

Immune Checkpoints and the Immunology of Liver Fibrosis


Abstract
1. Introduction
2. A Pathogenetic Overview of Liver Fibrosis
2.1. The Fibrotic Process
2.2. Cells Involved in Liver Fibrosis
2.2.1. Kupffer Cells and Liver Macrophages
2.2.2. Hepatic Stellate Cells
2.2.3. The Interplay Between KCs and HSCs
2.2.4. Liver Sinusoidal Endothelial Cells (LSECs)
2.2.5. Cytokines Involved in Liver Fibrosis
2.3. Resolution of Liver Fibrosis
3. Immunology of Liver Fibrosis
3.1. The Liver as an Immune Organ
3.2. Immune Factors Implicated in Liver Fibrosis
Innate and Adaptive Immunity
3.3. Innate Immunity in Liver Fibrosis
3.3.1. Cells Involved in Innate Immunity
3.3.2. LSECs as Gatekeepers of Innate and Adaptive Immunity
3.4. Adaptive Immunity T Cells
3.5. Unconventional T Cells
3.6. Extrahepatic Factors
3.7. The Interaction of Innate and Adaptive Immunity
4. Immune Checks in Liver Fibrosis
4.1. Immune Checkpoint
4.2. Association of Immune Checkpoints with Liver Fibrosis
4.2.1. The PD-1/PD-L1 Axis and CTLA-4
4.2.2. Other ICPs Involved in Liver Fibrosis
4.3. Therapeutic Implications of Checkpoint Inhibitors in Liver Fibrosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Ginès, P.; Krag, A.; Abraldes, J.G.; Solà, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef]
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666. [Google Scholar] [CrossRef]
- Ye, F.; Zhai, M.; Long, J.; Gong, Y.; Ren, C.; Zhang, D.; Lin, X.; Liu, S. The burden of liver cirrhosis in mortality: Results from the global burden of disease study. Front. Public. Health 2022, 10, 909455. [Google Scholar] [CrossRef]
- Jepsen, P.; Younossi, Z.M. The global burden of cirrhosis: A review of disability-adjusted life-years lost and unmet needs. J. Hepatol. 2021, 75, 3–13. [Google Scholar] [CrossRef]
- Smith, A.; Baumgartner, K.; Bositis, C. Cirrhosis: Diagnosis and Management. Am. Fam. Physician. 2019, 100, 759–770. [Google Scholar]
- Alberts, C.J.; Clifford, G.M.; Georges, D.; Negro, F.; Lesi, O.A.; Hutin, Y.J.; de Martel, C. Worldwide prevalence of hepatitis B virus and hepatitis C virus among patients with cirrhosis at country, region, and global levels: A systematic review. Lancet Gastroenterol. Hepatol. 2022, 7, 724–735. [Google Scholar] [CrossRef]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Zitvogel, L. Immune checkpoint inhibitors. J. Exp. Med. 2021, 218, 20201979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, J. Functions of Immune Checkpoint Molecules Beyond Immune Evasion. Adv. Exp. Med. Biol. 2020, 1248, 201–226. [Google Scholar]
- Pinheiro, D.; Dias, I.; Ribeiro Silva, K.; Stumbo, A.C.; Thole, A.; Cortez, E.; de Carvalho, L.; Weiskirchen, R.; Carvalho, S. Mechanisms Underlying Cell Therapy in Liver Fibrosis: An Overview. Cells 2019, 8, 1339. [Google Scholar] [CrossRef]
- Bourebaba, N.; Marycz, K. Hepatic stellate cells role in the course of metabolic disorders development—A molecular overview. Pharmacol. Res. 2021, 170, 105739. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Sun, B. The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application. Int. J. Mol. Sci. 2022, 23, 12572. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Res. 2018, 7, 921. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis--a common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Gao, C.C.; Bai, J.; Han, H.; Qin, H.Y. The versatility of macrophage heterogeneity in liver fibrosis. Front. Immunol. 2022, 13, 968879. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Zhu, R.T.; Sun, Y.L. Epithelial-mesenchymal transition in liver fibrosis. Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef]
- Park, J.H.; Park, B.; Park, K.K. Suppression of Hepatic Epithelial-to-Mesenchymal Transition by Melittin via Blocking of TGFβ/Smad and MAPK-JNK Signaling Pathways. Toxins 2017, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Li, T.Z.; Kim, S.M.; Hur, W.; Choi, J.E.; Kim, J.H.; Hong, S.W.; Lee, E.B.; Lee, J.H.; Yoon, S.K. Elk-3 Contributes to the Progression of Liver Fibrosis by Regulating the Epithelial-Mesenchymal Transition. Gut Liver. 2017, 11, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Lin, Y.; Shimizu-Hirota, R.; Hanada, S.; Neilson, E.G.; Greenson, J.K.; Weiss, S.J. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol. Cell Biol. 2011, 31, 2392–2403. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Liu, J.; Tu, X.; Zang, Y.; Zhu, J.; Chen, J.; Dong, L.; Zhang, J. miR-30 inhibits TGF-β1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting Snail1. Biochem. Biophys. Res. Commun. 2012, 417, 1100–1105. [Google Scholar] [CrossRef]
- Zhan, J.; Liu, S.; Meng, Y.; Yang, Q.; Wang, Z.; Zhang, S.; Ge, L.; Zhao, L.; Xu, X.; Zhao, Y.; et al. Systematic review of the mechanism and assessment of liver fibrosis in biliary atresia. Pediatr. Surg. Int. 2024, 40, 205. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Robertson, H.; Marshall, H.L.; Pekalski, M.; Zhao, L.; Booth, T.A.; Jones, D.E.; Burt, A.D.; Kirby, J.A. Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab. Investig. 2008, 88, 112–123. [Google Scholar] [CrossRef]
- Harada, K.; Sato, Y.; Ikeda, H.; Isse, K.; Ozaki, S.; Enomae, M.; Ohama, K.; Katayanagi, K.; Kurumaya, H.; Matsui, A.; et al. Epithelial-mesenchymal transition induced by biliary innate immunity contributes to the sclerosing cholangiopathy of biliary atresia. J. Pathol. 2009, 217, 654–664. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, H.; Song, G.; Liao, X.; Xian, Y.; Li, W. Regulation of epithelial-mesenchymal transition by tumor-associated macrophages in cancer. Am. J. Transl. Res. 2015, 7, 1699–1711. [Google Scholar]
- Yang, M.; Ma, B.; Shao, H.; Clark, A.M.; Wells, A. Macrophage phenotypic subtypes diametrically regulate epithelial-mesenchymal plasticity in breast cancer cells. BMC Cancer. 2016, 16, 419. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Toyoda, H.; Tanaka, M.; Kuroda, M.; Harada, Y.; Matsuda, F.; Tajima, A.; Kosaka, N.; Ochiya, T.; Shimotohno, K. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS ONE 2011, 6, 16081. [Google Scholar] [CrossRef]
- Li, Q.; Li, Z.; Lin, Y.; Che, H.; Hu, Y.; Kang, X.; Zhang, Y.; Wang, L.; Zhang, Y. High glucose promotes hepatic fibrosis via miR-32/MTA3-mediated epithelial-to-mesenchymal transition. Mol. Med. Rep. 2019, 19, 3190–3200. [Google Scholar] [CrossRef]
- Gao, Y.; Li, L.; Zhang, S.N.; Mang, Y.Y.; Zhang, X.B.; Feng, S.M. HepG2.2.15-derived exosomes facilitate the activation and fibrosis of hepatic stellate cells. World J. Gastroenterol. 2024, 30, 2553–2563. [Google Scholar] [CrossRef]
- Ezhilarasan, D. MicroRNA interplay between hepatic stellate cell quiescence and activation. Eur. J. Pharmacol. 2020, 885, 173507. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, W.; Yu, F.; Dong, P.; Chen, B.; Zhou, M.T. MicroRNA-30a Suppresses the Activation of Hepatic Stellate Cells by Inhibiting Epithelial-to-Mesenchymal Transition. Cell Physiol. Biochem. 2018, 46, 82–92. [Google Scholar] [CrossRef]
- Chen, J.; Yu, Y.; Li, S.; Liu, Y.; Zhou, S.; Cao, S.; Yin, J.; Li, G. MicroRNA-30a ameliorates hepatic fibrosis by inhibiting Beclin1-mediated autophagy. J. Cell Mol. Med. 2017, 21, 3679–3692. [Google Scholar] [CrossRef]
- Xu, W.; Mo, W.; Han, D.; Dai, W.; Xu, X.; Li, J.; Xu, X. Hepatocyte-derived exosomes deliver the lncRNA CYTOR to hepatic stellate cells and promote liver fibrosis. J. Cell Mol. Med. 2024, 28, 18234. [Google Scholar] [CrossRef]
- Dong, Z.; Li, S.; Wang, X.; Si, L.; Ma, R.; Bao, L.; Bo, A. lncRNA GAS5 restrains CCl4-induced hepatic fibrosis by targeting miR-23a through the PTEN/PI3K/Akt signaling pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Lin, H.; Chen, X.; Li, G.; Zhao, Y.; Zheng, L.; Shi, Z.; Zhang, K.; Hong, W.; Han, T. LncRNA Meg8 suppresses activation of hepatic stellate cells and epithelial-mesenchymal transition of hepatocytes via the Notch pathway. Biochem. Biophys. Res. Commun. 2020, 521, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Ou, Q.; Zhao, Y.; Zhou, J.; Wu, X. Comprehensive circular RNA expression profiles in a mouse model of nonalcoholic steatohepatitis. Mol. Med. Rep. 2019, 19, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Liu, J.; Deng, H.; Ma, R.; Liao, J.Y.; Liang, H.; Hu, J.; Li, J.; Guo, Z.; Cai, J.; et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell 2020, 183, 76–93. [Google Scholar] [CrossRef]
- Zollner, G.; Trauner, M. Nuclear receptors as therapeutic targets in cholestatic liver diseases. Br. J. Pharmacol. 2009, 156, 7–27. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear bile acid receptor farnesoid X receptor meets nuclear factor-kappaB: New insights into hepatic inflammation. Hepatology 2008, 48, 1383–1386. [Google Scholar] [CrossRef]
- Tran, M.; Liu, Y.; Huang, W.; Wang, L. Nuclear receptors and liver disease: Summary of the 2017 basic research symposium. Hepatol Commun. 2018, 2, 765–777. [Google Scholar] [CrossRef]
- Jin, L.; Li, Y. Structural and functional insights into nuclear receptor signaling. Adv. Drug Deliv. Rev. 2010, 62, 1218–1226. [Google Scholar] [CrossRef]
- Wu, W.B.; Chen, Y.Y.; Zhu, B.; Peng, X.M.; Zhang, S.W.; Zhou, M.L. Excessive bile acid activated NF-kappa B and promoted the development of alcoholic steatohepatitis in farnesoid X receptor deficient mice. Biochimie 2015, 115, 86–92. [Google Scholar] [CrossRef]
- Kong, B.; Luyendyk, J.P.; Tawfik, O.; Guo, G.L. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J. Pharmacol. Exp. Ther. 2009, 328, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Baghdasaryan, A.; Claudel, T.; Gumhold, J.; Silbert, D.; Adorini, L.; Roda, A.; Vecchiotti, S.; Gonzalez, F.J.; Schoonjans, K.; Strazzabosco, M.; et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2-/- (Abcb4-/-) mouse cholangiopathy model by promoting biliary HCO⁻₃ output. Hepatology 2011, 54, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A. Targeting nuclear receptors for NASH/MASH: From bench to bedside. Liver Res. 2024, 8, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Königshofer, P.; Brusilovskaya, K.; Petrenko, O.; Hofer, B.S.; Schwabl, P.; Trauner, M.; Reiberger, T. Nuclear receptors in liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166235. [Google Scholar] [CrossRef]
