Abstract
Nocturia is a common disease in patients with type 2 diabetes mellitus that can reduce the quality of life. Sodium glucose co-transporter 2 (SGLT2) inhibitors increase the urine volume and are often discontinued when polyuria occurs, although tofogliflozin, which has a short half-life in the blood, may improve nocturia by managing hyperglycemia and hypertension, without aggravating nocturia. As excessive sodium intake worsens nocturia and increases urine volume, sodium restriction is also effective in managing nocturia. This multicenter, open-label, randomized parallel-group trial will examine 80 patients with type 2 diabetes who experienced nocturia. After the baseline examination, the patients are randomly stratified into two groups and receive tofogliflozin treatment with or without sodium restriction for 12 weeks. The primary outcome is nocturia frequency at 12 weeks. The secondary outcomes are the frequency of daytime urine, changes in urine volume, and changes in home blood pressure.
1. Introduction
1.1. Background and Rationale
Nocturia is a condition of waking up at night to urinate one or more times during the night [1]. The frequency of nocturia in Japan increases with age, reaching 83.8% in men and 76.6% in women in their 60s [2]. Nocturia is caused by several factors, including nocturnal polyuria, cystourethral disturbance, sleep disturbance, and cardiovascular disease, and either one or a combination of these factors may be involved. In addition to age, diabetes mellitus, hypertension, stroke, heart disease, and obesity have been associated with nocturia [3,4,5,6,7,8]. Approximately 40% of patients with diabetes arise more than twice at night to void [9]. In our KAMOGAWA-DM cohort study [10], according to a questionnaire survey on the frequency of nocturia conducted in 396 patients, 80% had nocturia more than once, while 40% had nocturia more than twice. Nocturia is a common condition in patients with diabetes.
In addition, Hashimoto et al. [11] reported that sleep disorders were a major cause of poorer quality of life (QOL) in Japanese patients with type 2 diabetes (T2D), and more than half of them were frequently awakened by the urge to void. Nocturia is a risk factor for fractures and decreases the survival rate [12].
Sodium glucose co-transporter 2 (SGLT2) inhibitors can improve glycemic control, glycemic variability, and fat loss, as well as prevent heart and renal failure [13,14]. How-ever, concerns have been raised regarding the adverse events of SGLT2 inhibitors, not only dehydration and urinary tract infection, but also polyuria [15]. In fact, frequent uri-nation and polyuria have been the primary reasons for the discontinuation of SGLT2 inhibitor treatment [16].
One SGLT2 inhibitor, tofogliflozin, which has a short half-life, promotes urinary glu-cose excretion during daytime without worsening nocturia [17] and may improve hyper-glycemia and hypertension. As sodium retention exacerbates nocturia by causing non-dipping nocturnal hypertension, SGLT2 inhibitors with a short half-life could im-prove nocturia by increasing the daytime excretion of sodium [18].
Therefore, to correct and prevent the worsening of nocturia, not only medication but also restriction of dietary salt intake should be considered. Reduction in sodium intake can decrease the frequency of nocturia, nocturnal urine volume, and nocturnal polyuria index; patients with nocturia accompanied by nocturnal polyuria should control their sodium intake.
The observational study in Japan, nocturia volume, the frequency of nocturia, and nocturnal polyuria index were evaluated after 12 weeks of dietary guidance in patients who experienced one episode of nocturia, exceeding the maximum daily salt intake (8 g for men and 7 g for women). This study reported that nocturia frequency, urine volume, and nocturnal polyuria index improved in the group of participants who had been successful in restricting salt intake [19,20].
As mentioned above, various factors constitute nocturia, such as nocturnal hypergly- cemia, excessive salt intake, and nocturnal hypertension; in particular, non-dipper-type nocturnal hypertension and heart failure are the risk factors for nocturia [21,22,23].
Nocturia should be managed with both medication and salt intake restriction in order to correct and prevent the exacerbation of nocturia; however, no study has evaluated the effect of a combination of salt intake restriction and SGLT2 inhibitor treatment on nocturia. It would be significant to examine the effect of tofogliflozin, an SGLT2 inhibitor with the shortest half-life, on nocturia in patients with T2D, and whether tofogliflozin treatment could be effective in combination with sodium restriction.
