
Development and Validation of a Simple HPLC-UV Assay Method for Determination of Levetiracetam Concentrations in Human Plasma


Abstract
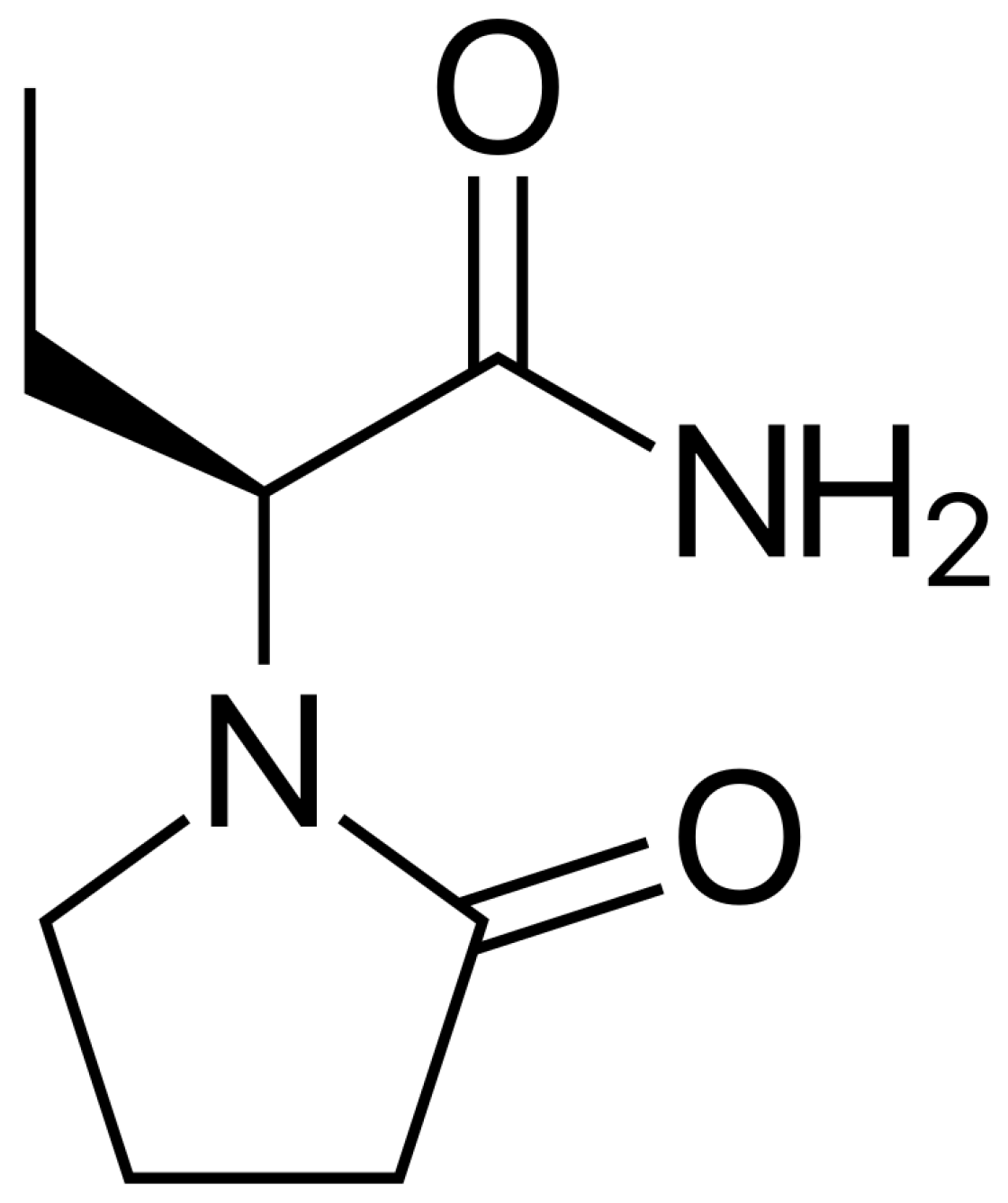
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrument
2.3. Chromatographic Conditions
2.4. Preparation of Standard and Working Solutions
2.5. Preparation of Calibration Concentrations and Quality Control (QC) Samples
2.6. Sample Preparation
2.7. Method Validation
2.7.1. Linearity
2.7.2. Selectivity and Sensitivity
2.7.3. Precision and Accuracy
2.7.4. Carry-Over
2.7.5. Dilution Integrity
2.7.6. Stability
2.7.7. Recovery
3. Results
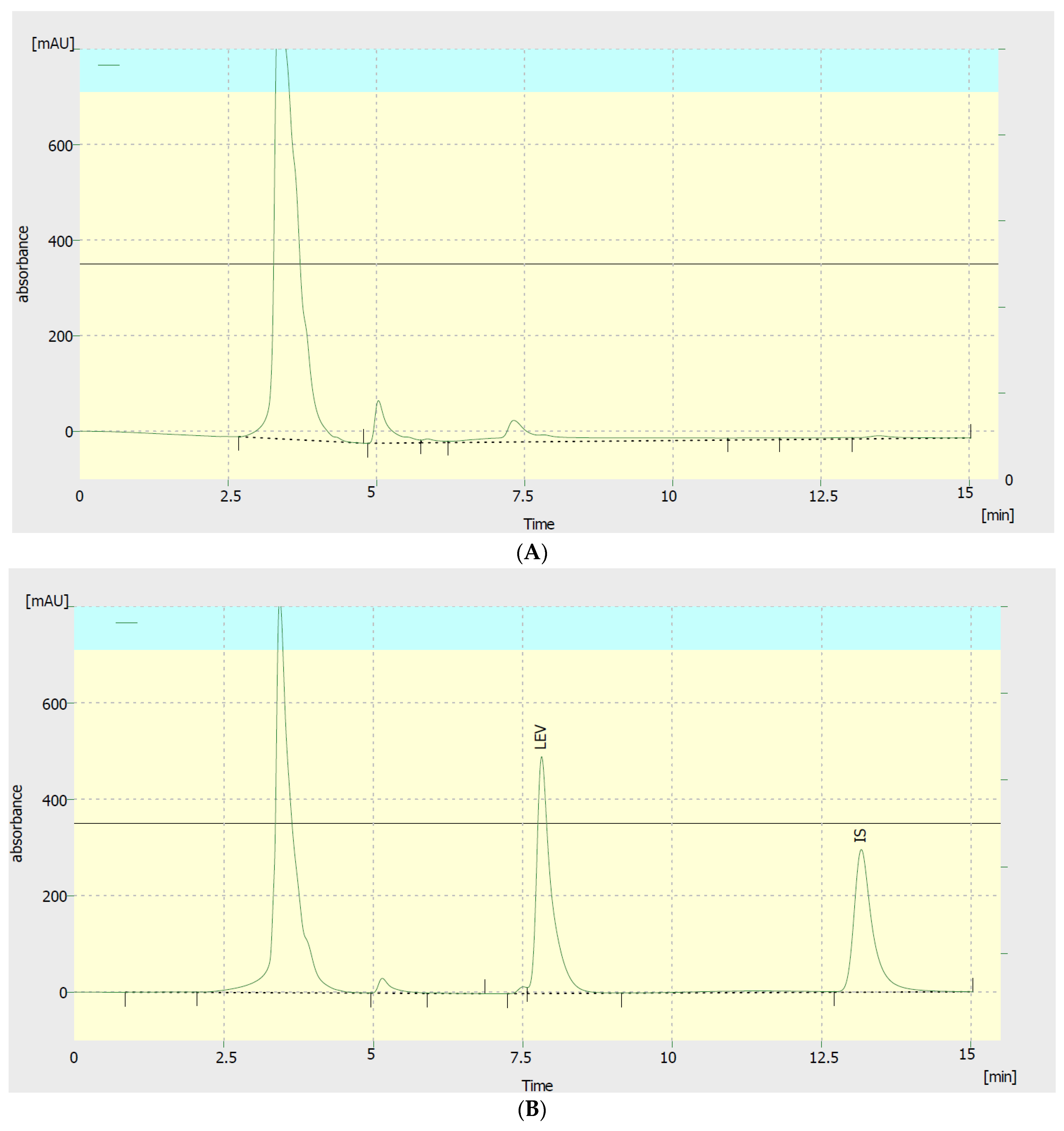
3.1. Optimization of Chromatographic Conditions
3.2. Method Validation
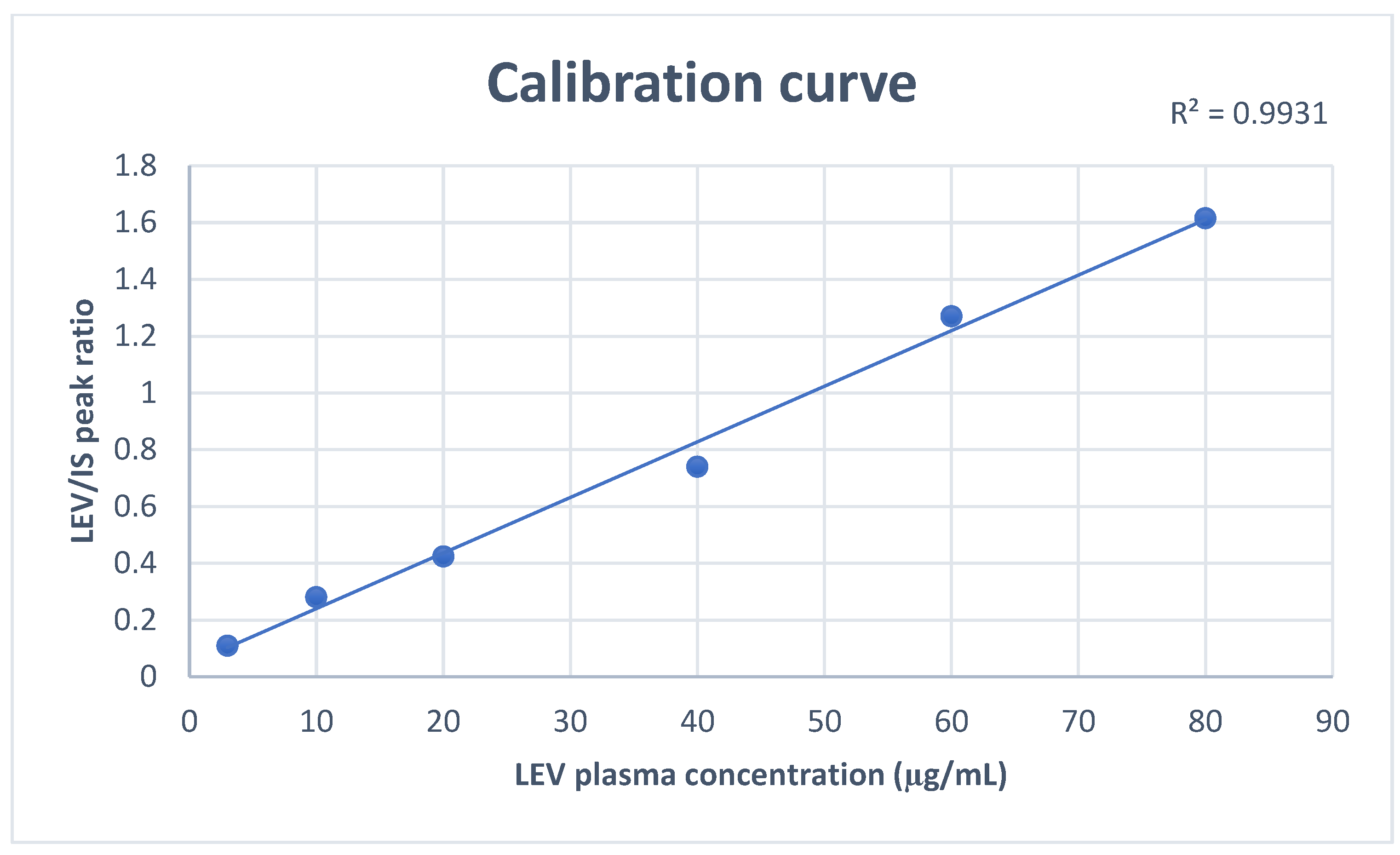
3.2.1. Linearity
3.2.2. Selectivity and Sensitivity
3.2.3. Precision and Accuracy
3.2.4. Carryover
3.2.5. Dilution integrity
3.2.6. Stability
3.2.7. Recovery
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patsalos, P. The pharmacokinetic characteristics of levetiracetam. Methods Find Exp. Clin. Pharmacol. 2003, 25, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, P. Pharmacokinetic profile of levetiracetam: Toward ideal characteristics. Pharmacol. Ther. 2000, 85, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Radtke, R.A. Pharmacokinetics