- Wang, X.C.; Song, K.; Tu, B.; Sun, H.; Zhou, Y.; Xu, S.S.; Lu, D.; Sha, J.M.; Tao, H. New aspects of the epigenetic regulation of EMT related to pulmonary fibrosis. Eur. J. Pharmacol. 2023, 956, 175959. [Google Scholar] [CrossRef]
- Sisto, M.; Lisi, S. Epigenetic Regulation of EMP/EMT-Dependent Fibrosis. Int. J. Mol. Sci. 2024, 25, 2775. [Google Scholar] [CrossRef]
- Liu, Y.; Wen, D.; Ho, C.; Yu, L.; Zheng, D.; O’Reilly, S.; Gao, Y.; Li, Q.; Zhang, Y. Epigenetics as a versatile regulator of fibrosis. J. Transl. Med. 2023, 21, 164. [Google Scholar] [CrossRef]
- Liu, R.; Li, Y.; Zheng, Q.; Ding, M.; Zhou, H.; Li, X. Epigenetic modification in liver fibrosis: Promising therapeutic direction with significant challenges ahead. Acta Pharm. Sin. B 2024, 14, 1009–1029. [Google Scholar] [CrossRef]
- Tao, S.; Duan, R.; Xu, T.; Hong, J.; Gu, W.; Lin, A.; Lian, L.; Huang, H.; Lu, J.; Li, T. Salvianolic acid B inhibits the progression of liver fibrosis in rats via modulation of the Hedgehog signaling pathway. Exp. Ther. Med. 2022, 23, 116. [Google Scholar] [CrossRef]
- Yu, F.; Lu, Z.; Chen, B.; Wu, X.; Dong, P.; Zheng, J. Salvianolic acid B-induced microRNA-152 inhibits liver fibrosis by attenuating DNMT1-mediated Patched1 methylation. J. Cell Mol. Med. 2015, 19, 2617–2632. [Google Scholar] [CrossRef]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Li, W.; Chang, N.; Li, L. Heterogeneity and Function of Kupffer Cells in Liver Injury. Front. Immunol. 2022, 13, 940867. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Park, M.D.; Silvin, A.; Ginhoux, F.; Merad, M. Macrophages in health and disease. Cell 2022, 185, 4259–4279. [Google Scholar] [CrossRef]
- Martrus, G.; Goebels, H.; Langeneckert, A.E.; Kah, J.; Flomm, F.; Ziegler, A.E.; Niehrs, A.; Löbl, S.M.; Russu, K.; Hess, L.U.; et al. CD49a Expression Identifies a Subset of Intrahepatic Macrophages in Humans. Front. Immunol. 2019, 10, 1247. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Fogg, D.K.; Sibon, C.; Miled, C.; Jung, S.; Aucouturier, P.; Littman, D.R.; Cumano, A.; Geissmann, F. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 2006, 311, 83–87. [Google Scholar] [CrossRef]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 3186–3195. [Google Scholar] [CrossRef]
- Traber, P.G.; Chou, H.; Zomer, E.; Hong, F.; Klyosov, A.; Fiel, M.I.; Friedman, S.L. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE 2013, 8, 75361. [Google Scholar] [CrossRef] [PubMed]
- Khambu, B.; Yan, S.; Huda, N.; Yin, X.M. Role of High-Mobility Group Box-1 in Liver Pathogenesis. Int. J. Mol. Sci. 2019, 20, 5314. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Hou, X.; Liu, W.; Han, Z.; Wei, L. Macrophages and hepatocellular carcinoma. Cell Biosci. 2019, 9, 79. [Google Scholar] [CrossRef]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Spiller, K.L.; Wrona, E.A.; Romero-Torres, S.; Pallotta, I.; Graney, P.L.; Witherel, C.E.; Panicker, L.M.; Feldman, R.A.; Urbanska, A.M.; Santambrogio, L.; et al. Differential gene expression in human, murine, and cell line-derived macrophages upon polarization. Exp. Cell Res. 2016, 347, 1–13. [Google Scholar] [CrossRef]
- Braga, T.T.; Agudelo, J.S.; Camara, N.O. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Ritz, T.; Krenkel, O.; Tacke, F. Dynamic plasticity of macrophage functions in diseased liver. Cell Immunol. 2018, 330, 175–182. [Google Scholar] [CrossRef]
- Trus, E.; Basta, S.; Gee, K. Who’s in charge here? Macrophage colony stimulating factor and granulocyte macrophage colony stimulating factor: Competing factors in macrophage polarization. Cytokine 2020, 127, 154939. [Google Scholar] [CrossRef]
- Petrina, M.; Alothaimeen, T.; Bouzeineddine, N.Z.; Trus, E.; Banete, A.; Gee, K.; Basta, S. Granulocyte macrophage colony stimulating factor exerts dominant effects over macrophage colony stimulating factor during macrophage differentiation in vitro to induce an inflammatory phenotype. Inflamm. Res. 2024, 73, 253–262. [Google Scholar] [CrossRef]
- Zheng, S.; Zhang, P.; Chen, Y.; Zheng, S.; Zheng, L.; Weng, Z. Inhibition of Notch Signaling Attenuates Schistosomiasis Hepatic Fibrosis via Blocking Macrophage M2 Polarization. PLoS ONE 2016, 11, e0166808. [Google Scholar] [CrossRef]
- Bansal, R.; van Baarlen, J.; Storm, G.; Prakash, J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015, 5, 18272. [Google Scholar] [CrossRef]
- Krenkel, O.; Hundertmark, J.; Abdallah, A.T.; Kohlhepp, M.; Puengel, T.; Roth, T.; Branco, D.P.P.; Mossanen, J.C.; Luedde, T.; Trautwein, C.; et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2020, 69, 551–563. [Google Scholar] [CrossRef]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell. 2019, 75, 644–660. [Google Scholar] [CrossRef]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wu, M.; Chen, B.; Wang, H. Myeloid cells in alcoholic liver diseases: Mechanism and prospect. Front. Immunol. 2022, 13, 971346. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Shen, H.; Li, X.; Wang, H. Endoplasmic reticulum stress in innate immune cells—A significant contribution to non-alcoholic fatty liver disease. Front. Immunol. 2022, 13, 951406. [Google Scholar] [CrossRef]
- Mentink-Kane, M.M.; Cheever, A.W.; Wilson, M.S.; Madala, S.K.; Beers, L.M.; Ramalingam, T.R.; Wynn, T.A. Accelerated and progressive and lethal liver fibrosis in mice that lack interleukin (IL)-10, IL-12p40, and IL-13Rα2. Gastroenterology 2011, 141, 2200–2209. [Google Scholar] [CrossRef]
- Li, F.; Li, Q.H.; Wang, J.Y.; Zhan, C.Y.; Xie, C.; Lu, W.Y. Effects of interferon-gamma liposomes targeted to platelet-derived growth factor receptor-beta on hepatic fibrosis in rats. J. Control Release. 2012, 159, 261–270. [Google Scholar] [CrossRef]
- Naim, A.; Pan, Q.; Baig, M.S. Matrix Metalloproteinases (MMPs) in Liver Diseases. J. Clin. Exp. Hepatol. 2017, 7, 367–372. [Google Scholar] [CrossRef]
- Robert, S.; Gicquel, T.; Bodin, A.; Lagente, V.; Boichot, E. Characterization of the MMP/TIMP Imbalance and Collagen Production Induced by IL-1β or TNF-α Release from Human Hepatic Stellate Cells. PLoS ONE 2016, 11, 0153118. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Pinzani, M. Epithelial-mesenchymal transition in chronic liver disease: Fibrogenesis or escape from death? J. Hepatol. 2011, 55, 459–465. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, Y.; Guo, D.Y.; Xu, B.; Shi, X.Y.; Li, J.T.; Duan, L.F. Study on the relationship between hepatic fibrosis and epithelial-mesenchymal transition in intrahepatic cells. Biomed. Pharmacother. 2020, 129, 110413. [Google Scholar] [CrossRef] [PubMed]
- Ribera, J.; Pauta, M.; Melgar-Lesmes, P.; Córdoba, B.; Bosch, A.; Calvo, M.; Rodrigo-Torres, D.; Sancho-Bru, P.; Mira, A.; Jiménez, W.; et al. A small population of liver endothelial cells undergoes endothelial-to-mesenchymal transition in response to chronic liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, 492–504. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Bocca, C.; Foglia, B.; Protopapa, F.; Maggiora, M.; Parola, M.; Cannito, S. Liver fibrogenesis: Un update on established and emerging basic concepts. Arch. Biochem. Biophys. 2020, 689, 108445. [Google Scholar] [CrossRef]
- Thoen, L.F.; Guimarães, E.L.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef]
- Mullan, L.A.; Mularczyk, E.J.; Kung, L.H.; Forouhan, M.; Wragg, J.M.; Goodacre, R.; Bateman, J.F.; Swanton, E.; Briggs, M.D.; Boot-Handford, R.P. Increased intracellular proteolysis reduces disease severity in an ER stress-associated dwarfism. J. Clin. Investig. 2017, 127, 3861–3865. [Google Scholar] [CrossRef]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010, 329, 229–232. [Google Scholar] [CrossRef]
- González-Rodríguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, 1179. [Google Scholar] [CrossRef] [PubMed]
- Bridle, K.R.; Popa, C.; Morgan, M.L.; Sobbe, A.L.; Clouston, A.D.; Fletcher, L.M.; Crawford, D.H. Rapamycin inhibits hepatic fibrosis in rats by attenuating multiple profibrogenic pathways. Liver Transpl. 2009, 15, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wei, B.; de Assuncao, T.M.; Liu, Z.; Hu, X.; Ibrahim, S.; Cooper, S.A.; Cao, S.; Shah, V.H.; Kostallari, E. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J. Hepatol. 2020, 73, 1144–1154. [Google Scholar] [CrossRef]
- Dobie, R.; Wilson-Kanamori, J.R.; Henderson, B.E.P.; Smith, J.R.; Matchett, K.P.; Portman, J.R.; Wallenborg, K.; Picelli, S.; Zagorska, A.; Pendem, S.V.; et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep. 2019, 29, 1832–1847. [Google Scholar] [CrossRef]
- Khan, M.A.; Fischer, J.; Harrer, L.; Schwiering, F.; Groneberg, D.; Friebe, A. Hepatic stellate cells in zone 1 engage in capillarization rather than myofibroblast formation in murine liver fibrosis. Sci. Rep. 2024, 14, 18840. [Google Scholar] [CrossRef]
- Du, K.; Jun, J.H.; Dutta, R.K.; Diehl, A.M. Plasticity, heterogeneity, and multifunctionality of hepatic stellate cells in liver pathophysiology. Hepatol. Commun. 2024, 8, 0411. [Google Scholar] [CrossRef]
- Zhang, W.J.; Chen, S.J.; Zhou, S.C.; Wu, S.Z.; Wang, H. Inflammasomes and Fibrosis. Front. Immunol. 2021, 12, 643149. [Google Scholar] [CrossRef]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef]
- Liu, C.; Chen, X.; Yang, L.; Kisseleva, T.; Brenner, D.A.; Seki, E. Transcriptional repression of the transforming growth factor β (TGF-β) Pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) by Nuclear Factor κB (NF-κB) p50 enhances TGF-β signaling in hepatic stellate cells. J. Biol. Chem. 2014, 289, 7082–7091. [Google Scholar] [CrossRef]
- Bartneck, M.; Koppe, C.; Fech, V.; Warzecha, K.T.; Kohlhepp, M.; Huss, S.; Weiskirchen, R.; Trautwein, C.; Luedde, T.; Tacke, F. Roles of CCR2 and CCR5 for Hepatic Macrophage Polarization in Mice With Liver Parenchymal Cell-Specific NEMO Deletion. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P.; Thompson, A.; Henderson, N.C. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim. Biophys. Acta 2013, 1832, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; de Minicis, S.; Inokuchi, S.; Taura, K.; Miyai, K.; van Rooijen, N.; Schwabe, R.F.; Brenner, D.A. CCR2 promotes hepatic fibrosis in mice. Hepatology 2009, 50, 185–197. [Google Scholar] [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Cai, X.; Li, Z.; Zhang, Q.; Qu, Y.; Xu, M.; Wan, X.; Lu, L. CXCL6-EGFR-induced Kupffer cells secrete TGF-β1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2 pathway in liver fibrosis. J. Cell Mol. Med. 2018, 22, 5050–5061. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Sasaki, R.; Devhare, P.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757. [Google Scholar] [CrossRef]