1.2. Objectives
1.2.1. Primary Objectives
The primary aim of this study is to evaluate the effect of tofogliflozin on nocturia in patients with T2D and to examine the efficacy of tofogliflozin with or without dietary sodium restriction on nocturia at 12 weeks after interventions.
1.2.2. Secondary Objectives
The secondary aim of this study is to assess below items at 12 weeks after interventions.
- Frequency of urination during the day
- Change in ratio of urinary volume at night to 24 h
- Change in urine volume at night
- Change in urine volume during the day
- Change in total urinary sodium excretion and other urinalysis
- Change in the results of blood tests
- Change in home blood pressure at night
- Change in body composition test
- Change in questionnaire score (The Diabetes Treatment Satisfaction Questionnaire, status version, DTSQs score; Diabetes Diet-Related Quality of Life Revised, DDRQOL-R; Brief-type Self-administered Diet History Questionnaire, BDHQ; Core Lower Urinary Tract Symptoms score, CLSS)
- Incidence of adverse events and diseases
2. Methods
2.1. Trial Design
This is a multicenter, open-label, randomized parallel group trial [Efficacy of dual therapy of TOfogliflozin and dietary instruction of sodium restriction in T2D patients with nocturia: A multicenter open-label randomized controlled superiority trial of the availability to reduce nocturnal urination frequency (TOP-STAR Study)] (Figure 1). Eighty patients with T2D and nocturia will be included in the study and randomly stratified into two groups.
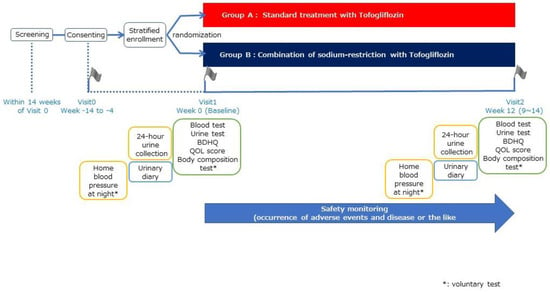
Figure 1.
Study design of TOP-STAR study.
2.2. Eligibility Criteria, Recruitment, and Sample Size
Inclusion and exclusion criteria were established according to the study objectives and to ensure participant safety (Table 1).

Table 1.
Inclusion and exclusion criteria.
This study will be conducted in 18 research institutions. In the previous study [20], the frequency of nocturia improved from 2.3 ± 0.9 times per day at baseline to 1.4 ± 1.0 times per day after 12 weeks in the group of participants who were successful in restricting salt intake. In the group of participants who were not successful in restricting salt intake, the frequency of nocturia did not improve (from 2.3 ± 1.1 days at baseline to 2.7 ± 1.1 days after 12 weeks). In this study, the frequency of nocturia was 2.3 ± 1.0 days in both groups at baseline, it remained unchanged (2.3 ± 1.0 days) in the tofogliflozin monotherapy group, and it was 1.3 ± 1.0 days in the combination group that received sodium restriction guidance after 12 weeks. Under these conditions and at a significance level of 5% in both groups, 10,000 tests were required to determine the differences in nocturia frequency between the groups after 12 weeks as shown in the results of Poisson regression analysis using data assuming a Poisson distribution. Therefore, the minimum number of patients required for this study was 36 patients per group. In addition, assuming a dropout rate of 10% during the study period, the target number of participants to be enrolled in this study was 40 patients per group or 80 patients in two groups.
2.3. Interventions
2.3.1. Random Grouping and Intervention Description
The participants are randomly assigned in different groups [Group A: standard treatment group]. Randomisation will be provided by a computer-generated program at the EviPRO Holdings Inc. (Tokyo, Japan). The study participants are administered with tofogliflozin 20 mg orally once a day before or after breakfast. The duration of tofogliflozin administration is 12 weeks (9–14 weeks) [Group B: group with dietary sodium restriction]. In addition to the oral administration of 20 mg of tofogliflozin once a day before or after breakfast, a nutritionist provides instructions on sodium restriction. The duration of tofogliflozin treatment is 12 weeks (9–14 weeks). SGLT2 inhibitor treatment is initiated on day 0 of the observation period. The study participants are requested to visit the research institutions at weeks 0 and 12, in addition to the date of obtaining consent.
