
A Robust Regression-Based Modeling to Predict Antiplasmodial Activity of Thiazolyl–Pyrimidine Hybrid Derivatives against Plasmodium falciparum †


Abstract
:1. Introduction
2. Methods
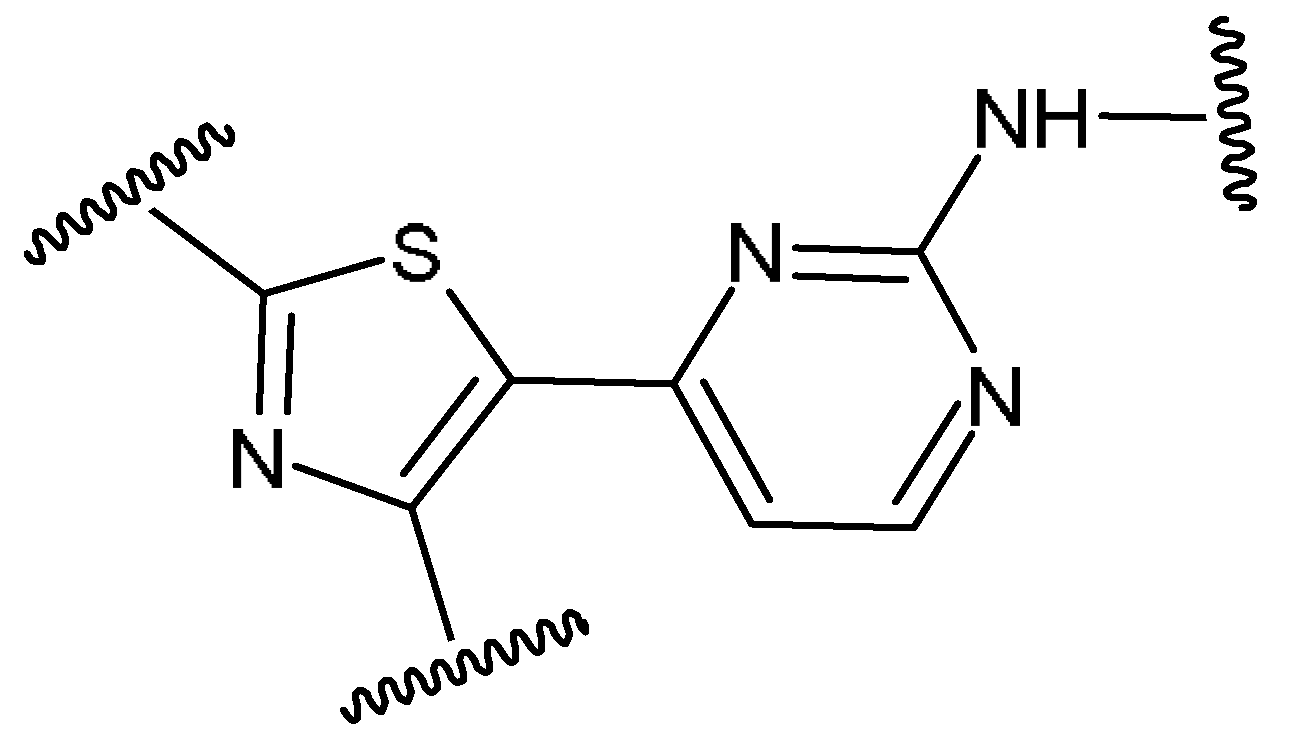
2.1. Chemical Data Set
2.2. Preparation of Data Set
2.3. Computation of Molecular Descriptors
2.4. Data Pretreatment
2.5. Selection of Relevant Descriptors
2.6. Data Splitting
2.7. Regression Modeling
2.8. Model Evaluation
3. Results and Discussion
3.1. Chemical Data Set
3.2. Selection of Significant Features
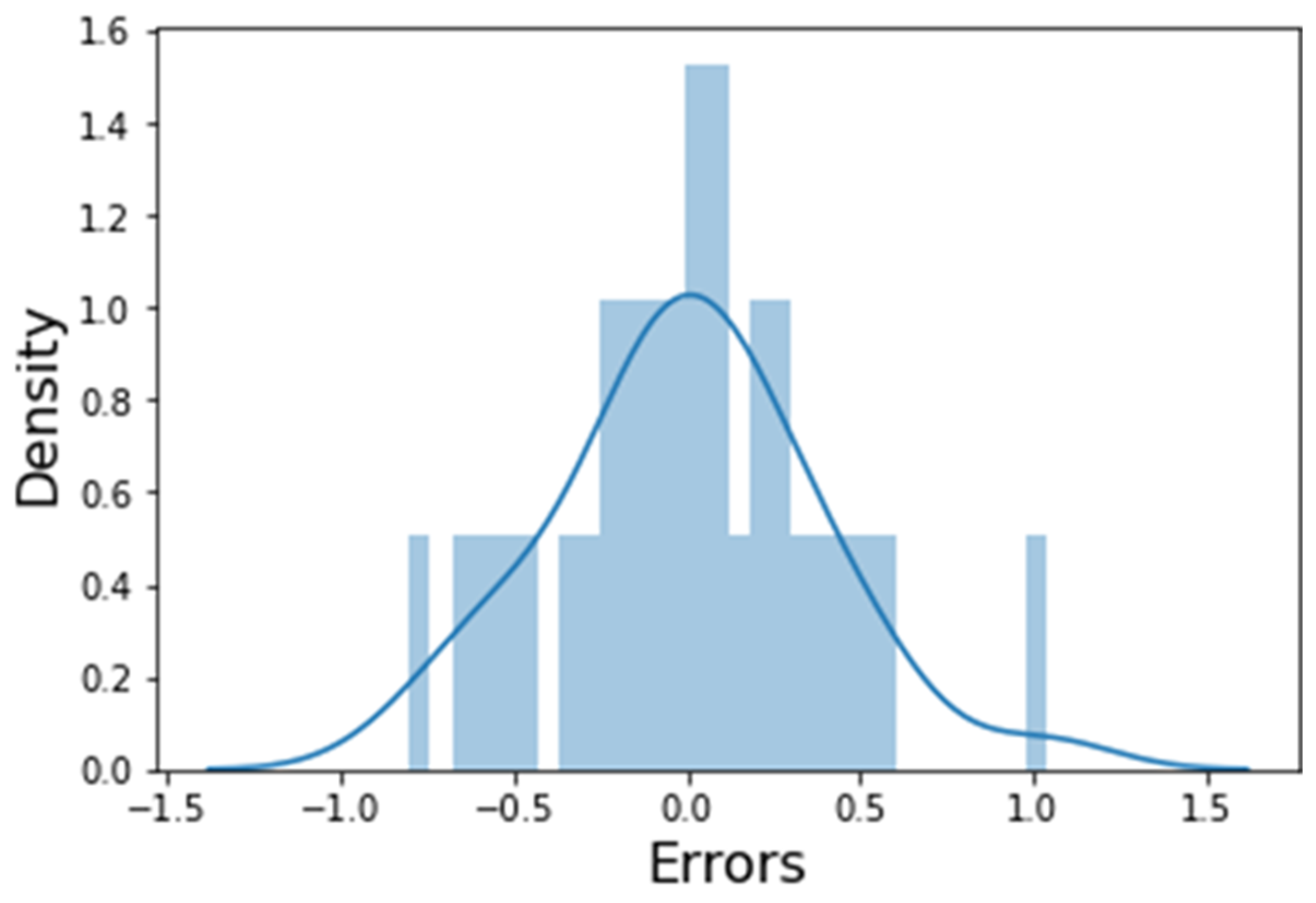
3.3. Residual Analysis of the Model
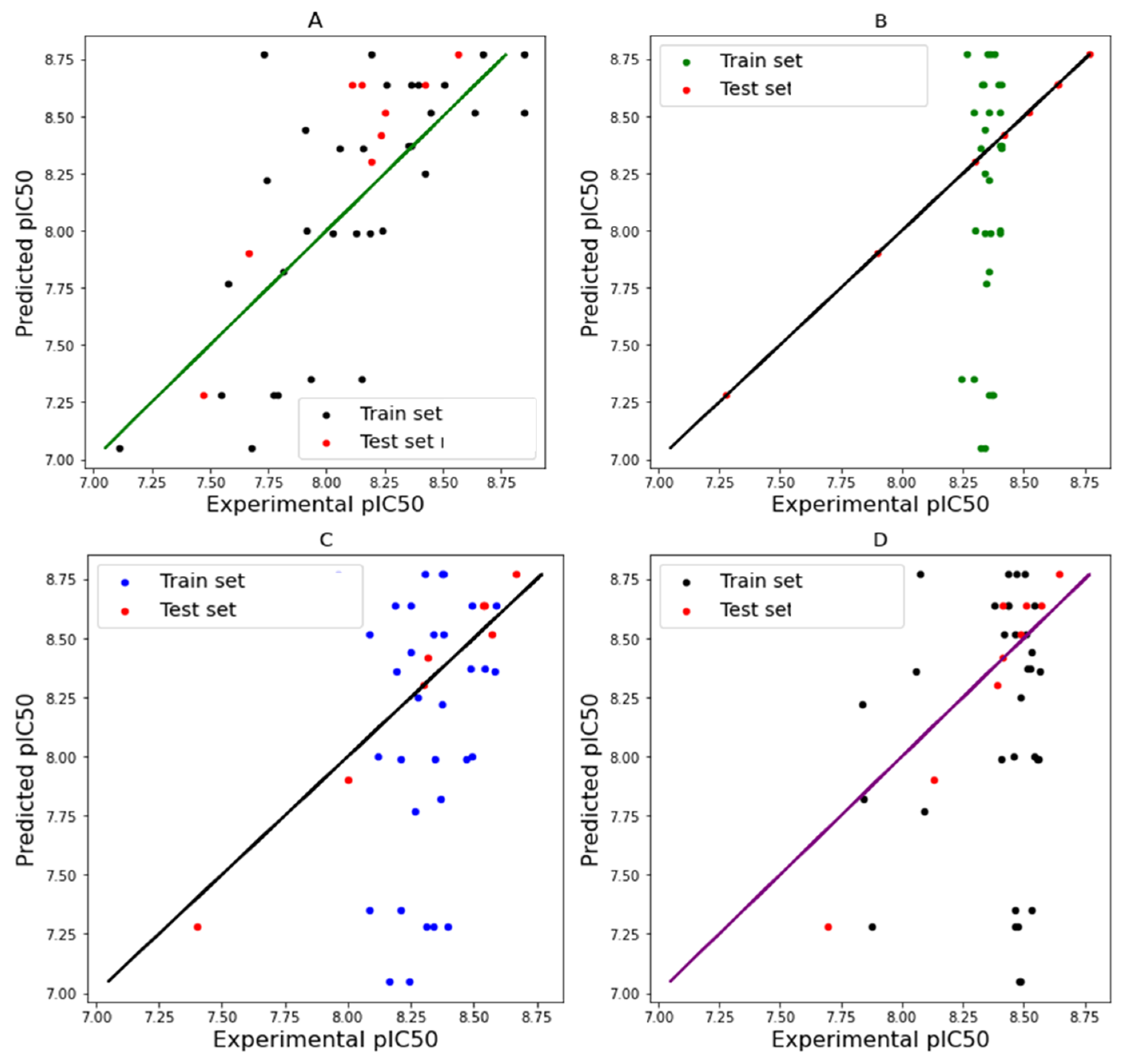
3.4. Model Building
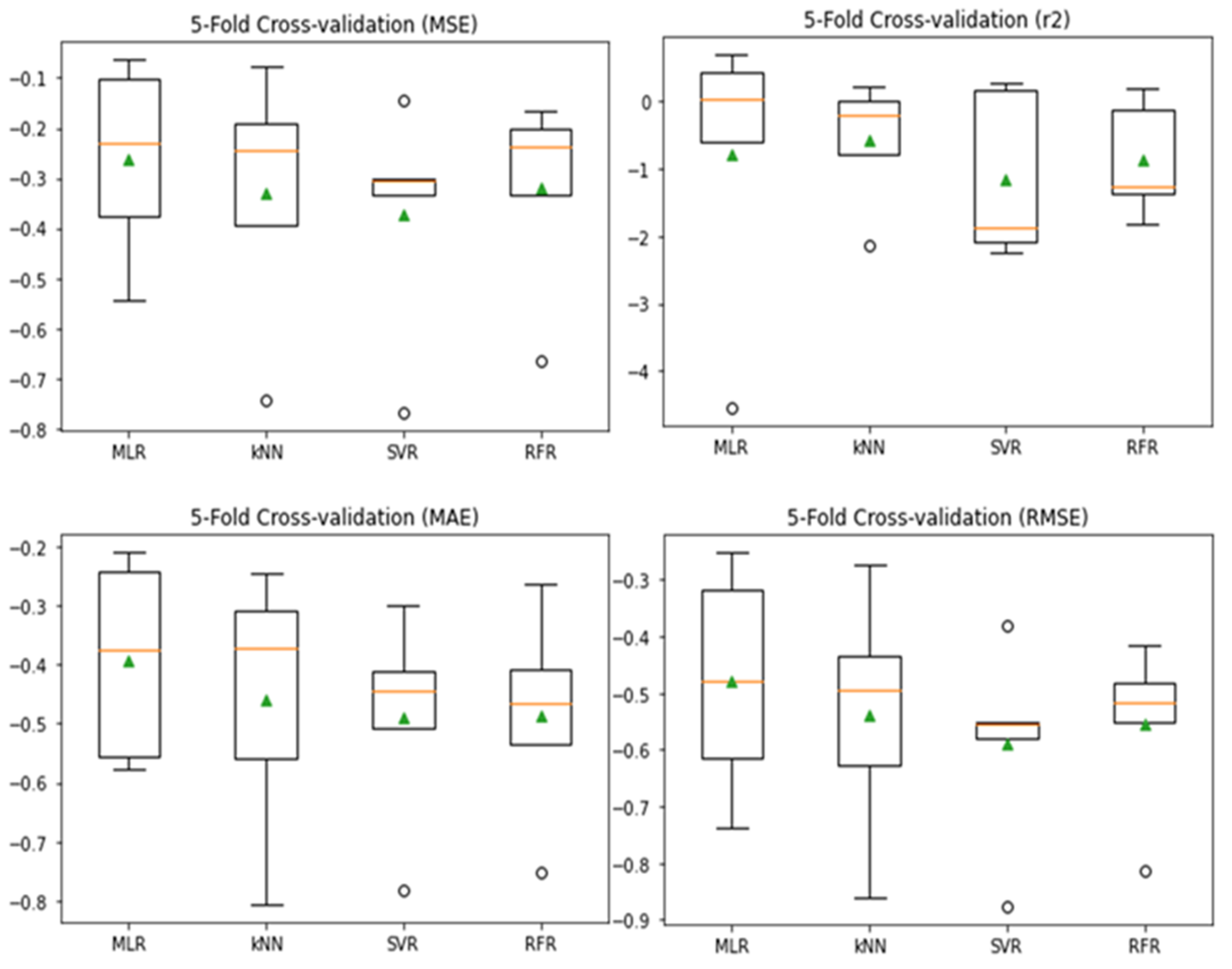
3.5. Model Evaluation and Comparison
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ikerionwu, C.; Ugwuishiwu, C.H.; Okpala, I.; James, I.; Okoronkwo, M.; Nnadi, C.; Orji, U.; Ebem, D.; Ike, A. Application of machine and deep learning algorithms in optical microscopic detection of Plasmodium: A malaria diagnostic tool for the future. Photodiagn. Photodyn. Ther. 2022, 40, 103198. [Google Scholar] [CrossRef] [PubMed]