- Roh, Y.S.; Seki, E. Chemokines and Chemokine Receptors in the Development of NAFLD. Adv. Exp. Med. Biol. 2018, 1061, 45–53. [Google Scholar]
- Matsuda, M.; Seki, E. Hepatic Stellate Cell-Macrophage Crosstalk in Liver Fibrosis and Carcinogenesis. Semin. Liver Dis. 2020, 40, 307–320. [Google Scholar] [CrossRef]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef]
- Hedger, M.P.; de Kretser, D.M. The activins and their binding protein, follistatin-Diagnostic and therapeutic targets in inflammatory disease and fibrosis. Cytokine Growth Factor. Rev. 2013, 24, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Youssef, S.S.; Mostafa, A.; Saad, A.; Omran, M.H.; El Zanaty, T.; Mohamed Seif, S. Impact of IL12B gene rs 3212227 polymorphism on fibrosis, liver inflammation, and response to treatment in genotype 4 Egyptian hepatitis C patients. Dis. Markers 2013, 35, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.M.; Modak, R.V.; Macdonald, F.; Li, F.; Smyth, D.J.; Dorward, D.A.; Blair, N.; Husovsky, C.; Muir, A.; Giampazolias, E.; et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 2019, 50, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Wang, L.; Li, Y.; Li, X. Liver sinusoidal endothelial cell: An important yet often overlooked player in the liver fibrosis. Clin. Mol. Hepatol. 2024, 30, 303–325. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Wojtalla, A.; Siegmund, S.V.; Endl, E.; Diehl, L.; Abdullah, Z.; Kurts, C.; Knolle, P.A. Murine hepatic stellate cells veto CD8 T cell activation by a CD54-dependent mechanism. Hepatology 2011, 54, 262–272. [Google Scholar] [CrossRef]
- Géraud, C.; Evdokimov, K.; Straub, B.K.; Peitsch, W.K.; Demory, A.; Dörflinger, Y.; Schledzewski, K.; Schmieder, A.; Schemmer, P.; Augustin, H.G.; et al. Unique cell type-specific junctional complexes in vascular endothelium of human and rat liver sinusoids. PLoS ONE 2012, 7, 34206. [Google Scholar] [CrossRef]
- Jophlin, L.L.; Cao, S.; Shah, V.H. The Transcriptome of Hepatic Fibrosis Revealed by Single-Cell RNA Sequencing. Hepatology 2020, 71, 1865–1867. [Google Scholar] [CrossRef]
- Liu, D.; Fu, X.; Wang, Y.; Wang, X.; Wang, H.; Wen, J.; Kang, N. Protein diaphanous homolog 1 (Diaph1) promotes myofibroblastic activation of hepatic stellate cells by regulating Rab5a activity and TGFβ receptor endocytosis. FASEB J. 2020, 34, 7345–7359. [Google Scholar] [CrossRef]
- Hammoutene, A.; Biquard, L.; Lasselin, J.; Kheloufi, M.; Tanguy, M.; Vion, A.C.; Mérian, J.; Colnot, N.; Loyer, X.; Tedgui, A.; et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J. Hepatol. 2020, 72, 528–538. [Google Scholar] [CrossRef]
- Li, Z.; Chen, B.; Dong, W.; Kong, M.; Fan, Z.; Yu, L.; Wu, D.; Lu, J.; Xu, Y. MKL1 promotes endothelial-to-mesenchymal transition and liver fibrosis by activating TWIST1 transcription. Cell Death Dis. 2019, 10, 899. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Yang, M.; Wang, Y. Roles of transforming growth factor-β signaling in liver disease. World J. Hepatol. 2024, 16, 973–979. [Google Scholar] [CrossRef]
- Carter, J.K.; Friedman, S.L. Hepatic Stellate Cell-Immune Interactions in NASH. Front. Endocrinol. 2022, 13, 867940. [Google Scholar] [CrossRef]
- Xiang, D.M.; Sun, W.; Ning, B.F.; Zhou, T.F.; Li, X.F.; Zhong, W.; Cheng, Z.; Xia, M.Y.; Wang, X.; Deng, X.; et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut 2018, 67, 1704–1715. [Google Scholar] [CrossRef]
- Kou, K.; Li, S.; Qiu, W.; Fan, Z.; Li, M.; Lv, G. Hypoxia-inducible factor 1α/IL-6 axis in activated hepatic stellate cells aggravates liver fibrosis. Biochem. Biophys. Res. Commun. 2023, 653, 21–30. [Google Scholar] [CrossRef]
- Eguchi, A.; Yan, R.; Pan, S.Q.; Wu, R.; Kim, J.; Chen, Y.; Ansong, C.; Smith, R.D.; Tempaku, M.; Ohno-Machado, L.; et al. Comprehensive characterization of hepatocyte-derived extracellular vesicles identifies direct miRNA-based regulation of hepatic stellate cells and DAMP-based hepatic macrophage IL-1β and IL-17 upregulation in alcoholic hepatitis mice. J. Mol. Med. 2020, 98, 1021–1034. [Google Scholar] [CrossRef]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci Signal. 2019, 12, eaao3469. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef]
- McConnell, M.J.; Kostallari, E.; Ibrahim, S.H.; Iwakiri, Y. The evolving role of liver sinusoidal endothelial cells in liver health and disease. Hepatology 2023, 78, 649–669. [Google Scholar] [CrossRef]
- Du, W.; Wang, L. The Crosstalk Between Liver Sinusoidal Endothelial Cells and Hepatic Microenvironment in NASH Related Liver Fibrosis. Front. Immunol. 2022, 13, 936196. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Furuta, K.; Islam, S.; Caporarello, N.; Kostallari, E.; Dielis, K.; Tschumperlin, D.J.; Hirsova, P.; Ibrahim, S.H. Liver sinusoidal endothelial cell expressed vascular cell adhesion molecule 1 promotes liver fibrosis. Front. Immunol. 2022, 13, 983255. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O.; Phillips, A.; Ruggiero, K.; Bartlett, A.; Dunbar, P.R. Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci. Rep. 2017, 7, 44356. [Google Scholar] [CrossRef]
- Wei, M.; Zhang, Y.; Zhang, H.; Huang, Z.; Miao, H.; Zhang, T.; Lu, B.; Ji, L. HMGB1 induced endothelial to mesenchymal transition in liver fibrosis: The key regulation of early growth response factor 1. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130202. [Google Scholar] [CrossRef]
- Ruan, B.; Duan, J.L.; Xu, H.; Tao, K.S.; Han, H.; Dou, G.R.; Wang, L. Capillarized liver sinusoidal endothelial cells undergo partial endothelial-mesenchymal transition to actively deposit sinusoidal ECM in liver fibrosis. Front. Cell Dev. Biol. 2021, 9, 671081. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- de Gouville, A.C.; Boullay, V.; Krysa, G.; Pilot, J.; Brusq, J.M.; Loriolle, F.; Gauthier, J.M.; Papworth, S.A.; Laroze, A.; Gellibert, F.; et al. Inhibition of TGF-beta signaling by an ALK5 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br. J. Pharmacol. 2005, 145, 166–177. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Q.; Tu, K.; Wang, Y.; Wang, X.; Liu, D.; Chen, C.; Liu, D.; Yang, R.; Qiu, W.; et al. Focal Adhesion Kinase Promotes Hepatic Stellate Cell Activation by Regulating Plasma Membrane Localization of TGFβ Receptor 2. Hepatol Commun. 2019, 4, 268–283. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef]
- Dong, X.; Zhao, B.; Iacob, R.E.; Zhu, J.; Koksal, A.C.; Lu, C.; Engen, J.R.; Springer, T.A. Force interacts with macromolecular structure in activation of TGF-β. Nature 2017, 542, 55–59. [Google Scholar] [CrossRef]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The αvβ1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288ra79. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef]
- Peng, Z.W.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Dixit, R.; Weinreb, P.H.; Violette, S.; Sheppard, D.; Schuppan, D.; et al. Integrin αvβ6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016, 63, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Dolinski, B.M.; Kikuchi, N.; Leone, D.R.; Peters, M.G.; Weinreb, P.H.; Violette, S.M.; Bissell, D.M. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology 2007, 46, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef]
- Ahmed, H.; Umar, M.I.; Imran, S.; Javaid, F.; Syed, S.K.; Riaz, R.; Hassan, W. TGF-β1 signaling can worsen NAFLD with liver fibrosis backdrop. Exp. Mol. Pathol. 2022, 124, 104733. [Google Scholar] [CrossRef]
- Wang, B.; Koh, P.; Winbanks, C.; Coughlan, M.T.; McClelland, A.; Watson, A.; Jandeleit-Dahm, K.; Burns, W.C.; Thomas, M.C.; Cooper, M.E.; et al. miR-200a Prevents renal fibrogenesis through repression of TGF-β2 expression. Diabetes 2011, 60, 280–287. [Google Scholar] [CrossRef]
- Voumvouraki, A.; Koulentaki, M.; Tzardi, M.; Sfakianaki, O.; Manousou, P.; Notas, G.; Kouroumalis, E. Increased ΤGF-β3 in primary biliary cirrhosis: An abnormality related to pathogenesis? World J. Gastroenterol. 2010, 16, 5057–5064. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Kurachi, M. CD8+ T cell exhaustion. Semin. Immunopathol. 2019, 41, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Li, T.Y.; Yang, Y.; Zhou, G.; Tu, Z.K. Immune suppression in chronic hepatitis B infection associated liver disease: A review. World J. Gastroenterol. 2019, 25, 3527–3537. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhai, N.; Wang, Z.; Song, H.; Yang, Y.; Cui, A.; Li, T.; Wang, G.; Niu, J.; Crispe, I.N.; et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut 2018, 67, 2035–2044. [Google Scholar] [CrossRef]
- Kiagiadaki, F.; Kampa, M.; Voumvouraki, A.; Castanas, E.; Kouroumalis, E.; Notas, G. Activin-A causes Hepatic stellate cell activation via the induction of TNFα and TGFβ in Kupffer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 891–899. [Google Scholar] [CrossRef]
- Tsuda, M.; Zhang, W.; Yang, G.X.; Tsuneyama, K.; Ando, Y.; Kawata, K.; Park, O.; Leung, P.S.; Coppel, R.L.; Ansari, A.A.; et al. Deletion of interleukin (IL)-12p35 induces liver fibrosis in dominant-negative TGFβ receptor type II mice. Hepatology 2013, 57, 806–816. [Google Scholar] [CrossRef]
- Masola, V.; Carraro, A.; Granata, S.; Signorini, L.; Bellin, G.; Violi, P.; Lupo, A.; Tedeschi, U.; Onisto, M.; Gambaro, G.; et al. In vitro effects of interleukin (IL)-1 beta inhibition on the epithelial-to-mesenchymal transition (EMT) of renal tubular and hepatic stellate cells. J. Transl. Med. 2019, 17, 12. [Google Scholar] [CrossRef]
- Sudo, K.; Yamada, Y.; Moriwaki, H.; Saito, K.; Seishima, M. Lack of tumor necrosis factor receptor type 1 inhibits liver fibrosis induced by carbon tetrachloride in mice. Cytokine 2005, 29, 236–244. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Karlsson, S. Inflammation and TGF beta 1: Lessons from the TGF beta 1 null mouse. Res. Immunol. 1997, 148, 453–456. [Google Scholar] [CrossRef]
- Tsomidis, I.; Notas, G.; Xidakis, C.; Voumvouraki, A.; Samonakis, D.N.; Koulentaki, M.; Kouroumalis, E. Enzymes of Fibrosis in Chronic Liver Disease. Biomedicines 2022, 10, 3179. [Google Scholar] [CrossRef] [PubMed]
