
Theoretical Study of the Addition Reaction of Arylazides to 1,3-Dicarbonyl Compounds †


Abstract
:1. Introduction
2. Methodology of Calculations
3. Results and Discussion
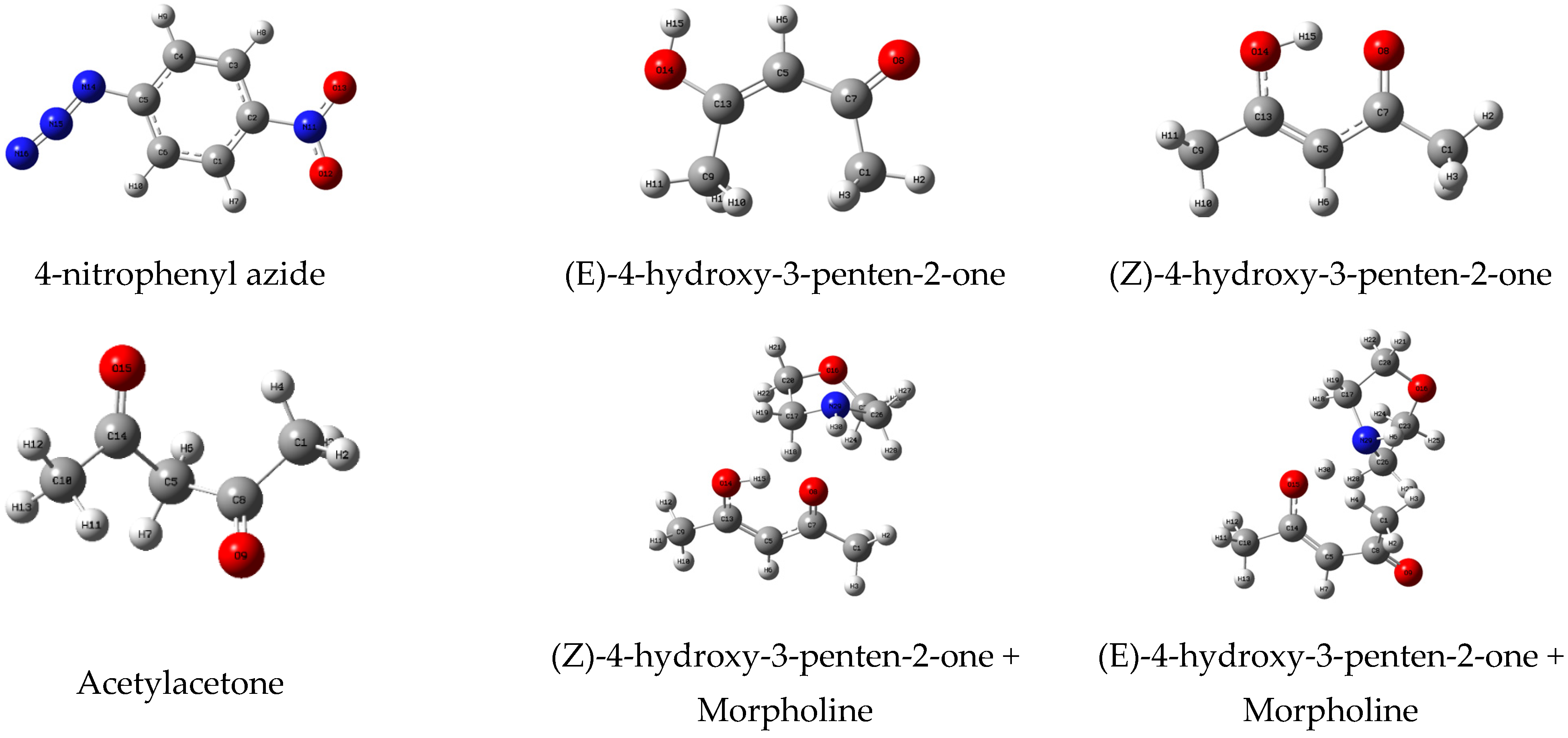
3.1. Prediction of the Relative Reactivity of the Reactants
3.2. DFT Study of the Domino Reactions of Acetylacetone Compounds with 4-Nitrophenyl Azide
- A.
- Tautomeric equilibrium between acetylacetone compounds 2 and their enol forms 4.
- B.
- The 1,3-dipolar cycloaddition (13DC) reaction between the enol compounds 4 and 4-nitrophenyl azide 1, resulting in the formation of 4,5-dihydro-1,2,3-triazoles 5.
- C.
- The dehydration process facilitated by the morpholine base to yield the final 1,2,3-triazoles 7 (see Scheme 2).
- A.
- Deprotonation of carbon atom C5: morpholine will deprotonate the carbon atom C5 of the cycloadduct, creating a carbanion on this carbon atom.