2.3.2. Details of Instruction on Sodium Restriction
The nutritionist provided dietary instructions, with a target sodium intake of 6 g/day. In addition, for both groups, the target weight and amount of energy and protein in the diet were based on the nutritional guidelines that the patients received.
2.3.3. Details of Other Dietary Instruction
In both groups, the patients are instructed to stay properly hydrated to prevent dehydration (avoid excessive drinking or excessive water conservation).
Energy level: 25–35 kcal/kg/day for physical activity multiplied by the target weight.
Protein restriction: 0.8–1.0 g/kg/day (stage 3 diabetic nephropathy) and 0.6–0.8 g/kg/day (stage 4 diabetic nephropathy).
2.3.4. Observation Items
A baseline examination will be conducted prior to the intervention. The observations and schedules are presented in Table 2 and Table 3. Generally, research participants will visit the research institution and undergo blood and urine tests at every visit.
Table 2.
Observation items.
Table 3.
Observation schedule.
Additionally, nocturnal home blood pressure measurements will be taken three times a day for five days. Next, the urinary frequency will be recorded for 7 days, and urine volume will be measured using a measuring cup for 3 days. In addition to the blood and urine tests, scores on the Japanese version of the Diabetes Treatment Satisfaction Questionnaire, status version (DTSQs) score, Diabetes Diet-Related Quality of Life Revised (DDRQOL-R), Brief-type Self-administered Diet History Questionnaire (BDHQ), and Core Lower Urinary Tract Symptoms score (CLSS) will be assessed using a patient questionnaire. DTSQs are questionnaires used for measuring the patients’ satisfaction to treatments specific for diabetes mellitus, are implemented worldwide, and consists of 8 questions [24]. DDRQOL-R consists of nine items. It is used to determine the diabetic patients’ level of satisfaction regarding their diet and quality of life in relation to the changes in their dietary habits [25]. BDHQ designed to quantitatively and precisely examine the status of nutrients and food intake [26]. CLSS is a 10-item questionnaire developed in Japan to investigate significant urinary tract symptoms [27].
2.4. Criteria for Discontinuing or Modifying Allocated Interventions
2.4.1. Criteria and Coping Strategies for Study Discontinuation
If the investigator judges that it is difficult to continue the clinical trial for any of the following reasons, the investigator will immediately take necessary measures such as discontinuing the administration of the study drug. Patients’ data will be used as data of a “study discontinuation case.” The investigator will note the date, when the study started, the reason for withdrawal, and the process on the card and on the case report form (CRF). At the time of discontinuation, the necessary tests will be conducted. The efficacy and safety the procedure will be assessed at this point. Moreover, investigators will evaluate the efficacy and safety of the treatment, following up on the endpoints and analyzing the safety of the medical treatment received.
2.4.2. Criteria for Discontinuation of Study in Each Participant
(1) A study participant voluntarily withdraws from the study or withdraws her or his consent.
(2) Discontinuation of the study is required due to the occurrence of adverse events and diseases.
(3) The continuous use of the study agent worsens the primary disease or causes complications.
(4) Patients have remarkably poor adherence with medication or sodium intake restrictions (the medication rate is estimated to be less than 60% or higher than 120% of the expected dosage).
(5) The study participant is found pregnant.
(6) A serious deviation from the research protocol occurs, which is judged to have a significant impact on the research results.
(7) The investigators have decided that the discontinuation of the study is appropriate due to other reasons.
2.4.3. Criteria for Discontinuation of Study
(1) When continuation of the study is difficult for any of the following reasons, the principal investigator will determine whether the study could be continued or not. When it is determined that continuation is inappropriate, the principal investigator shall inform the principal investigators of all collaborating institutions of the reasons for the discontinuation and how to deal with the participants, and have them take the necessary actions. The principal investigator shall inform the accreditation review committee in written form of the discontinuation of the study.
(1-1) Significant information regarding the efficacy, safety and quality of the study agent was obtained.
(1-2) Participant recruitment and the planned number of study participants were difficult to achieve.
(1-3) Protocol modification was instructed but was not executed.
(2) When discontinuing a study, the investigator should immediately discontinue the study and report the decision to the president of his/her institution. The principal investigator must also take appropriate action promptly and notify the participant of the decision to discontinue the study.