- Akkız, H.; Gieseler, R.K.; Canbay, A. Liver Fibrosis: From Basic Science towards Clinical Progress, Focusing on the Central Role of Hepatic Stellate Cells. Int. J. Mol. Sci. 2024, 25, 7873. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Tacke, F. Hepatic inflammatory responses in liver fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Park, O.; Suh, Y.G.; Byun, J.S.; Park, S.Y.; Choi, E.; Kim, J.K.; Ko, H.; Wang, H.; Miller, A.M.; et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology 2011, 53, 1342–1351. [Google Scholar] [CrossRef]
- Glässner, A.; Eisenhardt, M.; Krämer, B.; Körner, C.; Coenen, M.; Sauerbruch, T.; Spengler, U.; Nattermann, J. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab. Investig. 2012, 92, 967–977. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Wang, H.; Yin, S. Natural killer T cells in liver injury, inflammation and cancer. Expert. Rev. Gastroenterol. Hepatol. 2015, 9, 1077–1085. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Qiu, Y.; Shen, M.; Wang, L.; Shao, J.; Zhang, F.; Xu, X.; Zhang, Z.; Guo, M.; et al. Artesunate Induces Ferroptosis in Hepatic Stellate Cells and Alleviates Liver Fibrosis via the ROCK1/ATF3 Axis. J. Clin. Transl. Hepatol. 2024, 12, 36–51. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Campana, L.; Esser, H.; Huch, M.; Forbes, S. Liver regeneration and inflammation: From fundamental science to clinical applications. Nat. Rev. Mol. Cell Biol. 2021, 22, 608–624. [Google Scholar] [CrossRef]
- Vonderlin, J.; Chavakis, T.; Sieweke, M.; Tacke, F. The Multifaceted Roles of Macrophages in NAFLD Pathogenesis. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Puengel, T.; Loomba, R.; Friedman, S.L. An integrated view of anti-inflammatory and antifibrotic targets for the treatment of NASH. J. Hepatol. 2023, 79, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar]
- Zhang, L.; Schuppan, D. Traditional Chinese Medicine (TCM) for fibrotic liver disease: Hope and hype. J. Hepatol. 2014, 61, 166–168. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, J.F.; Ouyang, H. Progress on traditional Chinese medicine in improving hepatic fibrosis through inhibiting oxidative stress. World J. Hepatol. 2023, 15, 1091–1108. [Google Scholar] [CrossRef]
- Dai, Z.; Liao, X.; Wieland, L.S.; Hu, J.; Wang, Y.; Kim, T.H.; Liu, J.P.; Zhan, S.; Robinson, N. Cochrane systematic reviews on traditional Chinese medicine: What matters-the quantity or quality of evidence? Phytomedicine 2022, 98, 153921. [Google Scholar] [CrossRef]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef]
- Gong, J.; Tu, W.; Liu, J.; Tian, D. Hepatocytes: A key role in liver inflammation. Front. Immunol. 2023, 13, 1083780. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Schildberg, F.A.; Hegenbarth, S.I.; Schumak, B.; Scholz, K.; Limmer, A.; Knolle, P.A. Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur. J. Immunol. 2008, 38, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Diehl, L.; Schurich, A.; Grochtmann, R.; Hegenbarth, S.; Chen, L.; Knolle, P.A. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology 2008, 47, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N.; Giannandrea, M.; Klein, I.; John, B.; Sampson, B.; Wuensch, S. Cellular and molecular mechanisms of liver tolerance. Immunol. Rev. 2006, 213, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Isse, K.; Sato, Y.; Ozaki, S.; Nakanuma, Y. Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 2006, 26, 935–942. [Google Scholar] [CrossRef]
- Zheng, M.; Tian, Z. Liver-Mediated Adaptive Immune Tolerance. Front. Immunol. 2019, 10, 2525. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef]
- Yu, J.; Chen, Y.; Wu, Y.; Ye, L.; Lian, Z.; Wei, H.; Sun, R.; Tian, Z. The differential organogenesis and functionality of two liver-draining lymph nodes in mice. J. Autoimmun. 2017, 84, 109–121. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef]
- Jung, J.; Zeng, H.; Horng, T. Metabolism as a guiding force for immunity. Nat. Cell Biol. 2019, 21, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, H.; Yao, Y.; Zhang, X.; Guan, Y.; Zheng, F. CD4+ T cell activation and inflammation in NASH-related fibrosis. Front. Immunol. 2022, 13, 967410. [Google Scholar] [CrossRef]
- Alegre, F.; Pelegrin, P.; Feldstein, A.E. Inflammasomes in Liver Fibrosis. Semin. Liver Dis. 2017, 37, 119–127. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, R.; Wang, X.; Bao, X.; Lu, C. Roles of necroptosis in alcoholic liver disease and hepatic pathogenesis. Cell Prolif. 2022, 55, 13193. [Google Scholar] [CrossRef]
- Bataller, R.; Arab, J.P.; Shah, V.H. Alcohol-Associated Hepatitis. N. Engl. J. Med. 2022, 387, 2436–2448. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Shah, V.H. Gut microbiota in non-alcoholic fatty liver disease and alcohol-related liver disease: Current concepts and perspectives. Hepatol. Res. 2020, 50, 407–418. [Google Scholar] [CrossRef]
- Albillos, A.; Martin-Mateos, R.; Van der Merwe, S.; Wiest, R.; Jalan, R.; Álvarez-Mon, M. Cirrhosis-associated immune dysfunction. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 112–134. [Google Scholar] [CrossRef]
- Wiering, L.; Subramanian, P.; Hammerich, L. Hepatic Stellate Cells: Dictating Outcome in Nonalcoholic Fatty Liver Disease. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 1277–1292. [Google Scholar] [CrossRef]
- Zhangdi, H.J.; Su, S.B.; Wang, F.; Liang, Z.Y.; Yan, Y.D.; Qin, S.Y.; Jiang, H.X. Crosstalk network among multiple inflammatory mediators in liver fibrosis. World J. Gastroenterol. 2019, 25, 4835–4849. [Google Scholar] [CrossRef]
- Berumen, J.; Baglieri, J.; Kisseleva, T.; Mekeel, K. Liver fibrosis: Pathophysiology and clinical implications. WIREs Mech. Dis. 2021, 13, 1499. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kanai, T. Role of toll-like receptors in immune activation and tolerance in the liver. Front. Immunol. 2014, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Torre, P.; Motta, B.M.; Sciorio, R.; Masarone, M.; Persico, M. Inflammation and Fibrogenesis in MAFLD: Role of the Hepatic Immune System. Front. Med. 2021, 8, 781567. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, Y.; Wu, X.; Wang, X.; Niu, J. Identification of Immune Microenvironment Changes and the Expression of Immune-Related Genes in Liver Cirrhosis. Front. Immunol. 2022, 13, 918445. [Google Scholar] [CrossRef]
- Khanam, A.; Chua, J.V.; Kottilil, S. Immunopathology of Chronic Hepatitis B Infection: Role of Innate and Adaptive Immune Response in Disease Progression. Int. J. Mol. Sci. 2021, 22, 5497. [Google Scholar] [CrossRef]
- Martinon, F. Detection of immune danger signals by NALP3. J. Leukoc. Biol. 2008, 83, 507–511. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef]
- Kong, F.; You, H.; Zheng, K.; Tang, R.; Zheng, C. The crosstalk between pattern-recognition receptor signaling and calcium signaling. Int. J. Biol. Macromol. 2021, 192, 745–756. [Google Scholar] [CrossRef]
- Wree, A.; Holtmann, T.M.; Inzaugarat, M.E.; Feldstein, A.E. Novel Drivers of the Inflammatory Response in Liver Injury and Fibrosis. Semin. Liver Dis. 2019, 39, 275–282. [Google Scholar] [CrossRef]
- Jiang, Y.; Que, W.; Zhu, P.; Li, X.K. The Role of Diverse Liver Cells in Liver Transplantation Tolerance. Front. Immunol. 2020, 11, 1203. [Google Scholar] [CrossRef] [PubMed]
- Mühlbauer, M.; Fleck, M.; Schütz, C.; Weiss, T.; Froh, M.; Blank, C.; Schölmerich, J.; Hellerbrand, C. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J. Hepatol. 2006, 45, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Mooring, M.; Fowl, B.H.; Lum, S.Z.C.; Liu, Y.; Yao, K.; Softic, S.; Kirchner, R.; Bernstein, A.; Singhi, A.D.; Jay, D.G.; et al. Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator With PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology 2020, 71, 1813–1830. [Google Scholar] [CrossRef] [PubMed]
- Futakuchi, A.; Inoue, T.; Wei, F.Y.; Inoue-Mochita, M.; Fujimoto, T.; Tomizawa, K.; Tanihara, H. YAP/TAZ Are Essential for TGF-β2-Mediated Conjunctival Fibrosis. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3069–3078. [Google Scholar] [CrossRef] [PubMed]
- Filliol, A.; Schwabe, R.F. FoxM1 Induces CCl2 Secretion From Hepatocytes Triggering Hepatic Inflammation, Injury, Fibrosis, and Liver Cancer. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 555–556. [Google Scholar] [CrossRef]
- Kurahashi, T.; Yoshida, Y.; Ogura, S.; Egawa, M.; Furuta, K.; Hikita, H.; Kodama, T.; Sakamori, R.; Kiso, S.; Kamada, Y.; et al. Forkhead Box M1 Transcription Factor Drives Liver Inflammation Linking to Hepatocarcinogenesis in Mice. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 425–446. [Google Scholar] [CrossRef]
- Lu, J.G.; Iyasu, A.; French, B.; Tillman, B.; French, S.W. Overexpression of MHCII by hepatocytes in alcoholic hepatitis (AH) compared to non-alcoholic steatohepatitis (NASH) and normal controls. Alcohol 2020, 84, 27–32. [Google Scholar] [CrossRef]
- Huby, T.; Gautier, E.L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 2022, 22, 429–443. [Google Scholar] [CrossRef]
- Zhang, Q.; Qu, Y.; Zhang, Q.; Li, F.; Li, B.; Li, Z.; Dong, Y.; Lu, L.; Cai, X. Exosomes derived from hepatitis B virus-infected hepatocytes promote liver fibrosis via miR-222/TFRC axis. Cell Biol. Toxicol. 2023, 39, 467–481. [Google Scholar] [CrossRef]
- Francis, H.; Wu, N.; Alpini, G.; Meng, F. Hepatocyte Autophagy: Maintaining a Toxic-Free Environment. Hepatology 2020, 72, 371–374. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, S.G. Endoplasmic reticulum stress and autophagy dysregulation in alcoholic and non-alcoholic liver diseases. Clin. Mol. Hepatol. 2020, 26, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Kouroumalis, E.; Voumvouraki, A.; Augoustaki, A.; Samonakis, D.N. Autophagy in liver diseases. World J. Hepatol. 2021, 13, 6–65. [Google Scholar] [CrossRef]
- Shiode, Y.; Hikita, H.; Tanaka, S.; Shirai, K.; Doi, A.; Sakane, S.; Kai, Y.; Nakabori, T.; Yamada, R.; Kodama, T.; et al. Hepatitis C virus enhances Rubicon expression, leading to autophagy inhibition and intracellular innate immune activation. Sci. Rep. 2020, 10, 15290. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef]