- Oguike, E.; Ugwuishiwu, C.; Asogwa, C.; Nnadi, C.; Obonga, W.; Attama, A. A systematic review on the application of machine learning to quantitative structure-activity relationship modeling against Plasmodium falciparum. Mol. Divers. 2022, 26, 3447–3462. [Google Scholar] [CrossRef] [PubMed]
- Ikwuka, C.E.; Asogwa, C.M.; Ikwuka, O.J.; Ogbonna, J.E.; Onah, C.E.; Ohama, C.C.; Nnadi, C.O. Insights into the In-vivo Antiplasmodial Activity of Trisdimethylamino Pyrimidine Derivative in Plasmodium berghei Infected Mouse Model. J. Pharm. Res. Int. 2022, 34, 33–40. [Google Scholar] [CrossRef]
- Nnadi, C.O.; Ayoka, T.O.; Okorie, H.N. A ligand-based approach to lead optimization of N, N’-substituted diamines for leishmanicidal activity. Biointerface Res. Appl. Chem. 2022, 12, 7429–7437. [Google Scholar]
- Nnadi, C.O.; Althaus, J.B.; Nwodo, N.J.; Schmidt, T.J. A 3D-QSAR study on the antitrypanosomal and cytotoxic activities of steroid alkaloids by comparative molecular field analysis. Molecules 2018, 23, 1113. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chen, M.; Fei, Q.; Ge, Y.; Zhu, Y.; Chen, H.; Yang, M.; Ouyang, G. Synthesis and Bioactivities Study of Novel Pyridylpyrazol Amide Derivatives Containing Pyrimidine Motifs. Front. Chem. 2020, 8, 522. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group. Molecular Operating Environment (MOE) rel. 2011.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2014. [Google Scholar]
- Sydow, D.; Morger, A.; Driller, M.; Volkamer, A. TeachOpenCADD: A teaching platform for computer-aided drug design using open-source packages and data. J. Cheminform. 2019, 11, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Yang, H.; Lv, W.J.; Zhang, X.Y.; Zhai, H.L. prediction of the inhibitory concentrations of chloroquine derivatives using deep neural network models. J. Biomol. Struct. Dyn. 2021, 39, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Afuwape, A.A.; Xu, Y.; Anajemba, J.H.; Srivasta, G. Performance evaluation of secured network traffic classification using machine learning approach. Comput. Stand. Interfaces 2021, 78, 103545. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Coeff | SE | T | p-Value | 0.025–0.875 | VIF |
|---|---|---|---|---|---|---|
| Const | 8.0584 | 0.088 | 91.607 | 0.000 | 7.878–8.238 | - |
| vsurf_EDmin3 | 0.3627 | 0.271 | 1.341 | 0.190 | −0.191–0.916 | 39.46 |
| vsurf_D7 | −0.3807 | 0.266 | −1.430 | 0.163 | −0.925–0.164 | 9.16 |
| vsurf_D8 | 0.1435 | 0.255 | 0.562 | 0.578 | −0.379–0.665 | 8.42 |
| vsurf_EDmin1 | −0.3076 | 0.252 | −1.222 | 0.232 | −0.823–0.207 | 8.19 |
| FCASA- | 0.5262 | 0.159 | 3.305 | 0.003 | 0.201–0.852 | 3.28 |
| vsurf_G | 0.3665 | 0.147 | 2.495 | 0.019 | 0.066–0.667 | 2.79 |
| vsurf_HB1 | −0.3665 | 0.131 | −2.808 | 0.009 | −0.633–0.100 | 2.20 |
| E_str | −0.3877 | 0.127 | −3.044 | 0.005 | −0.648–0.127 | 2.10 |
| MNDO_LUMO | −0.3641 | 0.126 | −2.880 | 0.007 | −0.623–0.105 | 2.07 |
| vsurf_IW1 | −0.1351 | 0.123 | −1.101 | 0.280 | −0.386–0.116 | 1.94 |
| vsurf_IW2 | 0.0463 | 0.117 | 0.396 | 0.695 | −0.193–0.285 | 1.77 |