- B.
- Protonation of the ketone oxygen: The ketone oxygen in the cycloadduct will be protonated, usually by a proton from morpholine itself. This step will stabilize the negative charge formed during deprotonation.
- C.
- Elimination of a water molecule and formation of the C5=C4 double bond: Under dehydration conditions, a water molecule will be eliminated. This elimination reaction will lead to the formation of a double bond between the carbon atoms C5 and C4 of the cycloadduct (see Scheme 2).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Imperio, D.; Pirali, T.; Galli, U.; Pagliai, F.; Cafici, L.; Canonico, P.L.; Sorba, G.; Genazzani, A.A.; Tron, G.C. Replacement of the lactone moiety on podophyllotoxin and steganacin analogues with a 1,5-disubstituted 1,2,3-triazole via ruthenium-catalyzed click chemistry. Bioorg. Med. Chem. 2007, 15, 6748–6757. [Google Scholar] [CrossRef]
- Akselsen, Ø.W.; Odlo, K.; Cheng, J.J.; Maccari, G.; Botta, M.; Hansen, T.V. Synthesis, biological evaluation and molecular modeling of 1, 2, 3-triazole analogs of combretastatin A-1. Bioorg. Med. Chem. 2012, 20, 234–242. [Google Scholar] [CrossRef]
- Duan, Y.-C.; Ma, Y.C.; Zhang, E.; Shi, X.J.; Wang, M.M.; Ye, X.W.; Liu, H.M. Design and synthesis of novel 1,2,3-triazole-dithiocarbamate hybrids as potential anticancer agents. Eur. J. Med. Chem. 2013, 62, 11–19. [Google Scholar] [CrossRef]
- El Akri, K.; Bougrin, K.; Balzarini, J.; Faraj, A.; Benhida, R. Efficient synthesis and in vitro cytostatic activity of 4-substituted triazolyl-nucleosides. Bioorg. Med. Chem. Lett. 2007, 17, 6656–6659. [Google Scholar] [CrossRef]
- Hupe, D.J.; Boltz, R.; Cohen, C.; Felix, J.; Ham, E.; Miller, D.; Soderman, D.; Van Skiver, D. The inhibition of receptor-mediated and voltage-dependent calcium entry by the antiproliferative L-651,582. J. Biol. Chem. 1991, 266, 10136–10142. [Google Scholar] [CrossRef] [PubMed]
- Bascal, Z.; Holden-Dye, L.; Willis, R.; Smith, S.; Walker, R. Novel azole derivatives are antagonists at the inhibitory GABA receptor on the somatic muscle cells of the parasitic nematode Ascaris suum. Parasitology 1996, 112, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Pet, C.; Batta, G.; Györgydeák, Z.; Sztaricskai, F. Glycoside Synthesis with Anomeric 1-N-Glycobiosyl-1,2,3-triazoles1. J. Carbohydr. Chem. 1996, 15, 465–483. [Google Scholar]
- Buckle, D.R.; Rockell, C.J.; Smith, H.; Spicer, B.A. Studies on 1,2,3-triazoles. 13.(Piperazinylalkoxy)-[1] benzopyrano [2,3-d]-1, 2, 3-triazol-9 (1H)-ones with combined H1-antihistamine and mast cell stabilizing properties. J. Med. Chem. 1986, 29, 2262–2267. [Google Scholar] [CrossRef] [PubMed]
- Kurumi, M.; Sasaki, K.; Takata, H.; Nakayama, T. Synthesis and Chemiluminescent Activity of Pyridazino [4,5-b] indole-1,4 (2H, 3H)-diones. Heterocycles 2000, 53, 2809–2819. [Google Scholar]