2.4.4. Coping with Adverse Events
SGLT2 inhibitors can cause dehydration, urinary tract infections, normoglycemic ketoacidosis, and polyuria, and we carefully explain the possibility of these side effects at the recruitment of previous study. If a study participant suffers an adverse event during the study which may or may not be attributable to the SGLT2 inhibitor, the investigator will promptly take appropriate medical treatment. Investigators should report the adverse event to the responsible investigator and director of the institution and document the necessary information on the medical carte and CRF according to the study protocol. If it is necessary to interrupt the administration of study drug or medical treatment due to an adverse event, the study participant should be briefed on how to manage serious adverse events [SAEs].
2.5. Deviation from the Protocol
The investigator shall document in the carte and CRF any deviation or modification from the study protocol that is necessary to avoid immediate risk to study participants or for other compelling medical reasons, and the details and reasons for such deviation or modification shall be stated in the Carte and CRF. Study participants will be followed up throughout the study. If the investigator is unable to follow the protocol exactly, he/she should continue to collect sufficient information for the study. Data handling will be determined by the data handling committee in a blinded situation.
2.6. Management of Incompatibility
Incompatibility refers to non-compliance with legal regulations or operational protocols, fabrication and falsification of test data. The management of incompatibility shall be performed as follows:
- When the responsible investigator becomes aware of the incompatibility of present study, the responsible investigator must immediately report this fact to the principal investigator.
- When the investigator becomes aware of the non-conformity of the study, the investigator immediately reports this fact to the responsible investigator.
- When there are serious incompatibilities, the investigator must immediately ask the accreditation review committee members for their opinions.
2.7. Strategies to Improve Adherence to Interventions
2.7.1. Management of the Study Agent
No placebo will be used in this study. Both groups will use commercially available, approved drugs for the study. Additionally, this is an open-label study. Therefore, specific management of the study agent is not conducted, and the study agent is managed in the same manner as general drugs.
2.7.2. Outcomes
The investigators collect and enter the results of the examinations list in Table 2 in the CRF and send the CRF to the data center. Adverse events are considered as safety endpoints throughout the study. The items measured by the study participants themselves are recorded in specific documents and sent to the data center by the investigators.
2.8. Data Collection and Management
2.8.1. Plans for Assessment and Collection of Outcomes
Original Documents
The research institution preserves and manages the following information as original source documents, and responds to monitoring, audits, and certifying review committees’ requests.
- Original documents for all data items (medical data, nurse records, drug records, laboratory data, subject logbooks, CRFs, QOL questionnaires, etc.).
- Records of informed consent indicating the patient’s agreement to the study participation.
2.8.2. Documents to Be Served as Source Documents
In addition to the original source documents, the research institution preserves and controls the information described below as source documents and provides them to the monitoring, accreditation review committees, and audit.
- Withdrawal of consent form.
- Medication adherence information.
- Adverse event and disease information.
2.8.3. Data Management and Confidentiality
Central registry numbers will be used to identify study participants. When electronic data on subjects are transferred, the consent of the data management must be obtained. If data are transferred from an unsecured electronic network, encryption of the data must be performed at the source. If the data center needs to provide participant data to other research institutions, approval from the principal investigator and data management is required. Plan for collection, laboratory evaluation, and preservation of biospecimens for genetic or molecular analysis for the study/future use.
All involved individuals in this study are required to protect the personal information of the study participants. We will conduct this study in compliance with the Personal Information Protection Act and other applicable laws and regulations. Study participant’s unique information (medical record number, initials) will be securely contained within the research institution, and information that would allow someone external to the research organization to recognize the study participant (name, phone number, address, etc.) will not be included in the CRF or registry database.
The researcher will use the correspondence table to identify the research participant (anonymization) and will maintain it privately. Correspondence sheets will be securely stored and appropriately managed by the researcher for the retention period specified by the Clinical Trials Act (until the day after five years have elapsed from the date of study completion) or the retention period specified by each research institute, whichever is later. Anonymized data acquired for analysis will be preserved for any future secondary research, such as meta-analyses. Prior approval from the Ethics Review Committee is required before anonymized data will be used for further research.
Specific blood test samples will be assayed in laboratories made available by each research organization and will be disposed of after data acquisition under the responsibility and procedures of the respective companies.