| vsurf_DD12 | −0.2716 | 0.108 | −2.514 | 0.018 | −0.493–0.051 | 1.51 |
| vsurf_Wp6 | 0.0651 | 0.101 | 0.647 | 0.523 | −0.141–0.271 | 1.31 |
| Tzpd | Actual pIC50 | Predicted pIC50 |
|---|---|---|
| 25 | 8.37 | 8.380461 |
| 8 | 8.64 | 8.667578 |
| 27 | 7.35 | 6.915064 |
| 11 | 8.64 | 8.122377 |
| 22 | 8.77 | 7.905053 |
| 14 | 8.64 | 7.908562 |
| 6 | 8.77 | 8.151523 |
| 2 | 7.28 | 7.760386 |
| 7 | 8.42 | 8.064295 |
| ML Algorithms | kNN | SVR | RFR | MLR |
|---|---|---|---|---|
| Test MSE | 0.00 | 0.053 | 0.069 | 0.1453 |
| 5-fold cross-validation | 0.59 ± 0.41 | 0.67 ± 0.45 | 0.75 ± 0.29 | 0.091 ± 0.010 |
| Test R2 | 1.00 | 0.61 | 0.36 | 0.68 |
| 5-fold cross-validation | 0.36 ± 0.46 | 0.63 ± 0.62 | 0.59 ± 2.21 | 0.745 ± 0.281 |
| Test MAE | 0.00 | 0.174 | 0.209 | 0.290 |
| 5-fold cross-validation | 0.55 ± 0.18 | 0.58 ± 0.20 | 0.60 ± 0.60 | 0.270 ± 0.101 |
| Test RMSE | 0.00 | 0.230 | 0.262 | 0.381 |
| 5-fold cross-validation | 0.72 ± 0.27 | 0.77 ± 0.27 | 0.84 ± 0.18 | 0.302 ± 0.021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umoette, K.S.; Nnadi, C.O.; Obonga, W.O. A