
Pyrene-4,5,9,10-Tetrachalcogenone Derivatives: A Computational Study on Their Potential Use as Materials for Batteries †

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miao, L.; Liu, L.; Shang, Z.; Li, Y.; Lu, Y.; Cheng, F.; Chen, J. The structure–electrochemical property relationship of quinone electrodes for lithium-ion batteries. Phys. Chem. Chem. Phys. 2018, 20, 13478–13484. [Google Scholar] [CrossRef]
- Armand, M.; Tarascon, J.M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Sun, X.G.; Dai, S. Organic Cathode Materials for Lithium-Ion Batteries: Past, Present, and Future. Adv. Energy Sustainability Res. 2021, 2, 2000044. [Google Scholar] [CrossRef]
- Zhu, Z.; Hong, M.; Guo, D.; Shi, J.; Tao, Z.; Chen, J. All-Solid-State Lithium Organic Battery with Composite Polymer Electrolyte and Pillar [5]quinone Cathode. J. Am. Chem. Soc. 2014, 136, 16461–16464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Y.; Huang, W.; Zhang, Q. Recent Progress in Calix[n]quinone (n = 4, 6) and Pillar [5]quinone Electrodes for Secondary Rechargeable Batteries. Batter. Supercaps 2020, 3, 476–487. [Google Scholar] [CrossRef]
- Yoo, G.; Pyo, S.; Gong, Y.J.; Cho, J.; Kim, H.; Kim, Y.S.; Yoo, J.J.C. Highly reliable quinone-based cathodes and cellulose nanofiber separators: Toward eco-friendly organic lithium batteries. Cellulose 2020, 27, 6707–6717. [Google Scholar] [CrossRef]
- Shi, J.L.; Xiang, S.Q.; Su, D.J.; He, R.X.; Zhao, L.B. Revealing practical specific capacity and carbonyl utilization of multi-carbonyl compounds for organic cathode materials. Phys. Chem. Chem. Phys. 2021, 23, 13159–13169. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wang, M.; Dong, X.; Escobar, I.C.; Cheng, Y.-T. Lithium Ion Battery Electrodes Made Using Dimethyl Sulfoxide(DMSO) A Green Solvent. ACS Sustain. Chem. Eng. 2020, 8, 11046–11051. [Google Scholar] [CrossRef]
- Wang, D.; Huang, S.-P.; Wang, C.; Yue, Y.; Zhang, Q.-S. Computational Prediction for Oxidation and Reduction Potentials of Organic Molecules Used in Organic Light-Emitting Diodes. Org. Electron. 2019, 64, 216–222. [Google Scholar] [CrossRef]
- Shoaib, M.; Bibi, S.; Ullah, I.; Jamil, S.; Iqbal, J.; Alam, A.; Saeed, U.; Bai, F.Q. Theoretical Investigation of Perylene Diimide derivatives as Acceptors to Match with Benzodithiophene based Donors for Organic Photovoltaic Devices. Z. Phys. Chem. 2021, 235, 427–449. [Google Scholar] [CrossRef]
- Ji, L.-F.; Fan, J.-X.; Zhang, S.-F.; Ren, A.-M. Theoretical investigations into the charge transfer properties of thiophene α-substituted naphthodithiophene diimides: Excellent n-channel and ambipolar organic semiconductors. Phys. Chem. Chem. Phys. 2017, 19, 13978–13993. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.H. One-Electron and Two-Electron Transfers in Electrochemistry and Homogeneous Solution Reactions. Chem. Rev. 2008, 108, 2113–2144. [Google Scholar] [CrossRef] [PubMed]
- Organic Chemistry Portal. Available online: https://www.organic-chemistry.org/synthesis/C2S/thioketones.shtm (accessed on 17 October 2022).