2.9. Patient and Public Involvement Statement
Patients will not be included in the research design, selection of the study questions, or measurement of the results. No participants will be included in the analysis or publication of the results. Patients will receive a brief summary of the study results written in Japanese after the completion of the study.
2.10. Statistical Methods
2.10.1. Analysis of the Primary Endpoint
The primary and secondary endpoints are analyzed using the full analysis set and, if necessary, using the per-protocol set (PPS). The safety endpoints are analyzed in the safety analysis population. The two-sided significance level of the analysis was set at 5%. The person in charge of the statistical analysis is responsible for preparing a separate statistical analysis plan and specifying the details of the statistical method, including data handling. A statistical analysis plan is prepared prior to data fixation. If changes were made to the original analysis plan, the statistical analysis plan should be revised with a revision history, and the changes should be recorded.
The primary endpoint is the frequency of nocturia at 12 weeks, and the difference between groups is evaluated to determine its statistical significance. Poisson regression analysis (generalized linear model assuming a Poisson distribution) is performed with group as the fixed effect and the allocation adjustment factors and days of nocturnal voiding at baseline as covariates to test the following null hypothesis: that the days of nocturnal voiding in the two groups are equal. The summary statistics (number of cases, minimum, median, and maximum, mean, standard deviation) are calculated for each time point and each group.
2.10.2. Analysis of the Secondary Endpoints
With regard to the primary endpoint, the frequency of urination during the day is assessed for statistical significance using Poisson regression analysis with group as the fixed effect and the allocation adjustment factor and baseline value as covariates. For the other change endpoints, group differences are evaluated using analysis of covariance with the group as the fixed effect and allocation adjustment factors and baseline values as covariates. In addition, the summary statistics of the measurements and changes at each time point for each endpoint are calculated for each group. The incidence of adverse events and diseases are analyzed as part of the safety endpoints.
2.10.3. Methods for Additional Analyses (e.g., Subgroup Analyses)
For the primary and secondary endpoints, the combined salt intake restriction guidance group (tofogliflozin plus salt intake restriction guidance group) is subdivided into a successful salt reduction group and an unsuccessful salt reduction group, and the three groups are compared with the standard treatment group (tofogliflozin group). The details are described in a separate statistical analysis section.
Methods used for analyzing the protocol nonadherence and any statistical methods used for handle missing data
Data on protocol non-compliance will not be part of the per-protocol population analysis, but will be part of the full analysis.
2.11. Ethics and Dissemination
2.11.1. Data Handling Committee
The data handling committee will be responsible for processing all data, both missing data and data departing from the protocol, in a blinded manner prior to statistical analysis. The committee consist of the principal investigator, responsible officer, and a biomedical statistics expert.
2.11.2. Composition of the Data Monitoring Committee, Its Role, and Reporting Structure
Third-party institution (Evipro Holdings Corporation) will conduct the monitoring according to the standard operating procedures for monitoring. The responsible investigator will regularly monitor the persons in charge of data quality and investigate whether the study is being conducted in compliance with the research protocol, ethical guidelines for medicine, and the Clinical Trials Act, as well as the progress of the study by taking appropriate measures to ensure compliance with the protocol. Monitoring personnel is tasked to provide a monitoring report and submit it periodically to the principal investigator. The adverse events and diseases are monitored and promptly reported to ensure the safety of the study participants.
2.12. Adverse Event Reporting and Harms
2.12.1. Reporting of Adverse Events
Adverse events outside of SAEs will be reported by noting them on the CRF associated with those occurrences and sending it to the data center. The data center shall summarize the appropriate report and inform the responsible investigator and receive instructions on how to handle the event. The investigational drug manufacturer/distributor (Kowa Company, Ltd., Aichi Japan) will also be notified.
2.12.2. Frequency and Plans for Auditing Trial Conduct
EviPRO Holdings, a third-party institution, will conduct the audits. Audits will be performed according to the protocol and Standard Operating Procedures to ensure compliance with the study protocol. The results of the audit will be reported by the auditor to the investigator, other investigator.
2.12.3. Dissemination Plans
Results of the study will be reported in peer-reviewed international journals.