of levetiracetam. Epilepsia 2001, 42, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Jarvie, D.; Mahmoud, S.H. Therapeutic drug monitoring of levetiracetam in select populations. J. Pharm. Pharm. Sci. 2018, 21, 149s–176s. [Google Scholar] [CrossRef] [PubMed]
- Perucca, E.; Bialer, M. The clinical pharmacokinetics of the newer antiepileptic drugs. Clin. Pharmacokinet. 1996, 31, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Attallah, O.A.; Al-Ghobashy, M.A.; Ayoub, A.T.; Tuszynski, J.A.; Nebsen, M. Computer-aided design of magnetic molecularly imprinted polymer nanoparticles for solid-phase extraction and determination of levetiracetam in human plasma. RSC Adv. 2018, 8, 14280–14292. [Google Scholar] [CrossRef] [PubMed]
- Contin, M.; Mohamed, S.; Albani, F.; Riva, R.; Baruzzi, A. Simple and validated HPLC–UV analysis of levetiracetam in deproteinized plasma of patients with epilepsy. J. Chromatogr. B 2008, 873, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, S.N.; Dahmane, E.; Armahizer, M.; McCarthy, P.; Gopalakrishnan, M. Development and validation of a HPLC-UV assay for quantification of levetiracetam concentrations in critically ill patients undergoing continuous renal replacement therapy. Biomed. Chromatogr. 2018, 32, e4257. [Google Scholar] [CrossRef] [PubMed]