- Tavera-Mendoza, L.E.; Westerling, T.; Libby, E.; Marusyk, A.; Cato, L.; Cassani, R.; Cameron, L.A.; Ficarro, S.B.; Marto, J.A.; Klawitter, J.; et al. Vitamin D receptor regulates autophagy in the normal mammary gland and in luminal breast cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, 2186–2194. [Google Scholar] [CrossRef]
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver vitamin D receptor, CYP2R1, and CYP27A1 expression: Relationship with liver histology and vitamin D3 levels in patients with nonalcoholic steatohepatitis or hepatitis C virus. Hepatology 2012, 56, 2180–2187. [Google Scholar] [CrossRef]
- He, W.; Ni, W.; Zhao, L.; Wang, X.; Liu, L.; Fan, Z. MicroRNA-125a/VDR axis impaired autophagic flux and contributed to fibrosis in a CCL4-induced mouse model and patients with liver cirrhosis. Life Sci. 2021, 264, 118666. [Google Scholar] [CrossRef]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Mollica, M.P.; Lionetti, L.; Putti, R.; Cavaliere, G.; Gaita, M.; Barletta, A. From chronic overfeeding to hepatic injury: Role of endoplasmic reticulum stress and inflammation. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 222–230. [Google Scholar] [CrossRef]
- Aravinthan, A.; Pietrosi, G.; Hoare, M.; Jupp, J.; Marshall, A.; Verrill, C.; Davies, S.; Bateman, A.; Sheron, N.; Allison, M.; et al. Hepatocyte expression of the senescence marker p21 is linked to fibrosis and an adverse liver-related outcome in alcohol-related liver disease. PLoS ONE 2013, 8, 72904. [Google Scholar] [CrossRef]
- Aravinthan, A.; Scarpini, C.; Tachtatzis, P.; Verma, S.; Penrhyn-Lowe, S.; Harvey, R.; Davies, S.E.; Allison, M.; Coleman, N.; Alexander, G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol. 2013, 58, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.; Moch, A.; Saab, S. Relationship between Telomere Maintenance and Liver Disease. Gut Liver. 2019, 13, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Wijayasiri, P.; Astbury, S.; Kaye, P.; Oakley, F.; Alexander, G.J.; Kendall, T.J.; Aravinthan, A.D. Role of Hepatocyte Senescence in the Activation of Hepatic Stellate Cells and Liver Fibrosis Progression. Cells 2022, 11, 2221. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, X.J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef]
- Lee, Y.S.; Seki, E. In Vivo and In Vitro Models to Study Liver Fibrosis: Mechanisms and Limitations. Cell Mol. Gastroenterol. Hepatol. 2023, 16, 355–367. [Google Scholar] [CrossRef]
- David, B.A.; Rezende, R.M.; Antunes, M.M.; Santos, M.M.; Freitas Lopes, M.A.; Diniz, A.B.; Sousa Pereira, R.V.; Marchesi, S.C.; Alvarenga, D.M.; Nakagaki, B.N.; et al. Combination of Mass Cytometry and Imaging Analysis Reveals Origin, Location, and Functional Repopulation of Liver Myeloid Cells in Mice. Gastroenterology 2016, 15, 1176–1191. [Google Scholar] [CrossRef]
- Devisscher, L.; Scott, C.L.; Lefere, S.; Raevens, S.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Geerts, A.; Guilliams, M.; Van Vlierberghe, H. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell Immunol. 2017, 322, 74–83. [Google Scholar] [CrossRef]
- Ait Ahmed, Y.; Lafdil, F.; Tacke, F. Ambiguous Pathogenic Roles of Macrophages in Alcohol-Associated Liver Diseases. Hepat. Med. 2023, 15, 113–127. [Google Scholar] [CrossRef]
- Wang, Z.; Du, K.; Jin, N.; Tang, B.; Zhang, W. Macrophage in liver Fibrosis: Identities and mechanisms. Int. Immunopharmacol. 2023, 120, 110357. [Google Scholar] [CrossRef] [PubMed]
- An, S.Y.; Petrescu, A.D.; DeMorrow, S. Targeting Certain Interleukins as Novel Treatment Options for Liver Fibrosis. Front. Pharmacol. 2021, 12, 645703. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, Q.; Wu, W.; Huang, X.; Broering, R.; Werner, M.; Roggendorf, M.; Yang, D.; Lu, M. TLR2 Stimulation Strengthens Intrahepatic Myeloid-Derived Cell-Mediated T Cell Tolerance through Inducing Kupffer Cell Expansion and IL-10 Production. J. Immunol. 2018, 200, 2341–2351. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, H.; Chen, Z.; Chen, L.; Pan, F.; Zhou, Q. Proliferation of CD11b+ myeloid cells induced by TLR4 signaling promotes hepatitis B virus clearance. Cytokine 2022, 153, 155867. [Google Scholar] [CrossRef]
- Li, Y.; He, M.; Wang, Z.; Duan, Z.; Guo, Z.; Wang, Z.; Gong, R.; Chu, T.; Cai, J.; Gao, B. STING signaling activation inhibits HBV replication and attenuates the severity of liver injury and HBV-induced fibrosis. Cell Mol. Immunol. 2022, 19, 92–107. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, X.; Wang, Z.; Shu, W.; Li, L.; Li, Y.; Guo, Z.; Gao, B.; Xiong, S. Nuclear Sensor Interferon-Inducible Protein 16 Inhibits the Function of Hepatitis B Virus Covalently Closed Circular DNA by Integrating Innate Immune Activation and Epigenetic Suppression. Hepatology 2020, 71, 1154–1169. [Google Scholar] [CrossRef]
- Pose, E.; Coll, M.; Martínez-Sánchez, C.; Zeng, Z.; Surewaard, B.G.J.; Català, C.; Velasco-de Andrés, M.; Lozano, J.J.; Ariño, S.; Fuster, D.; et al. Programmed Death Ligand 1 Is Overexpressed in Liver Macrophages in Chronic Liver Diseases, and Its Blockade Improves the Antibacterial Activity Against Infections. Hepatology 2021, 74, 296–311. [Google Scholar] [CrossRef]
- Lough, J.; Rosenthall, L.; Arzoumanian, A.; Goresky, C.A. Kupffer cell depletion associated with capillarization of liver sinusoids in carbon tetrachloride-induced rat liver cirrhosis. J. Hepatol. 1987, 5, 190–198. [Google Scholar] [CrossRef]
- Buonomo, E.L.; Mei, S.; Guinn, S.R.; Leo, I.R.; Peluso, M.J.; Nolan, M.A.; Schildberg, F.A.; Zhao, L.; Lian, C.; Xu, S.; et al. Liver stromal cells restrict macrophage maturation and stromal IL-6 limits the differentiation of cirrhosis-linked macrophages. J. Hepatol. 2022, 76, 1127–1137. [Google Scholar] [CrossRef]
- Li, X.; Hollingshead, N.; Lampert, S.; Truong, C.D.; Li, W.; Niu, J.; Crispe, I.N.; Soysa, R. A conserved pathway of transdifferentiation in murine Kupffer cells. Eur. J. Immunol. 2021, 51, 2452–2463. [Google Scholar] [CrossRef]
- Vadasz, Z.; Kessler, O.; Akiri, G.; Gengrinovitch, S.; Kagan, H.M.; Baruch, Y.; Izhak, O.B.; Neufeld, G. Abnormal deposition of collagen around hepatocytes in Wilson’s disease is associated with hepatocyte specific expression of lysyl oxidase and lysyl oxidase like protein-2. J. Hepatol. 2005, 43, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Ding, J.; Wang, M.; Zhang, J.; Zhu, X.; Guan, W. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int. J. Biol. Sci. 2018, 14, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, F. Infusion of Kupffer Cells Expanded in Vitro Ameliorated Liver Fibrosis in a Murine Model of Liver Injury. Cell Transplant. 2021, 30, 9636897211004090. [Google Scholar] [CrossRef]
- Wu, H.; Chen, G.; Wang, J.; Deng, M.; Yuan, F.; Gong, J. TIM-4 interference in Kupffer cells against CCL4-induced liver fibrosis by mediating Akt1/Mitophagy signalling pathway. Cell Prolif. 2020, 53, 12731. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Bozorgzadeh, A.; Pierce, R.H.; Kurtis, J.; Crispe, I.N.; Orloff, M.S. TLR-dependent cross talk between human Kupffer cells and NK cells. J. Exp. Med. 2008, 205, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Boltjes, A.; van Montfoort, N.; Biesta, P.J.; Op den Brouw, M.L.; Kwekkeboom, J.; van der Laan, L.J.; Janssen, H.L.; Boonstra, A.; Woltman, A.M. Kupffer cells interact with hepatitis B surface antigen in vivo and in vitro, leading to proinflammatory cytokine production and natural killer cell function. J. Infect. Dis. 2015, 211, 1268–1278. [Google Scholar] [CrossRef]
- Hosomura, N.; Kono, H.; Tsuchiya, M.; Ishii, K.; Ogiku, M.; Matsuda, M.; Fujii, H. HCV-related proteins activate Kupffer cells isolated from human liver tissues. Dig. Dis. Sci. 2011, 56, 1057–1064. [Google Scholar] [CrossRef]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef]
- Jindal, A.; Bruzzì, S.; Sutti, S.; Locatelli, I.; Bozzola, C.; Paternostro, C.; Parola, M.; Albano, E. Fat-laden macrophages modulate lobular inflammation in nonalcoholic steatohepatitis (NASH). Exp. Mol. Pathol. 2015, 99, 155–162. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Morello, E.; Bocca, C.; Foglia, B.; Benetti, E.; Novo, E.; Chiazza, F.; Rogazzo, M.; Fantozzi, R.; Povero, D.; et al. Microvesicles released from fat-laden cells promote activation of hepatocellular NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS ONE 2017, 12, e0172575. [Google Scholar] [CrossRef] [PubMed]
- Morello, E.; Sutti, S.; Foglia, B.; Novo, E.; Cannito, S.; Bocca, C.; Rajsky, M.; Bruzzì, S.; Abate, M.L.; Rosso, C.; et al. Hypoxia-inducible factor 2α drives nonalcoholic fatty liver progression by triggering hepatocyte release of histidine-rich glycoprotein. Hepatology 2018, 67, 2196–2214. [Google Scholar] [CrossRef]
- Bartneck, M.; Fech, V.; Ehling, J.; Govaere, O.; Warzecha, K.T.; Hittatiya, K.; Vucur, M.; Gautheron, J.; Luedde, T.; Trautwein, C.; et al. Histidine-rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology 2016, 63, 1310–1324. [Google Scholar] [CrossRef]
- Suraweera, D.B.; Weeratunga, A.N.; Hu, R.W.; Pandol, S.J.; Hu, R. Alcoholic hepatitis: The pivotal role of Kupffer cells. World J. Gastrointest. Pathophysiol. 2015, 6, 90–98. [Google Scholar] [CrossRef]
- Ju, C.; Mandrekar, P. Macrophages and Alcohol-Related Liver Inflammation. Alcohol Res. 2015, 37, 251–262. [Google Scholar]
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Calmus, Y.; Poupon, R. Shaping macrophages function and innate immunity by bile acids: Mechanisms and implication in cholestatic liver diseases. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 550–556. [Google Scholar] [CrossRef]
- Gong, Z.; Zhou, J.; Zhao, S.; Tian, C.; Wang, P.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget 2016, 7, 83951–83963. [Google Scholar] [CrossRef]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Duwaerts, C.C.; Gehring, S.; Cheng, C.W.; van Rooijen, N.; Gregory, S.H. Contrasting responses of Kupffer cells and inflammatory mononuclear phagocytes to biliary obstruction in a mouse model of cholestatic liver injury. Liver Int. 2013, 33, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Hisamatsu, T.; Shimamura, K.; Yoneno, K.; Adachi, M.; Naruse, H.; Igarashi, T.; Higuchi, H.; Matsuoka, K.; Kitazume, M.T.; et al. Activated hepatic stellate cells mediate the differentiation of macrophages. Hepatol. Res. 2013, 43, 658–669. [Google Scholar] [CrossRef]
