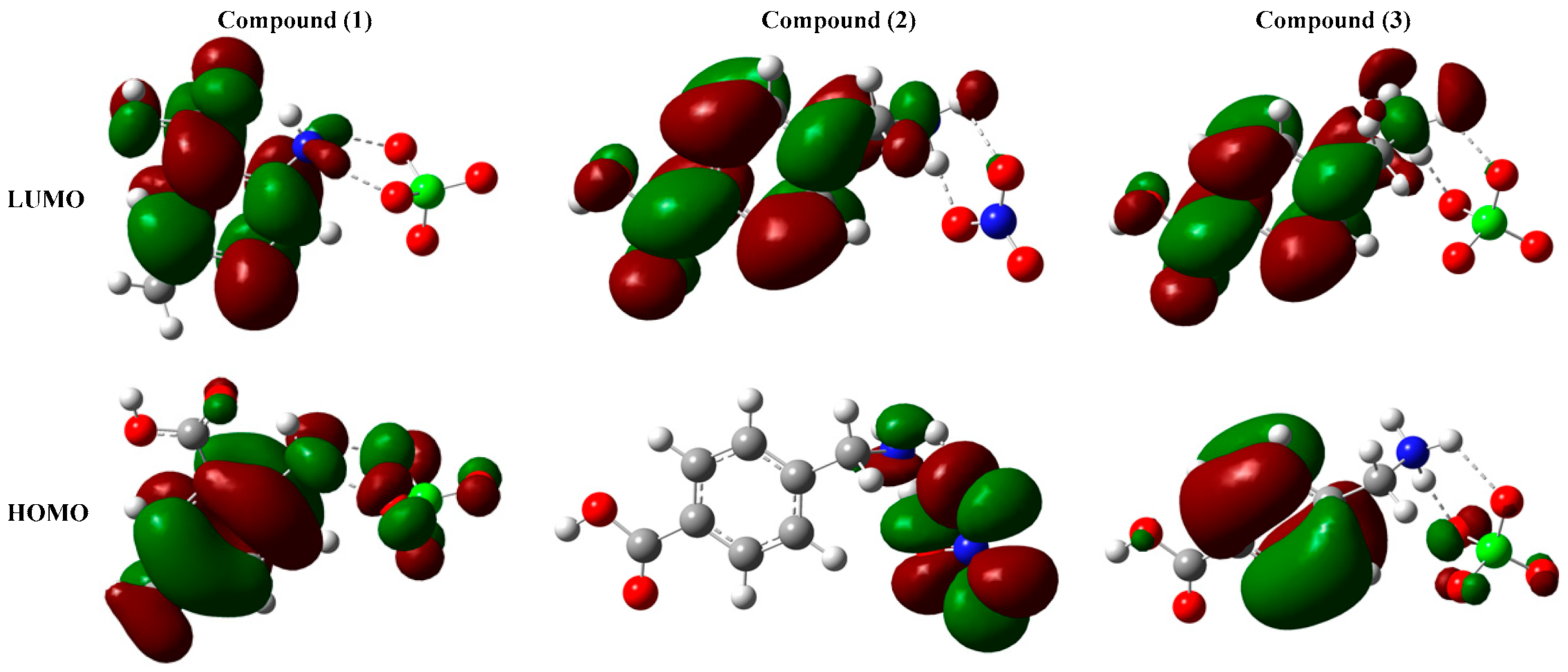
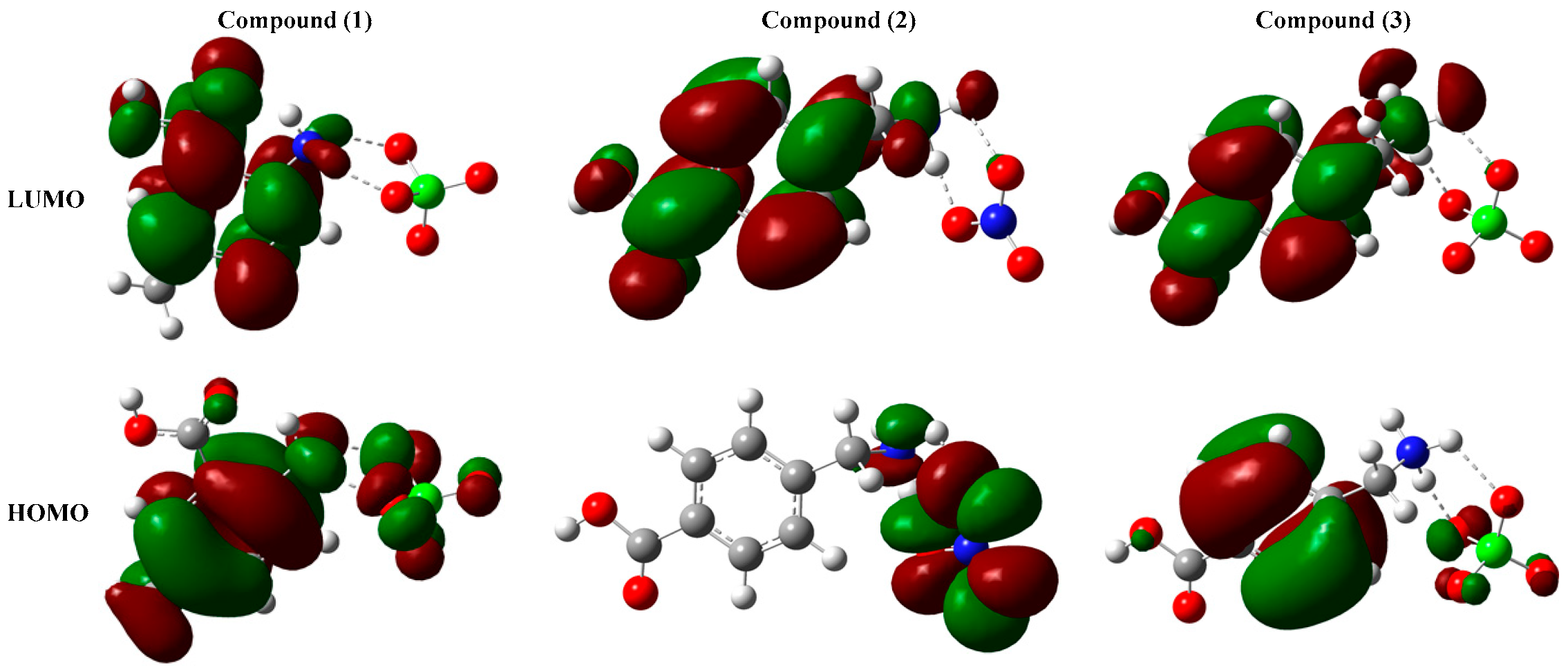
3.4. Non-Linear Optical Properties
4. Conclusions
Author Contributions
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, B.H.; Fang, Z.J.; Jiao, Y.; Jiang, Y.H. Composes a transfert de protons: Synthese, structure et etude des differentes interactions interatomiques. Jiangsu Chem. Ind. 2007, 55, 39–41. [Google Scholar]
- Cao, S.L.; Feng, Y.-P.; Jiang, Y.-Y.; Liu, S.Y.; Ding, G.Y.; Li, R.T. Synthesis and in vitro antitumor activity of 4(3H)-quinazolinone derivatives with dithiocarbamate side chains. Bioorg. Med. Chem. Lett. 2005, 15, 1915–1917. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.L.; Feng, Y.P.; Zheng, X.L.; Jiang, Y.Y.; Zhang, M.; Wang, Y.; Xu, M. Synthesis of substituted benzylamino- and heterocyclylmethylamino carbodithioate derivatives of 4-(3H)-quinazolinone and their cytotoxic activity. Arch. Pharm. 2006, 339, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.L.; Guo, Y.W.; Wang, X.B.; Zhang, M.; Feng, Y.P.; Jiang, Y.Y.; Wang, Y.; Gao, Q.; Ren, J. Synthesis and Cytotoxicity Screening of Piperazine-1-carbodithioate Derivatives of 2-Substituted Quinazolin-4(3H)-ones. Arch. Pharm. 2009, 342, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Steinmetzer, T.; Pilgram, O.; Wenzel, B.M.; Wiedemeyer, S.J.A. Fibrinolysis Inhibitors: Potential Drugs for the Treatment and Prevention of Bleeding. J. Med. Chem. 2020, 63, 1445–1472. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, F.; Neuland, P.; Klöcking, H.P. On the antifibrinolytic activity of the esters of 4-aminomethylbenzoic acid (PAMBA)]. Pharmazie 1966, 21, 345–348. [Google Scholar] [PubMed]
- Pilgrim, H.; Grosse, S. Proliferation of cultured endothelial cells under the effect of aprotinin and 4-aminomethylbenzoic acid. Biomed. Biochim. Acta 1986, 45, 1015–1019. [Google Scholar] [PubMed]