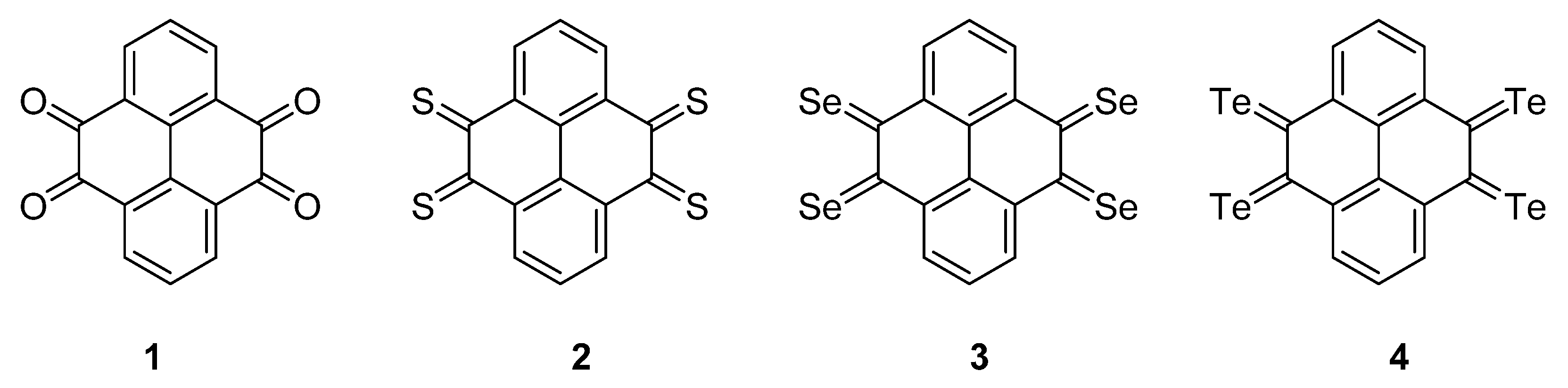

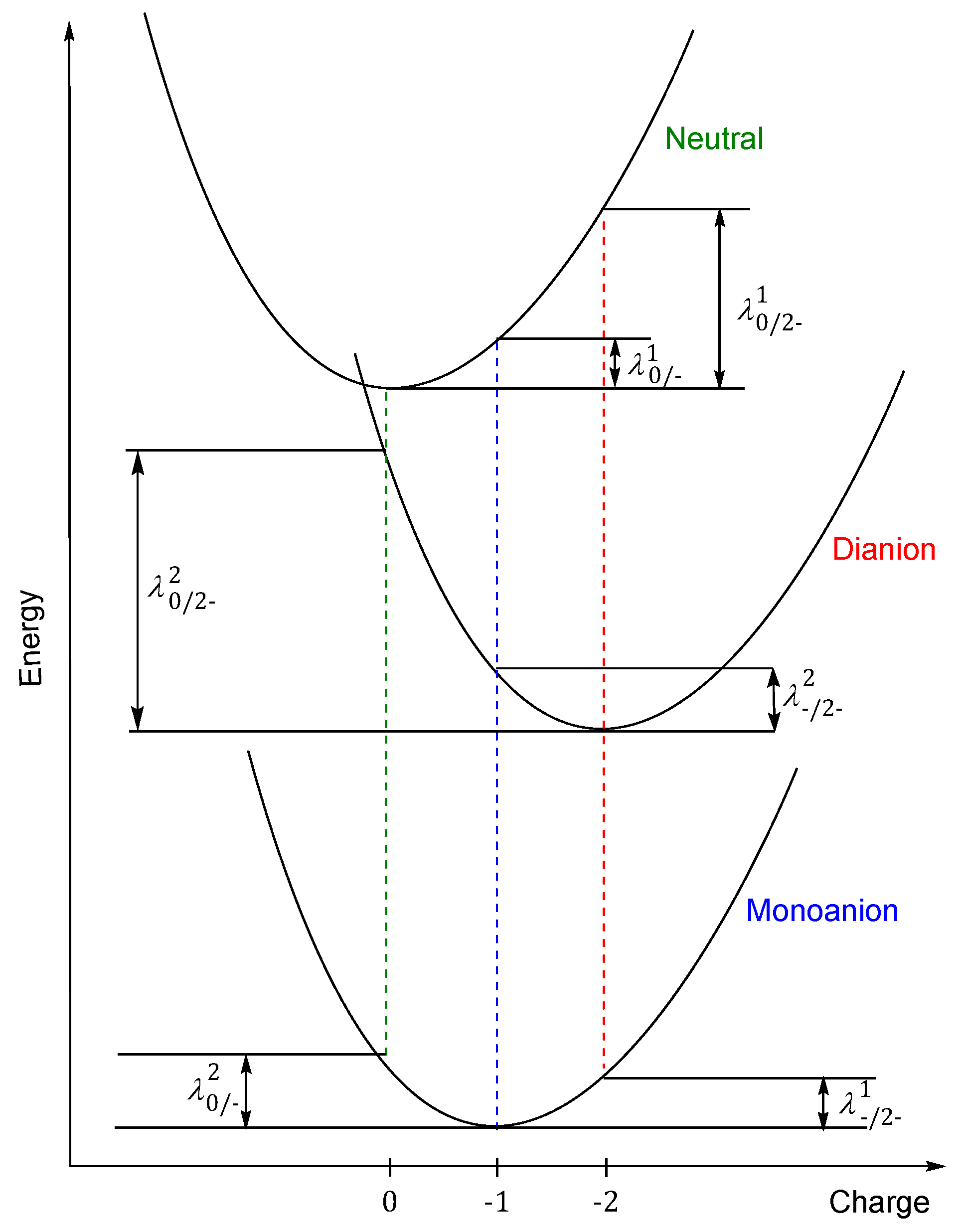
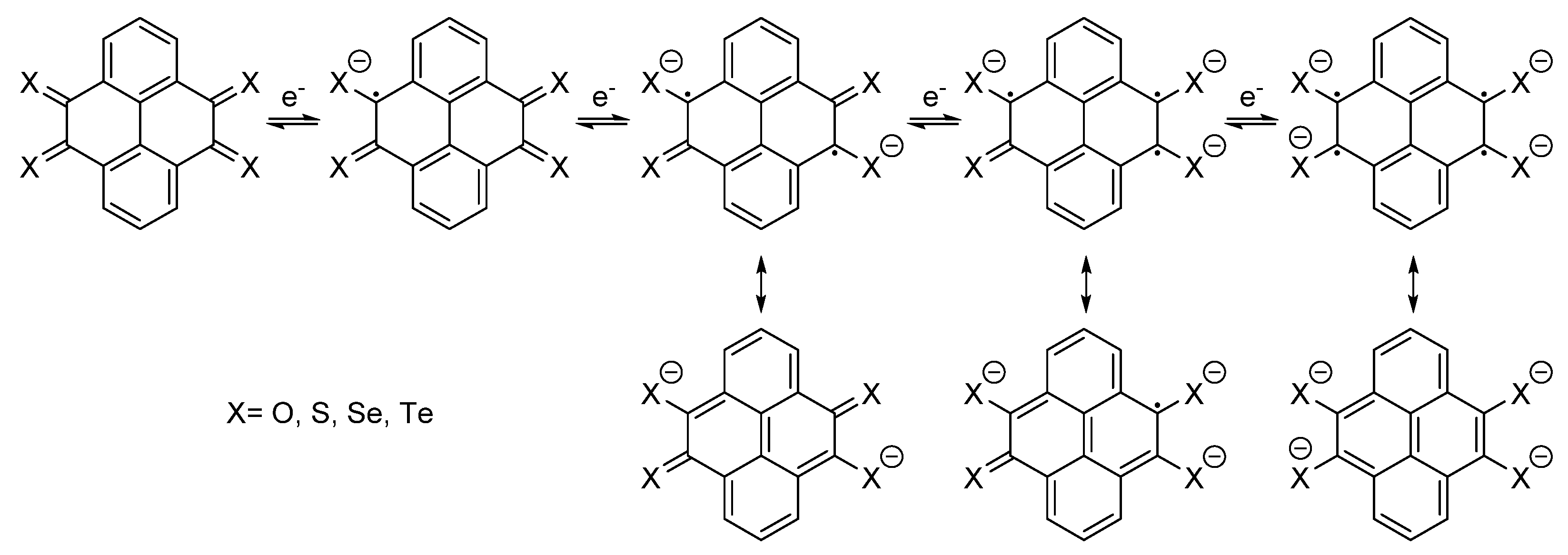
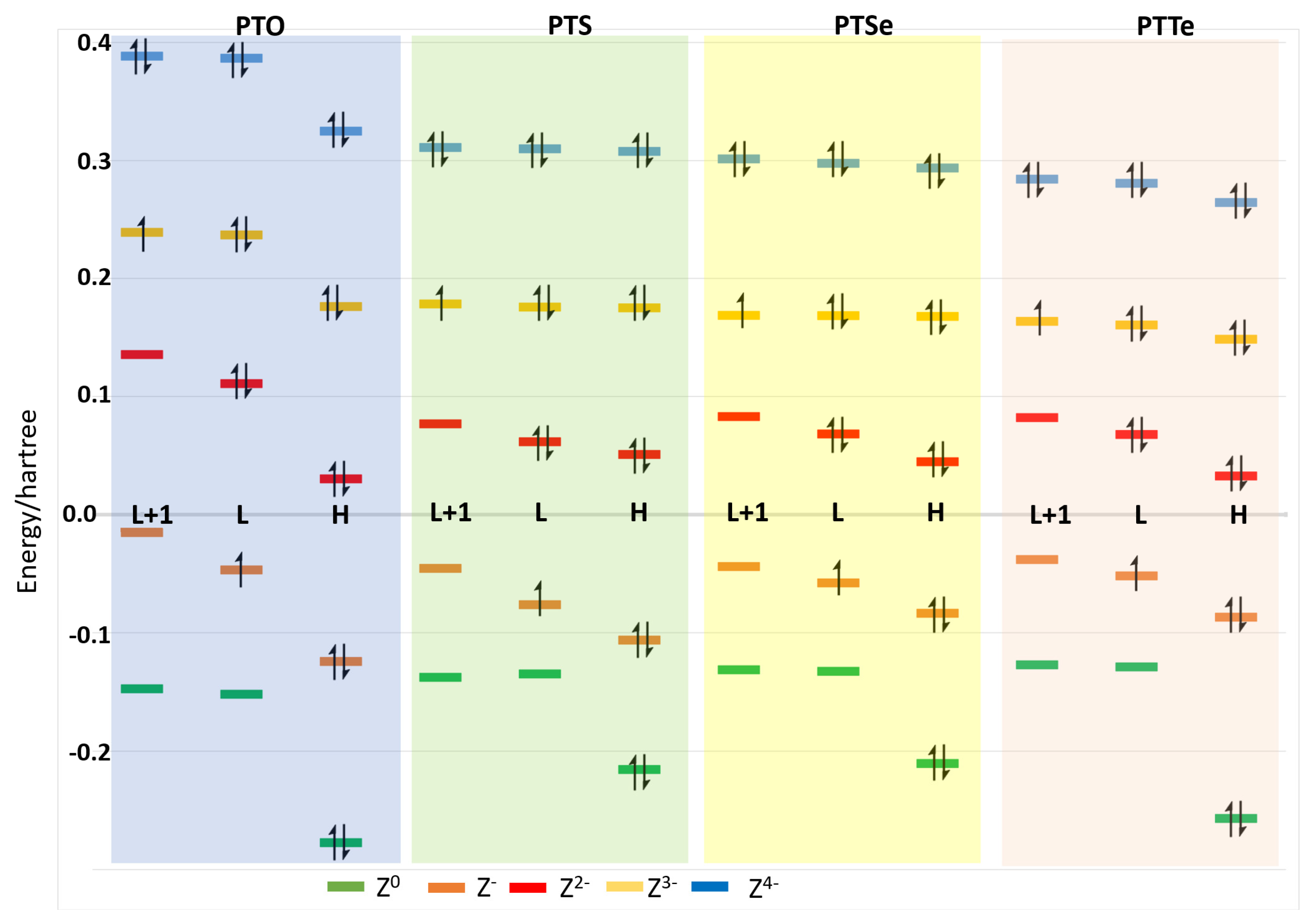
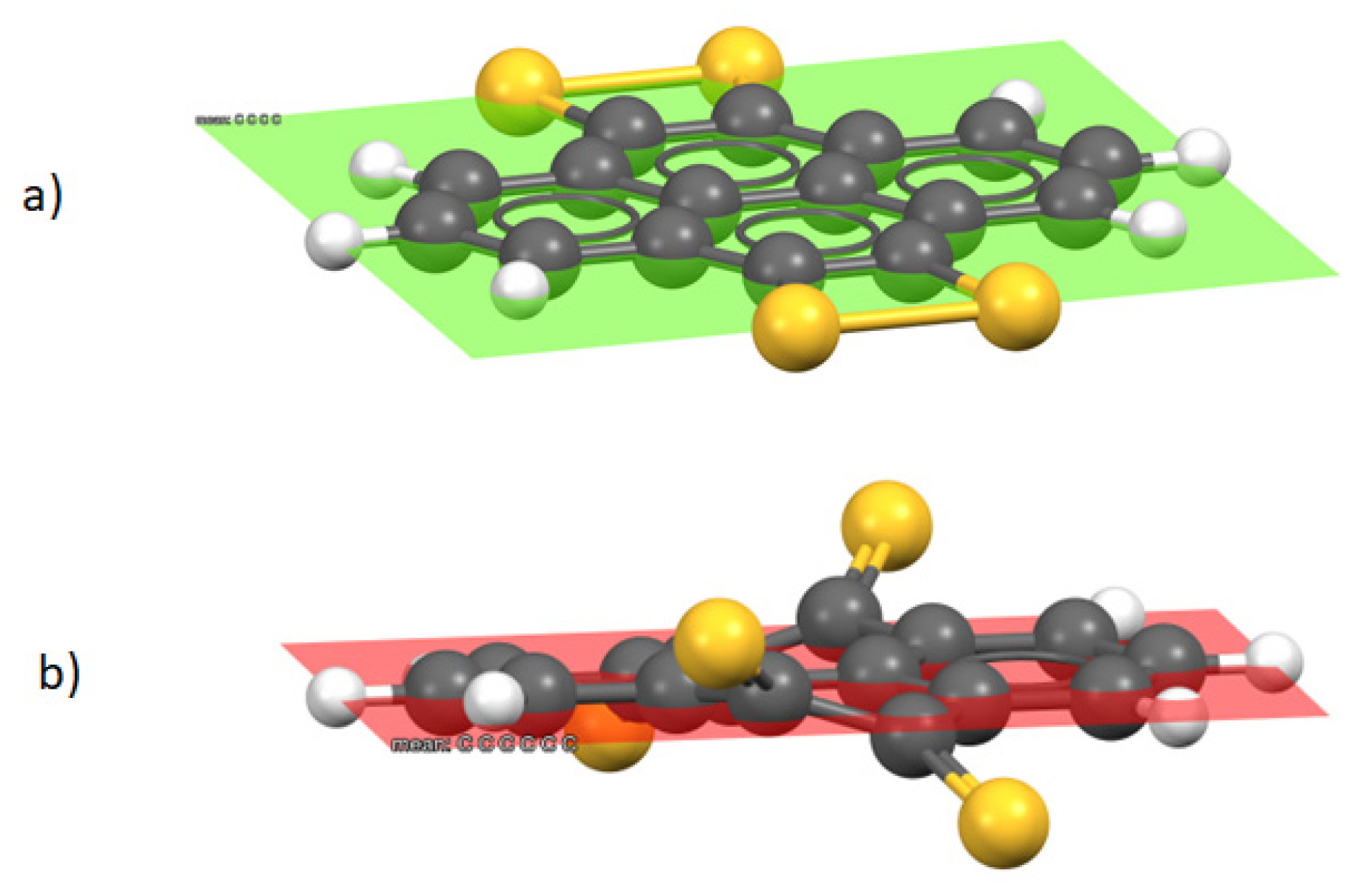
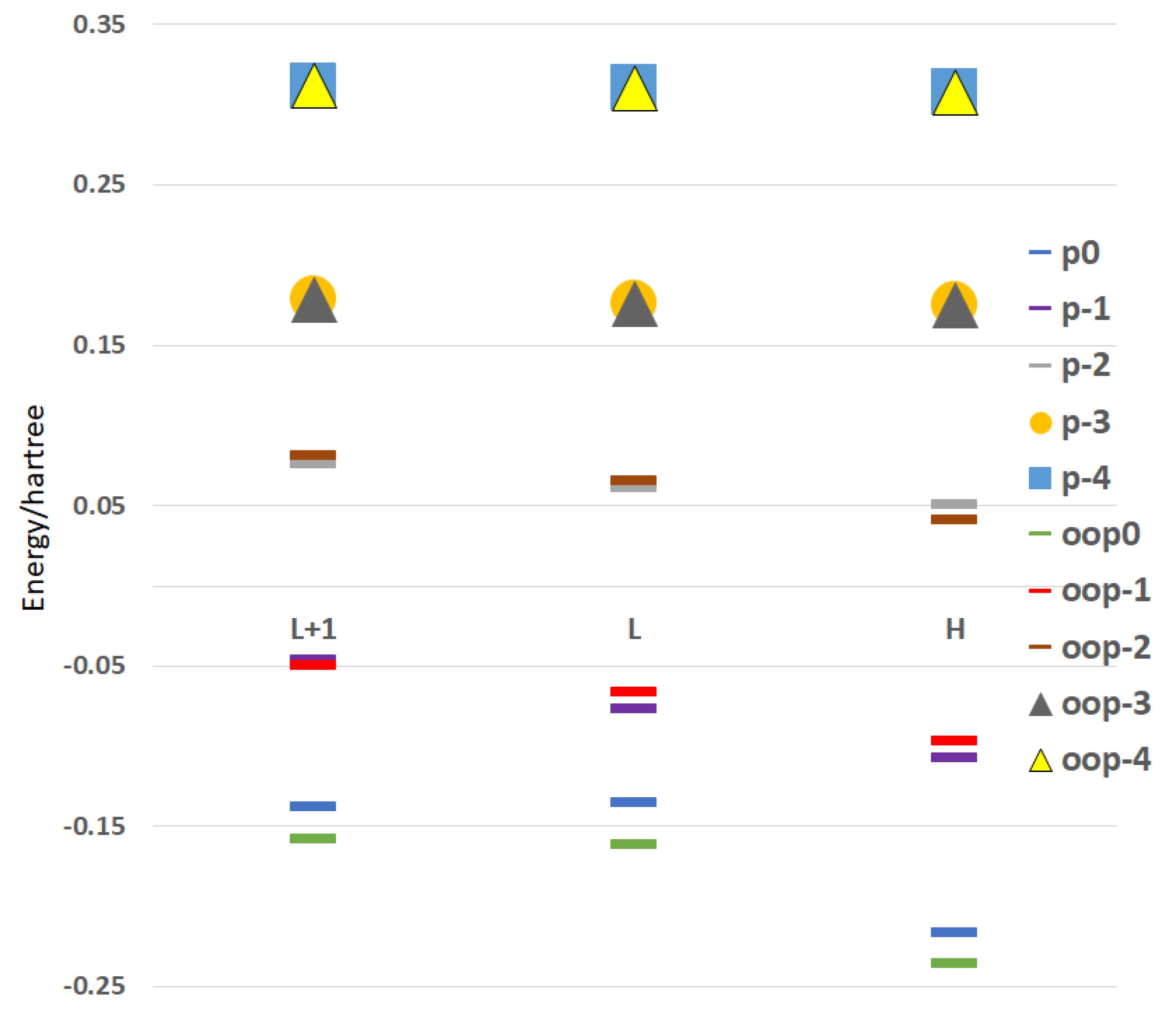

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PTO | PTS | PTSe | PTTe |
|---|---|---|---|---|
| [Z]0/− | −4.35 | −4.18 | −4.17 | −6.50 |