- Xiao, C.; Ghosh, S. NF-kappaB, an evolutionarily conserved mediator of immune and inflammatory responses. Adv. Exp. Med. Biol. 2005, 560, 41–45. [Google Scholar]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Twu, Y.C.; Lee, T.S.; Lin, Y.L.; Hsu, S.M.; Wang, Y.H.; Liao, C.Y.; Wang, C.K.; Liang, Y.C.; Liao, Y.J. Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation. Int. J. Mol. Sci. 2016, 17, 1122. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Usui, S.; Furuhashi, H.; Kimura, A.; Nishiyama, K.; et al. Acyl-CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J. Hepatol. 2014, 61, 98–106. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cai, B.; Yang, X.; Sonubi, O.O.; Zheng, Z.; Ramakrishnan, R.; Shi, H.; Valenti, L.; Pajvani, U.B.; Sandhu, J.; et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 969–986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yan, X.; Yang, C.; Zhan, Q.; Fu, Y.; Luo, H.; Luo, H. Intrahepatic T helper 17 cells recruited by hepatitis B virus X antigen-activated hepatic stellate cells exacerbate the progression of chronic hepatitis B virus infection. J. Viral Hepat. 2020, 27, 1138–1149. [Google Scholar] [CrossRef]
- Chou, H.S.; Hsieh, C.C.; Yang, H.R.; Wang, L.; Arakawa, Y.; Brown, K.; Wu, Q.; Lin, F.; Peters, M.; Fung, J.J.; et al. Hepatic stellate cells regulate immune response by way of induction of myeloid suppressor cells in mice. Hepatology 2011, 53, 1007–1019. [Google Scholar] [CrossRef]
- Jiang, G.; Yang, H.R.; Wang, L.; Wildey, G.M.; Fung, J.; Qian, S.; Lu, L. Hepatic stellate cells preferentially expand allogeneic CD4+ CD25+ FoxP3+ regulatory T cells in an IL-2-dependent manner. Transplantation 2008, 86, 1492–1502. [Google Scholar] [CrossRef]
- Li, Y.; Lu, L.; Qian, S.; Fung, J.J.; Lin, F. Hepatic Stellate Cells Directly Inhibit B Cells via Programmed Death-Ligand 1. J. Immunol. 2016, 196, 1617–1625. [Google Scholar] [CrossRef]
- Ding, X.; Saxena, N.K.; Lin, S.; Xu, A.; Srinivasan, S.; Anania, F.A. The roles of leptin and adiponectin: A novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am. J. Pathol. 2005, 166, 1655–1669. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Saxena, N.K.; Anania, F.A. Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef]
- Zhai, X.; Yan, K.; Fan, J.; Niu, M.; Zhou, Q.; Zhou, Y.; Chen, H.; Zhou, Y. The β-catenin pathway contributes to the effects of leptin on SREBP-1c expression in rat hepatic stellate cells and liver fibrosis. Br. J. Pharmacol. 2013, 169, 197–212. [Google Scholar] [CrossRef]
- Dong, Z.; Su, L.; Esmaili, S.; Iseli, T.J.; Ramezani-Moghadam, M.; Hu, L.; Xu, A.; George, J.; Wang, J. Adiponectin attenuates liver fibrosis by inducing nitric oxide production of hepatic stellate cells. J. Mol. Med. 2015, 93, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Smith, T.; Rahman, K.; Mells, J.E.; Thorn, N.E.; Saxena, N.K.; Anania, F.A. Adiponectin modulates focal adhesion disassembly in activated hepatic stellate cells: Implication for reversing hepatic fibrosis. FASEB J. 2014, 28, 5172–5183. [Google Scholar] [CrossRef] [PubMed]
- Ramezani-Moghadam, M.; Wang, J.; Ho, V.; Iseli, T.J.; Alzahrani, B.; Xu, A.; Van der Poorten, D.; Qiao, L.; George, J.; Hebbard, L. Adiponectin reduces hepatic stellate cell migration by promoting tissue inhibitor of metalloproteinase-1 (TIMP-1) secretion. J. Biol. Chem. 2015, 290, 5533–5542. [Google Scholar] [CrossRef]
- Tardelli, M.; Moreno-Viedma, V.; Zeyda, M.; Itariu, B.K.; Langer, F.B.; Prager, G.; Stulnig, T.M. Adiponectin regulates aquaglyceroporin expression in hepatic stellate cells altering their functional state. J. Gastroenterol. Hepatol. 2017, 32, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Zhang, Z.; Huang, B.; Cheng, X.; Wang, D.; la Gahu, Z.; Xue, Z.; Da, Y.; Li, D.; et al. Adiponectin-derived active peptide ADP355 exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury. Sci. Rep. 2016, 6, 19445. [Google Scholar] [CrossRef]
- Carambia, A.; Freund, B.; Schwinge, D.; Heine, M.; Laschtowitz, A.; Huber, S.; Wraith, D.C.; Korn, T.; Schramm, C.; Lohse, A.W.; et al. TGF-β-dependent induction of CD4⁺CD25⁺Foxp3⁺ Tregs by liver sinusoidal endothelial cells. J. Hepatol. 2014, 61, 594–599. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, C. The role of liver sinusoidal endothelial cells in cancer liver metastasis. Am. J. Cancer Res. 2021, 11, 1845–1860. [Google Scholar]
- Ishikawa, T.; Yokoyama, H.; Matsuura, T.; Fujiwara, Y. Fc gamma RIIb expression levels in human liver sinusoidal endothelial cells during progression of non-alcoholic fatty liver disease. PLoS ONE 2019, 14, 0211543. [Google Scholar] [CrossRef]
- Baiocchini, A.; Del Nonno, F.; Taibi, C.; Visco-Comandini, U.; D’Offizi, G.; Piacentini, M.; Falasca, L. Liver sinusoidal endothelial cells (LSECs) modifications in patients with chronic hepatitis C. Sci. Rep. 2019, 9, 8760, Erratum in: Sci. Rep. 2020, 10, 1420. [Google Scholar] [CrossRef]
- Maretti-Mira, A.C.; Wang, X.; Wang, L.; DeLeve, L.D. Incomplete Differentiation of Engrafted Bone Marrow Endothelial Progenitor Cells Initiates Hepatic Fibrosis in the Rat. Hepatology 2019, 69, 1259–1272. [Google Scholar] [CrossRef]
- Desroches-Castan, A.; Tillet, E.; Ricard, N.; Ouarné, M.; Mallet, C.; Belmudes, L.; Couté, Y.; Boillot, O.; Scoazec, J.Y.; Bailly, S.; et al. Bone Morphogenetic Protein 9 Is a Paracrine Factor Controlling Liver Sinusoidal Endothelial Cell Fenestration and Protecting Against Hepatic Fibrosis. Hepatology 2019, 70, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Desroches-Castan, A.; Tillet, E.; Ricard, N.; Ouarné, M.; Mallet, C.; Feige, J.J.; Bailly, S. Differential Consequences of Bmp9 Deletion on Sinusoidal Endothelial Cell Differentiation and Liver Fibrosis in 129/Ola and C57BL/6 Mice. Cells 2019, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, Y.; Zhu, L.; Yang, Z.; He, J.; Wang, L.; Shang, Q.; Pan, H.; Wang, H.; Ma, X.; et al. Targeting secreted cytokine BMP9 gates the attenuation of hepatic fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, Y.Y.; Nio, K.; Tang, H. Unveiling the Impact of BMP9 in Liver Diseases: Insights into Pathogenesis and Therapeutic Potential. Biomolecules 2024, 14, 1013. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Y.; Xie, X.; Wu, H.; Su, X.N.; Qi, J.; Xin, W.; Gao, L.; Zhang, Y.; Shah, V.H.; et al. Neuropilin-1 aggravates liver cirrhosis by promoting angiogenesis via VEGFR2-dependent PI3K/Akt pathway in hepatic sinusoidal endothelial cells. EBioMedicine 2019, 43, 525–536. [Google Scholar] [CrossRef]
- Kong, L.J.; Li, H.; Du, Y.J.; Pei, F.H.; Hu, Y.; Zhao, L.L.; Chen, J. Vatalanib, a tyrosine kinase inhibitor, decreases hepatic fibrosis and sinusoidal capillarization in CCl4-induced fibrotic mice. Mol. Med. Rep. 2017, 15, 2604–2610. [Google Scholar] [CrossRef]
- Ogawa, H.; Kaji, K.; Nishimura, N.; Takagi, H.; Ishida, K.; Takaya, H.; Kawaratani, H.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Lenvatinib prevents liver fibrosis by inhibiting hepatic stellate cell activation and sinusoidal capillarization in experimental liver fibrosis. J. Cell Mol. Med. 2021, 25, 4001–4013. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Z.; Xiu, A.Y.; Meng, D.X.; Wang, S.N.; Zhang, C.Q. Carvedilol attenuates carbon tetrachloride-induced liver fibrosis and hepatic sinusoidal capillarization in mice. Drug Des. Devel Ther. 2019, 13, 2667–2676. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, F.; Lei, Y.; Liu, P.; Liu, C.; Tao, Y. microRNA-322/424 promotes liver fibrosis by regulating angiogenesis through targeting CUL2/HIF-1α pathway. Life Sci. 2021, 266, 118819. [Google Scholar] [CrossRef]
- Meyer, J.; Balaphas, A.; Fontana, P.; Morel, P.; Robson, S.C.; Sadoul, K.; Gonelle-Gispert, C.; Bühler, L. Platelet Interactions with Liver Sinusoidal Endothelial Cells and Hepatic Stellate Cells Lead to Hepatocyte Proliferation. Cells 2020, 9, 1243. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, J.; Zhao, T.; Wang, L.; Wang, J. Modulation of the VEGF/AKT/eNOS signaling pathway to regulate liver angiogenesis to explore the anti-hepatic fibrosis mechanism of curcumol. J. Ethnopharmacol. 2021, 280, 114480. [Google Scholar] [CrossRef] [PubMed]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, R.; Wu, J.; Li, X. Self-eating: Friend or foe? The emerging role of autophagy in fibrotic diseases. Theranostics 2020, 10, 7993–8017. [Google Scholar] [CrossRef]
- Luo, X.; Wang, D.; Zhu, X.; Wang, G.; You, Y.; Ning, Z.; Li, Y.; Jin, S.; Huang, Y.; Hu, Y.; et al. Autophagic degradation of caveolin-1 promotes liver sinusoidal endothelial cells defenestration. Cell Death Dis. 2018, 9, 576. [Google Scholar] [CrossRef]
- Kohara, S.; Ogawa, K. Eph/Ephrin Promotes the Adhesion of Liver Tissue-Resident Macrophages to a Mimicked Surface of Liver Sinusoidal Endothelial Cells. Biomedicines 2022, 10, 3234. [Google Scholar] [CrossRef]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.M.; et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 2019, 51, 655–670. [Google Scholar] [CrossRef]
- Ganesan, L.P.; Mohanty, S.; Kim, J.; Clark, K.R.; Robinson, J.M.; Anderson, C.L. Rapid and efficient clearance of blood-borne virus by liver sinusoidal endothelium. PLoS Pathog. 2011, 7, 1002281. [Google Scholar] [CrossRef]
- Mates, J.M.; Yao, Z.; Cheplowitz, A.M.; Suer, O.; Phillips, G.S.; Kwiek, J.J.; Rajaram, M.V.; Kim, J.; Robinson, J.M.; Ganesan, L.P.; et al. Mouse Liver Sinusoidal Endothelium Eliminates HIV-Like Particles from Blood at a Rate of 100 Million per Minute by a Second-Order Kinetic Process. Front. Immunol. 2017, 8, 35. [Google Scholar] [CrossRef]
- Breiner, K.M.; Schaller, H.; Knolle, P.A. Endothelial cell-mediated uptake of a hepatitis B virus: A new concept of liver targeting of hepatotropic microorganisms. Hepatology 2001, 34, 803–808. [Google Scholar] [CrossRef]
- Cormier, E.G.; Tsamis, F.; Kajumo, F.; Durso, R.J.; Gardner, J.P.; Dragic, T. CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 2004, 101, 7270–7274. [Google Scholar] [CrossRef]