- Costa, M.S.; Boechat, N.; Rangel, E.A.; Da Silva, F.d.C.; De Souza, A.M.; Rodrigues, C.R.; Castro, H.C.; Junior, I.N.; Lourenço, M.C.S.; Wardell, S.M. Synthesis, tuberculosis inhibitory activity, and SAR study of N-substituted-phenyl-1,2,3-triazole derivatives. Bioorg. Med. Chem. 2006, 14, 8644–8653. [Google Scholar] [CrossRef]
- Arora, B.S.; Shafi, S.; Singh, S.; Ismail, T.; Kumar, H.S. A novel domino-click approach for the synthesis of sugar based unsymmetrical bis-1,2,3-triazoles. Carbohydr. Res. 2008, 343, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Boddy, I.K.; Briggs, G.G.; Harrison, R.P.; Jones, T.H.; O’Mahony, M.J.; Marlow, I.D.; Roberts, B.G.; John Willis, R.; Bardsley, R.; Reid, J. The Synthesis and Insecticidal Activity of a Series of 2-Aryl-1,2,3-triazoles. Pestic. Sci. 1996, 48, 189–196. [Google Scholar] [CrossRef]
- Buechel, K.; Gold, H.; Frohberger, P.; Kaspers, H. German Patent 2407305. Chem. Abstr. 1975, 83, 206290. [Google Scholar]
- Reisser, F. British Patent 8101239. Chem. Abstr. 1981, 96, 69006. [Google Scholar]
- Kaushik, C.; Kumar, K.; Singh, S.; Singh, D.; Saini, S. Synthesis and antimicrobial evaluation of 1, 4-disubstituted 1, 2, 3-triazoles with aromatic ester functionality. Arab. J. Chem. 2016, 9, 865–871. [Google Scholar] [CrossRef]
- Abdennabi, A.; Abdulhadi, A.; Abu-Orabi, S.; Saricimen, H. The inhibition action of 1 (benzyl) 1-H-4, 5-dibenzoyl-1, 2, 3-triazole on mild steel in hydrochloric acid media. Corros. Sci. 1996, 38, 1791–1800. [Google Scholar] [CrossRef]
- Voitekhovich, S.V.; Filippova, J.V.; Sukhanova, A.G.; Lyakhov, A.S.; Ivashkevich, L.S.; Sukhanov, G.T.; Grigoriev, Y.V. Selective complexation of 1-ethyl-5-nitro-1,2,3-triazole (entz) with copper (II) salts: Preparation and characterization of [Cu(entz)2Cl2] and [Cu(entz)4(H2O)2](ClO4)2. Inorg. Chem. Commun. 2012, 24, 77–80. [Google Scholar] [CrossRef]
- Gil, M.V.; Arévalo, M.J.; Lopez, O. Click chemistry-what’s in a name? Triazole synthesis and beyond. Synthesis 2007, 2007, 1589–1620. [Google Scholar] [CrossRef]
- Krivopalov, V.P.; Shkurko, O.P. 1,2,3-Triazole and its derivatives. Development of methods for the formation of the triazole ring. Russ. Chem. Rev. 2005, 74, 339. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Zhang, Y.; Singh, S.K. 1,2,3-Triazole formation under mild conditions via 1,3-dipolar cycloaddition of acetylenes with azides. Heterocycles Int. J. Rev. Commun. Heterocycl. Chem. 2003, 60, 1225–1239. [Google Scholar] [CrossRef]
- Anastas, P.; Warner, J. Green Chemistry: Theory Ant Practice; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Lancaster, M. Green Chemistry: An Introductory Text; Royal Society of Chemistry: London, UK, 2020. [Google Scholar]
- Totobenazara, J.; Burke, A.J. New click-chemistry methods for 1,2,3-triazoles synthesis: Recent advances and applications. Tetrahedron Lett. 2015, 56, 2853–2859. [Google Scholar] [CrossRef]
- Chassaing, S.; Bénéteau, V.; Pale, P. When CuAAC’click chemistry’goes heterogeneous. Catal. Sci. Technol. 2016, 6, 923–957. [Google Scholar] [CrossRef]