- Kazmirowski, H.G.; Neuland, P.; Landmann, H.; Markwardt, F. Synthesis of antiproteolytic active derivatives of 4-aminomethylbenzoic acid and other structurally similar compounds. Pharmazie 1967, 22, 465–470. [Google Scholar] [PubMed]
- Beyer, S.; Pilgrim, H. The inhibition of endothelial cell proliferation by esters of 4-aminomethylbenzoic acid (pamba). Pharmazie 1991, 46, 597–599. [Google Scholar] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Direm, A.; Altomare, A.; Moliterni, A.; Benali-Cherif, N. Intermolecular interactions of proton transfer compounds: Synthesis, crystal structure and Hirshfeld surface analysis. Acta Cryst. 2015, B71, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Dadda, N.; Nassour, A.; Guillot, B.; Benali-Cherif, N.; Jelsch, C. Charge-density analysis and electrostatic properties of 2-carboxy-4-methylanilinium chloride monohydrate obtained using a multipolar and a spherical-charges model. Acta Cryst. 2014, A68, 452–463. [Google Scholar] [CrossRef]
- Atria, A.M.; Garland, M.T.; Baggio, R. Crystal structure of zwitterionic 4-(ammonio methyl) benzoate: A simple molecule giving rise to a complex supra molecular structure. Acta Cryst. 2014, E70, 385–388. [Google Scholar]
- Sayin, K.; Karakaş, D. Determination of structural, spectral, electronic and biological properties of tosufloxacin boron complexes and investigation of substituent effect. J. Mol. Struct. 2017, 1146, 191–197. [Google Scholar] [CrossRef]
- Üngördü, A.; Tezer, N. The solvent (water) and metal effects on HOMO-LUMO gaps of guanine base pair: A computational study. J. Mol. Graph. Model. 2017, 74, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, R.; Thamaraichelvan, A.; Viswanathan, B. CO2 transformation on the active site of carbonic anhydrase enzyme leading to formation of H2CO3-A biomimetic model through computational study. Turk. Comput. Theor. Chem. 2017, 1, 17–26. [Google Scholar]