- Rowe, I.A.; Galsinh, S.K.; Wilson, G.K.; Parker, R.; Durant, S.; Lazar, C.; Branza-Nichita, N.; Bicknell, R.; Adams, D.H.; Balfe, P.; et al. Paracrine signals from liver sinusoidal endothelium regulate hepatitis C virus replication. Hepatology 2014, 59, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, S.; Kriss, M.; Golden-Mason, L.; Dobrinskikh, E.; Stone, A.E.; Soto-Gutierrez, A.; Mitchell, A.; Khetani, S.R.; Yamane, D.; Stoddard, M.; et al. Hepatitis C virus infection induces autocrine interferon signaling by human liver endothelial cells and release of exosomes, which inhibits viral replication. Gastroenterology 2015, 148, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chiu, S.M.; Motan, D.A.; Zhang, Z.; Chen, L.; Ji, H.L.; Tse, H.F.; Fu, Q.L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, 2062. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.D.; Kuo, T.K.; Whang-Peng, J.; Chung, Y.F.; Lin, C.T.; Chou, S.H.; Chen, J.R.; Chen, Y.P.; Lee, O.K. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology 2004, 40, 1275–1284. [Google Scholar] [CrossRef]
- Li, W.; Ren, G.; Huang, Y.; Su, J.; Han, Y.; Li, J.; Chen, X.; Cao, K.; Chen, Q.; Shou, P.; et al. Mesenchymal stem cells: A double-edged sword in regulating immune responses. Cell Death Differ. 2012, 19, 1505–1513. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Li, Q.; Liu, K.; Hou, J.; Shao, C.; Wang, Y. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat. Rev. Nephrol. 2018, 14, 493–507. [Google Scholar] [CrossRef]
- Viswanathan, S.; Shi, Y.; Galipeau, J.; Krampera, M.; Leblanc, K.; Martin, I.; Nolta, J.; Phinney, D.G.; Sensebe, L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT®) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019, 21, 1019–1024. [Google Scholar]
- Costa, L.A.; Eiro, N.; Fraile, M.; Gonzalez, L.O.; Saá, J.; Garcia-Portabella, P.; Vega, B.; Schneider, J.; Vizoso, F.J. Functional heterogeneity of mesenchymal stem cells from natural niches to culture conditions: Implications for further clinical uses. Cell Mol. Life Sci. 2021, 78, 447–467. [Google Scholar] [CrossRef]
- Han, Y.; Yang, J.; Fang, J.; Zhou, Y.; Candi, E.; Wang, J.; Hua, D.; Shao, C.; Shi, Y. The secretion profile of mesenchymal stem cells and potential applications in treating human diseases. Signal Transduct. Target. Ther. 2022, 7, 92. [Google Scholar] [CrossRef]
- Murakami, J.; Ishii, M.; Suehiro, F.; Ishihata, K.; Nakamura, N.; Nishimura, M. Vascular endothelial growth factor-C induces osteogenic differentiation of human mesenchymal stem cells through the ERK and RUNX2 pathway. Biochem. Biophys. Res. Commun. 2017, 484, 710–718. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, Y.; Ye, L.; Zhang, T.; Cheng, J.; Chen, G.; Zhang, Q.; Yang, Y. Umbilical Cord-derived Mesenchymal Stem Cells Instruct Monocytes Towards an IL10-producing Phenotype by Secreting IL6 and HGF. Sci. Rep. 2016, 6, 37566. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J. Concise review: Two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells 2013, 31, 2042–2046. [Google Scholar] [CrossRef] [PubMed]
- Ezquer, F.; Ezquer, M.; Contador, D.; Ricca, M.; Simon, V.; Conget, P. The antidiabetic effect of mesenchymal stem cells is unrelated to their transdifferentiation potential but to their capability to restore Th1/Th2 balance and to modify the pancreatic microenvironment. Stem Cells 2012, 30, 1664–1674. [Google Scholar] [CrossRef]
- Tao, H.; Liu, Q.; Zeng, A.; Song, L. Unlocking the potential of Mesenchymal stem cells in liver Fibrosis: Insights into the impact of autophagy and aging. Int. Immunopharmacol. 2023, 21, 110497. [Google Scholar] [CrossRef]
- Giacomini, C.; Granéli, C.; Hicks, R.; Dazzi, F. The critical role of apoptosis in mesenchymal stromal cell therapeutics and implications in homeostasis and normal tissue repair. Cell Mol. Immunol. 2023, 20, 570–582. [Google Scholar] [CrossRef]
- Li, P.; Ou, Q.; Shi, S.; Shao, C. Immunomodulatory properties of mesenchymal stem cells/dental stem cells and their therapeutic applications. Cell Mol. Immunol. 2023, 20, 558–569. [Google Scholar] [CrossRef]
- Zhou, J.; Shi, Y. Mesenchymal stem/stromal cells (MSCs): Origin, immune regulation, and clinical applications. Cell Mol. Immunol. 2023, 20, 555–557. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Melhem, A.; Muhanna, N.; Bishara, A.; Alvarez, C.E.; Ilan, Y.; Bishara, T.; Horani, A.; Nassar, M.; Friedman, S.L.; Safadi, R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J. Hepatol. 2006, 45, 60–71. [Google Scholar] [CrossRef]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef]
- Baroni, G.S.; D’Ambrosio, L.; Curto, P.; Casini, A.; Mancini, R.; Jezequel, A.M.; Benedetti, A. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology 1996, 23, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.M.; Ryu, T.; Lee, J.H.; Shim, Y.R.; Kim, M.H.; Kim, H.H.; Kim, Y.E.; Yang, K.; Kim, K.; Choi, S.E.; et al. Metabotropic Glutamate Receptor 5 in Natural Killer Cells Attenuates Liver Fibrosis by Exerting Cytotoxicity to Activated Stellate Cells. Hepatology 2021, 74, 2170–2185. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, N.; Seki, S.; Hatsuse, K.; Ohkawa, T.; Koike, Y.; Aihara, T.; Habu, Y.; Nakagawa, R.; Ami, K.; Hiraide, H.; et al. Decrease of CD56(+)T cells and natural killer cells in cirrhotic livers with hepatitis C may be involved in their susceptibility to hepatocellular carcinoma. Hepatology 2000, 32, 962–969. [Google Scholar] [CrossRef]
- Morishima, C.; Paschal, D.M.; Wang, C.C.; Yoshihara, C.S.; Wood, B.L.; Yeo, A.E.; Emerson, S.S.; Shuhart, M.C.; Gretch, D.R. Decreased NK cell frequency in chronic hepatitis C does not affect ex vivo cytolytic killing. Hepatology 2006, 43, 573–580. [Google Scholar] [CrossRef]
- Muhanna, N.; Doron, S.; Wald, O.; Horani, A.; Eid, A.; Pappo, O.; Friedman, S.L.; Safadi, R. Activation of hepatic stellate cells after phagocytosis of lymphocytes: A novel pathway of fibrogenesis. Hepatology 2008, 48, 963–977. [Google Scholar] [CrossRef]
- Gan, J.; Mao, X.R.; Zheng, S.J.; Li, J.F. Invariant natural killer T cells: Not to be ignored in liver disease. J. Dig. Dis. 2021, 22, 136–142. [Google Scholar] [CrossRef]
- Sajid, M.; Liu, L.; Sun, C. The Dynamic Role of NK Cells in Liver Cancers: Role in HCC and HBV Associated HCC and Its Therapeutic Implications. Front. Immunol. 2022, 13, 887186. [Google Scholar] [CrossRef]
- Ma, Q.; Dong, X.; Liu, S.; Zhong, T.; Sun, D.; Zong, L.; Zhao, C.; Lu, Q.; Zhang, M.; Gao, Y.; et al. Hepatitis B e Antigen Induces NKG2A+ Natural Killer Cell Dysfunction via Regulatory T Cell-Derived Interleukin 10 in Chronic Hepatitis B Virus Infection. Front. Cell Dev. Biol. 2020, 8, 421. [Google Scholar] [CrossRef]
- Diedrich, T.; Kummer, S.; Galante, A.; Drolz, A.; Schlicker, V.; Lohse, A.W.; Kluwe, J.; Eberhard, J.M.; Schulze Zur Wiesch, J. Characterization of the immune cell landscape of patients with NAFLD. PLoS ONE 2020, 15, 0230307. [Google Scholar] [CrossRef]
- Gu, M.; Zhang, Y.; Lin, Z.; Hu, X.; Zhu, Y.; Xiao, W.; Jia, X.; Chen, W.; Lu, G.; Gong, W. Decrease in UCP1 by sustained high lipid promotes NK cell necroptosis to exacerbate nonalcoholic liver fibrosis. Cell Death Dis. 2024, 15, 518. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, G.; Neumann, K.; Berkhout, L.K.; Weidemann, S.; Langeneckert, A.E.; Schwinge, D.; Poch, T.; Huber, S.; Schiller, B.; Hess, L.U.; et al. Interferon-γ-dependent immune responses contribute to the pathogenesis of sclerosing cholangitis in mice. J. Hepatol. 2019, 71, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Siwicki, M.; Kubes, P. Neutrophils in host defense, healing, and hypersensitivity: Dynamic cells within a dynamic host. J. Allergy Clin. Immunol. 2023, 151, 634–655. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Koyama, Y.; Wang, P.; Liang, S.; Iwaisako, K.; Liu, X.; Xu, J.; Zhang, M.; Sun, M.; Cong, M.; Karin, D.; et al. Mesothelin/mucin 16 signaling in activated portal fibroblasts regulates cholestatic liver fibrosis. J. Clin. Investig. 2017, 127, 1254–1270. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Moles, A.; Murphy, L.; Wilson, C.L.; Chakraborty, J.B.; Fox, C.; Park, E.J.; Mann, J.; Oakley, F.; Howarth, R.; Brain, J.; et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J. Hepatol. 2014, 60, 782–791. [Google Scholar] [CrossRef]
- Peiseler, M.; Kubes, P. More friend than foe: The emerging role of neutrophils in tissue repair. J. Clin. Investig. 2019, 129, 2629–2639. [Google Scholar] [CrossRef]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwälder, M.; Tacke, F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease—Novel insights into cellular communication circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef]
- Wan, Y.; Li, X.; Slevin, E.; Harrison, K.; Li, T.; Zhang, Y.; Klaunig, J.E.; Wu, C.; Shetty, A.K.; Dong, X.C.; et al. Endothelial dysfunction in pathological processes of chronic liver disease during aging. FASEB J. 2022, 36, e22125. [Google Scholar] [CrossRef]
- Inverso, D.; Iannacone, M. Spatiotemporal dynamics of effector CD8+ T cell responses within the liver. J. Leukoc. Biol. 2016, 99, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Wohlleber, D.; Knolle, P.A. The role of liver sinusoidal cells in local hepatic immune surveillance. Clin. Transl. Immunol. 2016, 5, 117. [Google Scholar] [CrossRef] [PubMed]
- Pennington, D.J.; Vermijlen, D.; Wise, E.L.; Clarke, S.L.; Tigelaar, R.E.; Hayday, A.C. The integration of conventional and unconventional T cells that characterizes cell-mediated responses. Adv. Immunol. 2005, 87, 27–59. [Google Scholar] [PubMed]
- Godfrey, D.I.; Uldrich, A.P.; McCluskey, J.; Rossjohn, J.; Moody, D.B. The burgeoning family of unconventional T cells. Nat. Immunol. 2015, 16, 1114–1123. [Google Scholar] [CrossRef]
- Nguyen, Q.P.; Deng, T.Z.; Witherden, D.A.; Goldrath, A.W. Origins of CD4+ circulating and tissue-resident memory T-cells. Immunology 2019, 157, 3–12. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Wang, H.; Luo, H.; Wan, X.; Fu, X.; Mao, Q.; Xiang, X.; Zhou, Y.; He, W.; Zhang, J.; Guo, Y.; et al. TNF-α/IFN-γ profile of HBV-specific CD4 T cells is associated with liver damage and viral clearance in chronic HBV infection. J. Hepatol. 2020, 72, 45–56. [Google Scholar] [CrossRef]
- Li, Y.; You, Z.; Tang, R.; Ma, X. Tissue-resident memory T cells in chronic liver diseases: Phenotype, development and function. Front. Immunol. 2022, 13, 967055. [Google Scholar] [CrossRef]
- Herkel, J.; Jagemann, B.; Wiegard, C.; Lazaro, J.F.; Lueth, S.; Kanzler, S.; Blessing, M.; Schmitt, E.; Lohse, A.W. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocyutes. Hepatology 2003, 37, 1079–1085. [Google Scholar] [CrossRef]