- De Proft, F.; Geerlings, P. Conceptual and computational DFT in the study of aromaticity. Chem. Rev. 2001, 101, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chem. Acc. Theory Comput. Model. (Theor. Chim. Acta) 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Hadj Mokhtar, H. Application de la Réaction Multicomposés à la Synthèse de Triazolines. Ph.D. Thesis, University of Oran 1, Oran, Algeria, 2018. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
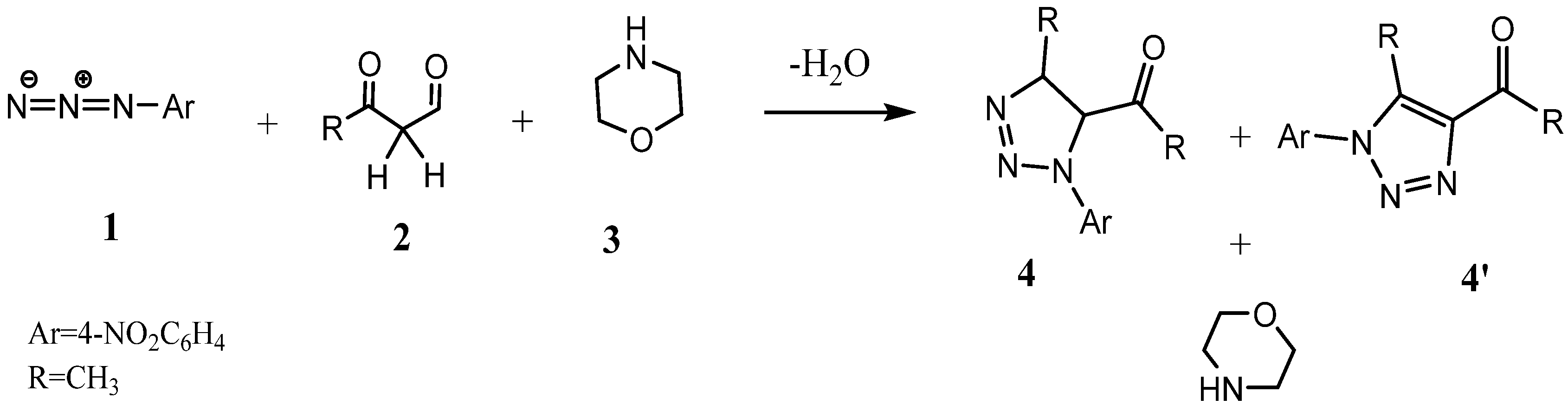




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LUMO | HOMO | HOMO * −LUMO | LUMO * −HOMO | μ (a.u.) | η (eV) | ω (eV) | N (eV) | |
|---|---|---|---|---|---|---|---|---|
| 4-nitrophenyl azide | −0.116 | −0.263 | 0.195 | 0.107 | −0.190 | 0.147 | 3.341 | 2.019 |
| (Z)-4-hydroxy-3-penten-2-one + Morpholine | −0.068 | −0.224 | 0.217 | 0.122 | −0.146 | 0.156 | 1.856 | 3.092 |
| (E)-4-hydroxy-3-penten-2-one + Morpholine | −0.051 | −0.241 | 0.223 | 0.125 | −0.146 | 0.190 | 1.523 | 2.616 |
| (E)-4-hydroxy-3-penten-2-one | −0.054 | −0.256 | 0.202 | 0.139 | −0.155 | 0.201 | 1.626 | 2.220 |
| (Z)-4-hydroxy-3-penten-2-one | −0.061 | −0.256 | 0.199 | 0.152 | −0.158 | 0.195 | 1.751 | 2.216 |
| Acetylacetone | −0.064 | −0.268 | 0.195 | 0.107 | −0.166 | 0.205 | 1.832 | 1.875 |
| Reactants | ΔE (kcal/mol) | Reactants | ΔE (kcal/mol) |
|---|---|---|---|
| Acetylacetone + Morpholine | 0.00 | Acetylacetone | 0.00 |
| (Z)-4-hydroxy-3-penten-2-one + Morpholine | −0.99 | (Z)-4-hydroxy-3-penten-2-one | −3.63 |
| (E)-4-hydroxy-3-penten-2-one + Morpholine | 0.75 | (E)-4-hydroxy-3-penten-2-one | 7.96 |
| ΔE | ΔG | ΔE≠ | ΔG≠ | ||
|---|---|---|---|---|---|
| Reactant | 0.00 | 0.00 | 10.85 | 12.54 | |
| Enolization | TS1 | 10.85 | 12.54 | ||
| MIN1 | 2.55 | 3.81 | 26.21 18.17 | 26.52 18.09 | |
| Cycloaddition | TS2a TS2b | 28.76 20.72 | 30.33 21.90 | ||
| MIN2a MIN2b | −10.92 −11.51 | −8.65 −9.42 | 11.77 17.1 | 24.68 18.33 | |
| Dehydration | TS3a TS3b | 0.85 5.59 | 16.03 8.91 | ||
| Product-a Product-b | −38.14 −42.20 | −27.53 −29.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelghani, A.; Hadj Mokhtar, H.; Boumaza, O.; Naous, A. Theoretical Study of the Addition Reaction of Arylazides to 1,3-Dicarbonyl Compounds. Chem. Proc. 2023, 14, 60. https://doi.org/10.3390/ecsoc-27-16160
Abdelghani A, Hadj Mokhtar H, Boumaza O, Naous A. Theoretical Study of the Addition Reaction of Arylazides to 1,3-Dicarbonyl Compounds. Chemistry Proceedings. 2023; 14(1):60. https://doi.org/10.3390/ecsoc-27-16160
Chicago/Turabian StyleAbdelghani, Adda, Halima Hadj Mokhtar, Ouda Boumaza, and Abderrahmane Naous. 2023. "Theoretical Study of the Addition Reaction of Arylazides to 1,3-Dicarbonyl Compounds" Chemistry Proceedings 14, no. 1: 60. https://doi.org/10.3390/ecsoc-27-16160
APA StyleAbdelghani, A., Hadj Mokhtar, H., Boumaza, O., & Naous, A. (2023). Theoretical Study of the Addition Reaction of Arylazides to 1,3-Dicarbonyl Compounds. Chemistry Proceedings, 14(1), 60. https://doi.org/10.3390/ecsoc-27-16160