- Mirzaei, M. 5–Fluorouracil: Computational Studies of Tautomers and NMR Properties. Turk. Comput. Theor. Chem. 2017, 1, 27–34. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. (Eds.) GaussView, version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. (Eds.) Gaussian 09; revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths (Å) | Bond Angles (°) | ||||
|---|---|---|---|---|---|
| Experimental | Calculated | Experimental | Calculated | ||
| Compound (1) (R2 = 0.9998) | |||||
| O1-C7 | 1.316 | 1.333 | O1-C7-O2 | 122.5 | 121.6 |
| O2-C7 | 1.203 | 1.212 | O1-C7-C1 | 113.7 | 113.4 |
| N1-C2 | 1.465 | 1.459 | O2-C7-C1 | 123.8 | 124.9 |
| C1-C2 | 1.396 | 1.398 | C2-C1-C6 | 118.7 | 118.7 |
| C1-C7 | 1.499 | 1.486 | C2-C1-C7 | 120.8 | 121.2 |
| C5-C8 | 1.507 | 1.505 | C1-C2-N1 | 121.8 | 121.3 |
| Compound (2) (R2 = 0.9998) | |||||
| O1-C7 | 1.311 | 1.346 | O1-C7-O2 | 123.3 | 122.6 |
| O2-C7 | 1.213 | 1.200 | O1-C7-C1 | 113.9 | 112.7 |
| N1-C8 | 1.486 | 1.494 | O2-C7-C1 | 122.9 | 124.6 |
| C1-C2 | 1.386 | 1.392 | C2-C1-C6 | 119.4 | 120.4 |
| C1-C7 | 1.485 | 1.491 | C2-C1-C7 | 121.2 | 121.7 |
| C4-C8 | 1.505 | 1.507 | C4-C8-N1 | 112.1 | 110.2 |
| Compound (3) (R2 = 0.9993) | |||||
| O1A-C7A | 1.302 | 1.346 | O1A-C7A-O2A | 123.7 | 122.7 |
| O2A-C7A | 1.241 | 1.200 | O1A-C7A-C1A | 115.3 | 112.7 |
| N1A-C8A | 1.499 | 1.503 | O2A-C7A-C1A | 120.9 | 124.6 |
| C1A-C2A | 1.388 | 1.393 | C2A-C1A-C6A | 119.8 | 120.5 |
| C1A-C7A | 1.494 | 1.492 | C2A-C1A-C7A | 119.3 | 121.6 |
| C4A-C8A | 1.516 | 1.504 | C4A-C8A-N1A | 110.6 | 109.5 |
| Moiety | Total Energy (a.u.) | Compound | Total Energy (a.u.) |
|---|---|---|---|
| (A) | −515.608 | Compound (1) | −1276.528 |
| (B) | −515.593 | Compound (2) | −796.100 |
| (C) | −760.764 | Compound (3) | −1276.522 |
| (D) | −280.326 |
| Compound (1) | Compound (2) | Compound (3) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Label | Calculated | Experimental | Mode a | Calculated | Experimental | Mode a | Calculated | Experimental | Mode a |
| 1 | 3832 | 3446 | νOH | 3843 | 3338 | νOH | 3841 | 3337 | νOH |
| 2 | 3212 | 3030 | νNH | 3106 | 3119 | νNH | 3153 | 3118 | νNH |