3. Discussion
This study was designed based on the hypothesis that SGLT2 inhibitors, which are anti-diabetic drugs with cardioprotective and renal protective effects, may relieve nocturia not only by improving hyperglycemia and hypertension, but also by increasing daytime sodium excretion, with a focus on managing polyuria and nocturia, which are common complaints of patients with type 2 diabetes. In addition, comparing the efficacy of SGLT2 inhibitors alone with that of salt restriction combined with SGLT2 inhibitors is meaningful as it will enable the researchers to examine the efficacy and superiority of each treatment for nocturia. In addition, the long-term intention of this study is to improve health quality of life through the improvement of nocturia, hoping that this will lead to an improvement in the overall health of the patients.
In this pilot study, the frequency of nocturia is the primary endpoint, while the frequency of urination during the day and change in urine volume as secondary endpoints, which provides insight into the effect of SGLT2 inhibitors and sodium restriction on urination frequency and volume. This finding will be essential for developing future randomized trials.
In previous observational studies, the frequency of nocturia, urine volume, and nocturnal polyuria index improved in patients who successfully restricted salt intake, but nocturia did not improve in patients with unsuccessfully restricted salt intake by nearly 70% of the time. SGLT2 inhibitors are effective in preventing the progression of macrovascular disease and diabetic complications, and their use is expected to increase in the future. However, SGLT2 inhibitors have some disadvantages, such as nocturia; hence, we hope that this study will expand the possibilities of SGLT2 inhibitors. In previous observational studies, the frequency of nocturia, urine volume, and nocturnal polyuria index improved in patients who successfully restricted salt intake, but nocturia did not improve in patients with unsuccessfully restricted salt intake by nearly 70% of the time. SGLT2 inhibitors are effective in preventing the progression of macrovascular disease and diabetic complications, and their use is expected to increase in the future. However, SGLT2 inhibitors have some disadvantages, such as nocturia; hence, we hope that this study will expand the possibilities of SGLT2 inhibitors.
Author Contributions
H.N. led the drafting of the manuscript; H.O., M.H. and M.F. reviewed the manuscript and study design and contributed to the final draft. The other authors recruited the participants and contributed to the final draft. All authors have read and agreed to the published version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The TOP-STAR study, including the article processing charge, is funded by Kowa Co., Ltd. The grant number is not applicable. No drugs will be donated or funded by the sponsor. The funding bodies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
This study was registered with the Japan Clinical Trial Registry (jRCTs051210212) and was approved by the ethics committees of the Kyoto Prefectural University of Medicine (CRB5200001). The TOP-STAR study is to be conducted in accordance with the Declaration of Helsinki.
Informed Consent Statement
Written informed consent has been obtained from all the participants.
Data Availability Statement
Data will not be publicly available.
Acknowledgments
We would like to thank all the clinical staff for their assistance with the execution of the clinical trial and EviPRO Holdings. Inc. for their technical assistance in the launch and execution of this trial.
Conflicts of Interest
H.O. received personal fees from MSD K.K., Mitsubishi Tanabe Pharma Corporation, Sumitomo Dainippon Pharma Co., Ltd., Novo Nordisk Pharma Ltd., Daiichi Sankyo Co., Ltd., Eli Lilly Japan K.K., Kyowa Hakko Kirin Company Ltd., Kissei Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Kowa Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd. and Sanofi K.K. M.H. received personal fees from Ono Pharma Co. Ltd., AstraZeneca K.K., Oishi Kenko Inc., Yamada Bee Farm, Sumitomo Dainippon Pharma Co., Ltd., Eli Lilly, Japan, Daiichi Sankyo Co. Ltd., Mitsubishi Tanabe Pharma Corp., Sanofi K.K. and Kowa Pharma Co. Ltd., outside the submitted work. M.F. received personal fees from Oishi Kenko inc., Yamada Bee Farm, Sanofi K.K., Terumo Corp., Nippon Chemiphar Co., Ltd., Johnson & Johnson K.K. Medical Co., Abbott Japan Co. Ltd., Kissei Pharma Co., Ltd., Sumitomo Dainippon Pharma Co. Ltd., Mitsubishi Tanabe Pharma Corp., Daiichi Sankyo Co. Ltd., Sanofi K.K., Astellas Pharma Inc., MSD K.K., Kyowa Kirin Co. Ltd., Taisho Pharma Co., Ltd., Kowa Pharma Co. Ltd., Mochida Pharma Co. Ltd., Novo Nordisk Pharma Ltd., Ono Pharma Co. Ltd., Sanwa Kagaku Kenkyusho Co. Ltd., Eli Lilly Japan K.K., Takeda Pharma Co. Ltd., Bayer Yakuhin, Ltd., AstraZeneca K.K., Nippon Boehringer Ingelheim Co., Ltd., Teijin Pharma Ltd., Medtronic Japan Co. Ltd., Arkray Inc. and Nipro Corp. outside the submitted work. The other authors have nothing to disclose.