| [Z]0/2− | −7.55 | −9.42 | −7.77 | −9.69 |
| [Z]0/3− | −10.61 | −12.91 | −11.88 | −13.49 |
| [Z]0/4− | −12.58 | −16.11 | −14.96 | −16.46 |
| 0.14 | 2.22 | 0.87 | 0.58 | |
| 0.99 | 3.51 | 3.13 | 2.12 | |
| 1.80 | 4.58 | 5.39 | 4.32 | |
| 2.92 | 8.17 | 8.70 | 7.04 |
| Compound | PTS | Out-of-Plane PTS |
|---|---|---|
| [Z]0/− | −4.18 | −4.59 |
| [Z]0/2− | −9.42 | −8.54 |
| [Z]0/3− | −12.91 | −12.61 |
| [Z]0/4− | −16.11 | −15.91 |
| 2.22 | 0.26 | |
| 3.51 | 0.91 | |
| 4.58 | 1.92 | |
| 8.17 | 3.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Tato, M.P.; Meijide, F.; Fraga, F.; Tato, J.V.; Seijas, J.A. Pyrene-4,5,9,10-Tetrachalcogenone Derivatives: A Computational Study on Their Potential Use as Materials for Batteries. Chem. Proc. 2022, 12, 76. https://doi.org/10.3390/ecsoc-26-13554
Vázquez-Tato MP, Meijide F, Fraga F, Tato JV, Seijas JA. Pyrene-4,5,9,10-Tetrachalcogenone Derivatives: A Computational Study on Their Potential Use as Materials for Batteries. Chemistry Proceedings. 2022; 12(1):76. https://doi.org/10.3390/ecsoc-26-13554
Chicago/Turabian StyleVázquez-Tato, M. Pilar, Francisco Meijide, Francisco Fraga, José Vázquez Tato, and Julio A. Seijas. 2022. "Pyrene-4,5,9,10-Tetrachalcogenone Derivatives: A Computational Study on Their Potential Use as Materials for Batteries" Chemistry Proceedings 12, no. 1: 76. https://doi.org/10.3390/ecsoc-26-13554
APA StyleVázquez-Tato, M. P., Meijide, F., Fraga, F., Tato, J. V., & Seijas, J. A. (2022). Pyrene-4,5,9,10-Tetrachalcogenone Derivatives: A Computational Study on Their Potential Use as Materials for Batteries. Chemistry Proceedings, 12(1), 76. https://doi.org/10.3390/ecsoc-26-13554