| 3 | 3024 | νNH | 2696 | νNH | 2873 | νNH | |||
| 4 | 1817 | 1698 | νC = O | 1868 | 1688 | νC = O | 1868 | 1689 | νC = O |
| 5 | 1620 | 1507 | αNH2 | 1618 | 1516 | αNH2, | 1677 | 1514 | αNH2 |
| 1385 | νN = O | ||||||||
| 6 | 1150 | 111512551091 | νCl-O, ωCH, ωNH | 1355 | 1283 | ωCH | 1148 | 111512901086 | νCl-O, ωCH, ωNH |
| Assignments | 1H-NMR | Assignments | 13C-NMR | ||||
|---|---|---|---|---|---|---|---|
| (1) | (2) | (3) | (1) | (2) | (3) | ||
| C1 | 135.8 | 151.7 | 152.8 | C2H | - | 9.03 | 9.07 |
| C2 | 151.5 | 153.0 | 153.5 | C3H | 9.52 | 8.07 | 8.10 |
| C3 | 150.0 | 149.1 | 148.3 | C4H | 8.79 | - | - |
| C4 | 163.6 | 160.5 | 158.7 | C5H | - | 9.64 | 9.80 |
| C5 | 164.3 | 154.7 | 157.7 | C6H | 9.15 | 9.47 | 9.50 |
| C6 | 155.5 | 157.4 | 157.3 | C8H′ | 3.04 | 5.03 | 5.51 |
| C7 | 185.6 | 178.3 | 179.0 | C8H″ | 2.67 | 3.67 | 3.66 |
| C8 | 24.1 | 49.4 | 50.7 | C8H′″ | 2.05 | - | - |
| N1H′ | 10.88 | 16.65 | 11.90 | ||||
| N1H″ | 10.79 | 7.02 | 7.30 | ||||
| N1H′″ | 10.72 | 3.19 | 4.29 | ||||
| O1H | 6.48 | 6.64 | 6.34 | ||||
| Compound | µ 1 | α 2 | Δα 2 | β 3 |
|---|---|---|---|---|
| Urea | 1.76 | 2.83 | 10.46 | 5.09 × 10−28 |
| (1) | 4.88 | 16.48 | 32.52 | 1.80 × 10−27 |
| (2) | 4.04 | 15.56 | 30.68 | 1.03 × 10−27 |
| (3) | 4.40 | 16.22 | 33.04 | 8.88 × 10−28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Direm, A.; Sayın, K. A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates. Chem. Proc. 2021, 3, 102. https://doi.org/10.3390/ecsoc-24-08099
Direm A, Sayın K. A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates. Chemistry Proceedings. 2021; 3(1):102. https://doi.org/10.3390/ecsoc-24-08099
Chicago/Turabian StyleDirem, Amani, and Koray Sayın. 2021. "A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates" Chemistry Proceedings 3, no. 1: 102. https://doi.org/10.3390/ecsoc-24-08099
APA StyleDirem, A., & Sayın, K. (2021). A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates. Chemistry Proceedings, 3(1), 102. https://doi.org/10.3390/ecsoc-24-08099