References
- Abrams, P.; Cardozo, L.; Fall, M.; Griffiths, D.; Rosier, P.; Ulmsten, U.; van Kerrebroeck, P.; Victor, A.; Wein, A. The standardisation of terminology of lower urinary tract function: Report from the standardisation sub-committee of the international continence society. Am. J. Obstet. Gynecol. 2002, 187, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokoyama, O. Pathogenesis and management of nocturia in the elderly. Jpn. J. Geriatr. 2013, 50, 434–439. (In Japanese) [Google Scholar]
- Yoshimura, K.; Terada, N.; Matsui, Y.; Terai, A.; Kinukawa, N.; Arai, Y. Prevalence of and risk factors for nocturia: Analysis of a health screening program. Int. J. Urol. 2004, 11, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, H.; Xu, K.; Zhang, X.; Wang, X.; Na, Y.; Kang, X. Prevalence, risk factors, and symptom bother of nocturia: A population-based survey in China. World J. Urol. 2015, 33, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Wen, Y.B.; Wang, Z.M.; Wen, J.G.; Li, Z.Z.; Shang, X.P.; Liu, Z.S.; Jia, L.H.; Qin, G.J.; Heesakkers, J.; et al. Risk Factors of Nocturia (Two or More Voids Per Night) in Chinese People Older Than 40 Years. Neurourol. Urodyn. 2015, 570, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Madhu, C.; Coyne, K.; Hashim, H.; Chapple, C.; Milsom, I.; Kopp, Z. Nocturia: Risk factors and associated comorbidities; findings from the EpiLUTS study. Int. J. Clin. Pr. 2015, 69, 1508–1516. [Google Scholar] [CrossRef]
- Chow, P.M.; Liu, S.P.; Chuang, Y.C.; Lee, K.S.; Yoo, T.K.; Liao, L.; Wang, J.; Liu, M.; Sumarsono, B.; Jong, J.J. The prevalence and risk factors of nocturia in China, South Korea, and Taiwan: Results from a cross-sectional, population-based study International Index of Erectile Function. World J. Urol. 2018, 36, 1853–1862. [Google Scholar] [CrossRef]
- Hirayama, A.; Torimoto, K.; Mastusita, C.; Okamoto, N.; Morikawa, M.; Tanaka, N.; Yoshida, K.; Fujimoto, K.; Hirao, Y.; Kurumatani, N. Evaluation of Factors Influencing the Natural History of Nocturia in Elderly Subjects: Results of the Fujiwara-kyo Study. J. Urol. 2013, 189, 980–986. [Google Scholar] [CrossRef]
- Furukawa, S. Smoking and prevalence of nocturia in Japanese patients with type 2 diabetes mellitus: A post-hoc analysis of The Dogo Study. Neurourol. Urodyn. 2017, 36, 1336–1341. [Google Scholar] [CrossRef]
- Kawano, R.; Takahashi, F.; Hashimoto, Y.; Okamura, T.; Miki, A.; Kaji, A.; Sakai, R.; Kitagawa, N.; Senmaru, T.; Majima, S.; et al. Short energy intake is associated with muscle mass loss in older patients with type 2 diabetes: A prospective study of the KAMOGAWA-DM cohort. Clin. Nutr. 2021, 40, 1613–1620. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Sakai, R.; Ikeda, K.; Fukui, M. Association between sleep disorder and quality of life in patients with type 2 diabetes: A cross-sectional study. BMC Endocr. Disord. 2020, 4, 98. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Niu, K.; Hozawa, A.; Ikeda, Y.; Kaiho, Y.; Ohmori-Matsuda, K.; Nakaya, N.; Kuriyama, S.; Ebihara, S.; Nagatomi, R.; et al. Impact of Nocturia on Bone Fracture and Mortality in Older Individuals: A Japanese Longitudinal Cohort Study. J. Urol. 2010, 184, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.C.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- The Japan Diabetes Society. Recommendations for the Appropriate Use of SGLT2 Inhibitors. Available online: http://www.fa.kyorin.co.jp/jds/uploads/recommendation_SGLT2.pdf (accessed on 22 February 2022). (In Japanese).