- Olah, E.; Bacsói, G.; Fekete, J.; Sharma, V. Determination of ng/mL levetiracetam using ultra-high-performance liquid chromatography–photodiode absorbance. J. Chromatogr. Sci. 2012, 50, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, F.A.; El-Yazbi, A.F.; Barary, M.A.; Wagih, M.M. Sensitive inexpensive HPLC determination of four antiepileptic drugs in human plasma: Application to PK studies. Bioanalysis 2016, 8, 2219–2234. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Mitre, M.P.; Medellín-Garibay, S.E.; Rodríguez-Leyva, I.; Rodríguez-Pinal, C.J.; Zarazúa, S.; Jung-Cook, H.H.; Roberts, J.A.; Romano-Moreno, S.; del Carmen Milán-Segovia, R. Population pharmacokinetics and dosing recommendations of levetiracetam in adult and elderly patients with epilepsy. J. Pharm. Sci. 2020, 109, 2070–2078. [Google Scholar] [CrossRef] [PubMed]
- Zufia, L.; Aldaz, A.; Ibanez, N.; Giraldez, J.; Viteri, C. LC method for therapeutic drug monitoring of levetiracetam: Evaluation of the assay performance and validation of its application in the routine area. Clin. Biochem. 2010, 43, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Antonilli, L.; Brusadin, V.; Filipponi, F.; Guglielmi, R.; Nencini, P. Development and validation of an analytical method based on high performance thin layer