Robust Regression-Based Modeling to Predict Antiplasmodial Activity of Thiazolyl–Pyrimidine Hybrid Derivatives against Plasmodium falciparum. Chem. Proc. 2023, 14, 52. https://doi.org/10.3390/ecsoc-27-16167
Umoette KS, Nnadi CO, Obonga WO. A Robust Regression-Based Modeling to Predict Antiplasmodial Activity of Thiazolyl–Pyrimidine Hybrid Derivatives against Plasmodium falciparum. Chemistry Proceedings. 2023; 14(1):52. https://doi.org/10.3390/ecsoc-27-16167
Chicago/Turabian StyleUmoette, Kevin S., Charles O. Nnadi, and Wilfred O. Obonga. 2023. "A Robust Regression-Based Modeling to Predict Antiplasmodial Activity of Thiazolyl–Pyrimidine Hybrid Derivatives against Plasmodium falciparum" Chemistry Proceedings 14, no. 1: 52. https://doi.org/10.3390/ecsoc-27-16167
APA StyleUmoette, K. S., Nnadi, C. O., & Obonga, W. O. (2023). A Robust Regression-Based Modeling to Predict Antiplasmodial Activity of Thiazolyl–Pyrimidine Hybrid Derivatives against Plasmodium falciparum. Chemistry Proceedings, 14(1), 52. https://doi.org/10.3390/ecsoc-27-16167