- Takegoshi, S.; Miyata, Y.; Ogata, N.; Saito, T. Availability and efficacy of 20-mg tofogliflozin administered every other day to type 2 diabetic patients. Prog. Med. 2015, 35, 1077–1088. (In Japanese) [Google Scholar]
- Takeishi, S.; Tsuboi, H.; Takekoshi, S. Comparison of tofogliflozin 20 mg and ipragliflozin 50 mg used together with insulin glargine 300 U/ml using continuous glucose monitoring (CGM): A randomized crossover study. Endocr. J. 2017, 64, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Kawasoe, S.; Maruguchi, Y.; Kajiya, S.; Uenomachi, H.; Miyata, M.; Kawasoe, M.; Kubozono, T.; Ohishi, M. Mechanism of the blood pressure-lowering effect of sodium-glucose cotransporter 2 inhibitors in obese patients with type 2 diabetes. BMC Pharmacol. Toxicol. 2017, 18, 23. [Google Scholar] [CrossRef]
- Matsuo, T.; Miyata, Y.; Sakai, H. Effect of salt intake reduction on nocturia in patients with excessive salt intake. Neurourol. Urodyn. 2019, 38, 927–933. [Google Scholar] [CrossRef]
- Ushigome, E.; Oyabu, C.; Iwai, K.; Kitagawa, N.; Kitae, A.; Kimura, T.; Yokota, I.; Ushigome, H.; Hamaguchi, M.; Asano, M.; et al. Effects of dietary salt restriction on home blood pressure in diabetic patients with excessive salt intake: A pilot study. J. Clin. Biochem. Nutr. 2019, 65, 252–257. [Google Scholar] [CrossRef]
- Matsuo, T.; Miyata, Y.; Sakai, H. Daily salt intake is an independent risk factor for pollakiuria and nocturia. Int. J. Urol. 2017, 24, 384–389. [Google Scholar] [CrossRef]
- Matsumoto, T.; Tabara, Y.; Murase, K.; Setoh, K.; Kawaguchi, T.; Nagashima, S.; Kosugi, S.; Nakayama, T.; Wakamura, T.; Hirai, T.; et al. Nocturia and increase in nocturnal blood pressure: The Nagahama study. J. Hypertens. 2018, 36, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Obayashi, K.; Tai, Y.; Yamagami, Y.; Negoro, H.; Kataoka, H.; Kurumatani, N.; Saeki, K. Association between NT-proBNP and nocturia among community-dwelling elderly males and females: A cross-sectional analysis of the HEIJO-KYO study. Neurourol. Urodyn. 2021, 40, 112–119. [Google Scholar] [CrossRef]
- Ishii, H. The Japanese version of the Diabetes Treatment Satisfaction Questionnaire (DTSQ): Translation and clinical evaluation. J. Clin. Exp. Med. 2000, 192, 809–814. [Google Scholar]
- Sato, E.; Ochiai, R.; Shibayama, T.; Nishigaki, M.; Abe, Y.; Sawa, T.; Suzukamo, Y.; Kazuma, K. Reliability and validity of revised and short form versions of diabetes diet-related quality of life scale. Diabetol. Int. 2017, 8, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Honda, S.; Murakami, K.; Sasaki, S.; Okubo, H.; Hirota, N.; Notsu, A.; Fukui, M.; Date, C. Both comprehensive and brief self-administered diet history questionnaires satisfactorily rank nutrient intakes in Japanese adults. J. Epidemiol. 2012, 22, 151–159. [Google Scholar] [CrossRef]
- Homma, Y.; Yoshida, M.; Yamanishi, T.; Gotoh, M. Core lowerurinary tract symptom (CLSS) questionnaire: A reliable tool in the overall assessment of lower urinary tract symptoms. Int. J. Urol. 2008, 15, 816–820. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).