chromatography for the simultaneous determination of lamotrigine, zonisamide and levetiracetam in human plasma. J. Pharm. Biomed. Anal. 2011, 56, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Pucci, V.; Bugamelli, F.; Mandrioli, R.; Ferranti, A.; Kenndler, E.; Raggi, M.A. High-performance liquid chromatographic determination of Levetiracetam in human plasma: Comparison of different sample clean-up procedures. Biomed. Chromatogr. 2004, 18, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Engelbrecht, L.; Grobler, C.; Rheeders, M. A simple and cost-effective HPLC-UV method for the detection of levetiracetam in plasma/serum of patients with epilepsy. Biomed. Chromatogr. 2017, 31, e3969. [Google Scholar] [CrossRef] [PubMed]
- Lancelin, F.; Franchon, E.; Kraoul, L.; Garciau, I.; Brovedani, S.; Tabaouti, K.; Landré, E.; Chassoux, F.; Paubel, P.; Piketty, M.-L. Therapeutic drug monitoring of levetiracetam by high-performance liquid chromatography with photodiode array ultraviolet detection: Preliminary observations on correlation between plasma concentration and clinical response in patients with refractory epilepsy. Ther. Drug Monit. 2007, 29, 576–583. [Google Scholar]
- Guideline on Bioanalytical Method Validation. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 10 October 2022).
- Harden, C. Safety profile of levetiracetam. Epilepsia 2001, 42, 36–39. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| QC Concentration (μg/mL) | Observed Concentration (μg/mL) Mean ± SD | Precision, CV (%) | Accuracy, Error (%) | |
|---|---|---|---|---|
| Intraday | 3 | 3.14 ± 0.36 | 11.35 | −4.74 |
| 30 | 29.02 ± 0.84 | 2.88 | 3.28 | |
| 50 | 47.76 ± 3.2 | 6.72 | 4.48 | |
| 70 | 69.82 ± 2.67 | 3.82 | 0.25 | |
| Interday | 3 | 3.47 ± 0.3 | 8.77 | −15.78 |
| 30 | 31.28 ± 2.3 | 7.38 | −4.27 | |
| 50 | 48.69 ± 1.99 | 4.08 | 2.62 | |
| 70 | 67.92 ± 1.66 | 2.45 | 2.96 |
| Stability Test | Concentration (μg/mL) | Mean Calculated Concentrations (μg/mL) ± SD | CV (%) | % Error |
|---|---|---|---|---|
| Stock solution stability at −80 °C (1 month) | 60 | 60.06 ± 6.53 | 10.87 | −0.11 |
| Stock solution stability at −80 °C (2 month) | 60 | 63.63 ± 0.24 | 0.38 | −6.06 |
| Stock solution stability at 4 °C (1 month) | 60 | 63.63 ± 2.61 | 4.1 | −6.03 |
| Long term stability at room temperature (1 month) | 60 | 60.92 ± 2.81 | 3.95 | −1.5 |
| Long term stability at −80 °C (1 month) | 60 | 66.75 ± 1.35 | 2.02 | −11.26 |
| Free-thaw stability | 60 | 64.87 ± 1.16 | 1.79 | −8.12 |
| Bench-top stability (12 h) | 60 | 66.51 ± 0.79 | 1.19 | −10.85 |
| Reinjection stability | 60 | 52.02 ± 0.64 | 1.49 | 13.28 |
| Drug | Concentration (μg/mL) | Recovery, Mean ± SD (%) | CV (%) |
|---|---|---|---|
| Levetiracetam | 30 | 80.38 ± 6.43 | 8.0 |
| 50 | 77.36 ± 1.60 | 2.06 | |
| 70 | 76.75 ± 1.64 | 2.13 | |
| Caffeine (IS) | 50 | 83.39 ± 1.8 | 2.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kharouba, M.; Mahmoud, S.H. Development and Validation of a Simple HPLC-UV Assay Method for Determination of Levetiracetam Concentrations in Human Plasma. Analytica 2023, 4, 1-9. https://doi.org/10.3390/analytica4010001
Kharouba M, Mahmoud SH. Development and Validation of a Simple HPLC-UV Assay Method for Determination of Levetiracetam Concentrations in Human Plasma. Analytica. 2023; 4(1):1-9. https://doi.org/10.3390/analytica4010001
Chicago/Turabian StyleKharouba, Maged, and Sherif Hanafy Mahmoud. 2023. "Development and Validation of a Simple HPLC-UV Assay Method for Determination of Levetiracetam Concentrations in Human Plasma" Analytica 4, no. 1: 1-9. https://doi.org/10.3390/analytica4010001
APA StyleKharouba, M., & Mahmoud, S. H. (2023). Development and Validation of a Simple HPLC-UV Assay Method for Determination of Levetiracetam Concentrations in Human Plasma. Analytica, 4(1), 1-9. https://doi.org/10.3390/analytica4010001