- Koch, K.S.; Leffert, H.L. Hypothesis: Targeted Ikkβ deletion upregulates MIF signaling responsiveness and MHC class II expression in mouse hepatocytes. Hepat. Med. 2010, 2010, 39–47. [Google Scholar] [CrossRef]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; Pereira da Costa, M.; Reis e Sousa, C. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef] [PubMed]
- Eckert, C.; Klein, N.; Kornek, M.; Lukacs-Kornek, V. The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front. Immunol. 2015, 6, 179. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.H.; Aloman, C. Dendritic cells and liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Doherty, D.G. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J. Autoimmun. 2016, 66, 60–75. [Google Scholar] [CrossRef]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thoné, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396. [Google Scholar] [CrossRef]
- Haas, J.T.; Vonghia, L.; Mogilenko, D.A.; Verrijken, A.; Molendi-Coste, O.; Fleury, S.; Deprince, A.; Nikitin, A.; Woitrain, E.; Ducrocq-Geoffroym, L.; et al. Transcriptional Network Analysis Implicates Altered Hepatic Immune Function in NASH development and resolution. Nat. Metab. 2019, 1, 604–614. [Google Scholar] [CrossRef]
- Kambayashi, T.; Laufer, T.M. Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nat. Rev. Immunol. 2014, 14, 719–730. [Google Scholar] [CrossRef]
- Alcover, A.; Alarcón, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef]
- Elhai, M.; Avouac, J.; Hoffmann-Vold, A.M.; Ruzehaji, N.; Amiar, O.; Ruiz, B.; Brahiti, H.; Ponsoye, M.; Fréchet, M.; Burgevin, A.; et al. OX40L blockade protects against inflammation-driven fibrosis. Proc. Natl. Acad. Sci. USA 2016, 113, 3901–3910. [Google Scholar] [CrossRef]
- Sun, G.; Jin, H.; Zhang, C.; Meng, H.; Zhao, X.; Wei, D.; Ou, X.; Wang, Q.; Li, S.; Wang, T.; et al. OX40 Regulates Both Innate and Adaptive Immunity and Promotes Nonalcoholic Steatohepatitis. Cell Rep. 2018, 25, 3786–3799. [Google Scholar] [CrossRef]
- Webb, G.J.; Hirschfield, G.M.; Lane, P.J. OX40, OX40L and Autoimmunity: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 312–332. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T cell receptor (TCR) signaling in health and disease. Signal Transduct. Target. Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Hu, Y.; Zhang, M.; Lin, C.; Zhang, W.; Li, Y.; Zhu, P.; Xue, P.; Chen, Y.; Li, Q.; et al. The T Cell Receptor Immune Repertoire Protects the Liver From Reconsitution. Front. Immunol. 2020, 11, 584979. [Google Scholar] [CrossRef] [PubMed]
- Safadi, R.; Ohta, M.; Alvarez, C.E.; Fiel, M.I.; Bansal, M.; Mehal, W.Z.; Friedman, S.L. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology 2004, 127, 870–882. [Google Scholar] [CrossRef]
- Tang, L.; Chen, C.; Gao, X.; Zhang, W.; Yan, X.; Zhou, Y.; Guo, L.; Zheng, X.; Wang, W.; Yang, F.; et al. Interleukin 21 Reinvigorates the Antiviral Activity of Hepatitis B Virus (HBV)-Specific CD8+ T Cells in Chronic HBV Infection. J. Infect. Dis. 2019, 219, 750–759. [Google Scholar] [CrossRef]
- Liu, H.; Hu, B.; Huang, J.; Wang, Q.; Wang, F.; Pan, F.; Chen, L. Endoplasmic Reticulum Aminopeptidase 1 Is Involved in Anti-viral Immune Response of Hepatitis B Virus by Trimming Hepatitis B Core Antigen to Generate 9-Mers Peptides. Front. Microbiol. 2022, 13, 829241. [Google Scholar] [CrossRef]
- Li, C.; Yu, T.; Shi, X.; Yu, J. Interleukin-33 Reinvigorates Antiviral Function of Viral-Specific CD8+ T Cells in Chronic Hepatitis B Virus Infection. Viral Immunol. 2022, 35, 41–49. [Google Scholar] [CrossRef]
- Roger, P.M.; Chaillou, S.; Breittmayer, J.P.; Dahman, M.; St Paul, M.C.; Chevallier, P.; Benzaken, S.; Ticchioni, M.; Bernard, A.; Dellamonica, P.; et al. Intrahepatic CD4 T-Cell apoptosis is related to METAVIR score in patients with chronic hepatitis C virus. Scand. J. Immunol. 2005, 62, 168–175. [Google Scholar] [CrossRef]
- Glässner, A.; Eisenhardt, M.; Kokordelis, P.; Krämer, B.; Wolter, F.; Nischalke, H.D.; Boesecke, C.; Sauerbruch, T.; Rockstroh, J.K.; Spengler, U.; et al. Impaired CD4⁺ T cell stimulation of NK cell anti-fibrotic activity may contribute to accelerated liver fibrosis progression in HIV/HCV patients. J. Hepatol. 2013, 59, 427–433. [Google Scholar] [CrossRef]
- Her, Z.; Tan, J.H.L.; Lim, Y.S.; Tan, S.Y.; Chan, X.Y.; Tan, W.W.S.; Liu, M.; Yong, K.S.M.; Lai, F.; Ceccarello, E.; et al. CD4+ T Cells Mediate the Development of Liver Fibrosis in High Fat Diet-Induced NAFLD in Humanized Mice. Front. Immunol. 2020, 11, 580968. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A.; Robinson, D. Development and function of T helper 1 cells. Adv. Immunol. 2004, 83, 133–162. [Google Scholar] [PubMed]
- Luo, X.Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.K. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, 891–899. [Google Scholar] [CrossRef]
- Ferreyra Solari, N.E.; Inzaugarat, M.E.; Baz, P.; De Matteo, E.; Lezama, C.; Galoppo, M.; Galoppo, C.; Cherñavsky, A.C. The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J. Clin. Immunol. 2012, 32, 611–621. [Google Scholar] [CrossRef]
- Inzaugarat, M.E.; Ferreyra Solari, N.E.; Billordo, L.A.; Abecasis, R.; Gadano, A.C.; Cherñavsky, A.C. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J. Clin. Immunol. 2011, 31, 1120–1130. [Google Scholar] [CrossRef]
- Rolla, S.; Alchera, E.; Imarisio, C.; Bardina, V.; Valente, G.; Cappello, P.; Mombello, C.; Follenzi, A.; Novelli, F.; Carini, R. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin. Sci. 2016, 130, 193–203. [Google Scholar] [CrossRef]
- Nakayama, T.; Hirahara, K.; Onodera, A.; Endo, Y.; Hosokawa, H.; Shinoda, K.; Tumes, D.J.; Okamoto, Y. Th2 Cells in Health and Disease. Annu. Rev. Immunol. 2017, 35, 53–84. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Feng, K.; Fan, D.; Xue, W.; Lu, J. ’Repair’ Treg Cells in Tissue Injury. Cell Physiol. Biochem. 2017, 43, 2155–2169. [Google Scholar] [CrossRef]
- Shimamura, T.; Fujisawa, T.; Husain, S.R.; Kioi, M.; Nakajima, A.; Puri, R.K. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J. Immunol. 2008, 181, 4656–4665. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, Y.; Yang, M.; Guo, X.; Zhang, M.; Li, H.; Li, J.; Zhao, J. IL-33 treatment attenuated diet-induced hepatic steatosis but aggravated hepatic fibrosis. Oncotarget 2016, 7, 33649–33661. [Google Scholar] [CrossRef] [PubMed]
- Lafoz, E.; Ruart, M.; Anton, A.; Oncins, A.; Hernández-Gea, V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells 2020, 9, 929. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Xu, M.; Hu, J.; Huang, X.; Lin, N.; Deng, M. Inhibiting Th1/2 cells influences hepatic capillarization by adjusting sinusoidal endothelial fenestrae through Rho-ROCK-myosin pathway. Aging 2021, 13, 5069–5086. [Google Scholar] [CrossRef] [PubMed]
- Kremer, M.; Hines, I.N.; Milton, R.J.; Wheeler, M.D. Favored T helper 1 response in a mouse model of hepatosteatosis is associated with enhanced T cell-mediated hepatitis. Hepatology 2006, 44, 216–227. [Google Scholar] [CrossRef]
- Molina, M.F.; Abdelnabi, M.N.; Fabre, T.; Shoukry, N.H. Type 3 cytokines in liver fibrosis and liver cancer. Cytokine 2019, 124, 154497. [Google Scholar] [CrossRef]
- Fabre, T.; Kared, H.; Friedman, S.L.; Shoukry, N.H. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J. Immunol. 2014, 193, 3925–3933. [Google Scholar] [CrossRef]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776. [Google Scholar] [CrossRef]
- Tan, Z.; Qian, X.; Jiang, R.; Liu, Q.; Wang, Y.; Chen, C.; Wang, X.; Ryffel, B.; Sun, B. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J. Immunol. 2013, 191, 1835–1844. [Google Scholar] [CrossRef]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Cappelletti, M.; Huppert, S.S.; Iwakura, Y.; Dong, C.; Shanmukhappa, S.K.; Divanovic, S. Regulation of Inflammation by IL-17A and IL-17F Modulates Non-Alcoholic Fatty Liver Disease Pathogenesis. PLoS ONE 2016, 11, 0149783. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Weyler, J.; Kwanten, W.J.; Dirinck, E.L.; De Winter, B.Y.; Francque, S.M.; Vonghia, L. The Differential Roles of T Cells in Non-alcoholic Fatty Liver Disease and Obesity. Front. Immunol. 2019, 10, 82. [Google Scholar] [CrossRef]
- Gomes, A.L.; Teijeiro, A.; Burén, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell. 2016, 30, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Tao, A.; Zhang, S.; Zhang, M. Neutralization of interleukin-17 attenuates high fat diet-induced non-alcoholic fatty liver disease in mice. Acta Biochim. Biophys. Sin. 2013, 45, 726–733. [Google Scholar] [CrossRef]
- Harley, I.T.; Stankiewicz, T.E.; Giles, D.A.; Softic, S.; Flick, L.M.; Cappelletti, M.; Sheridan, R.; Xanthakos, S.A.; Steinbrecher, K.A.; Sartor, R.B.; et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014, 59, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhang, T.; Tang, L.; Li, Y. Cytokines and Chemokines in HBV Infection. Front. Mol. Biosci. 2021, 8, 805625. [Google Scholar] [CrossRef]
- Ge, D.; You, Z. Expression of interleukin-17RC protein in normal human tissues. Int. Arch. Med. 2008, 1, 19. [Google Scholar] [CrossRef]
- Lemmers, A.; Moreno, C.; Gustot, T.; Maréchal, R.; Degré, D.; Demetter, P.; de Nadai, P.; Geerts, A.; Quertinmont, E.; Vercruysse, V.; et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 2009, 49, 646–657. [Google Scholar] [CrossRef]
- Paquissi, F.C. Immunity and Fibrogenesis: The Role of Th17/IL-17 Axis in HBV and HCV-induced Chronic Hepatitis and Progression to Cirrhosis. Front. Immunol. 2017, 8, 1195. [Google Scholar] [CrossRef]
- Buschow, S.I.; Jansen, D.T.S.L. CD4+ T Cells in Chronic Hepatitis B and T Cell-Directed Immunotherapy. Cells 2021, 10, 1114. [Google Scholar] [CrossRef]
- Zhu, L.; Li, J.; Xu, J.; Chen, F.; Wu, X.; Zhu, C. Significance of T-Cell Subsets for Clinical Response to Peginterferon Alfa-2a Therapy in HBeAg-Positive Chronic Hepatitis B Patients. Int. J. Gen. Med. 2022, 15, 4441–4451. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, W.; Zhang, L.; Wang, J.; Chen, M.; Peng, M.; Ren, H.; Hu, P. Th17/Treg imbalance and increased interleukin-21 are associated with liver injury in patients with chronic severe hepatitis B. Int. Immunopharmacol. 2017, 46, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yang, G.; Xiao, F.; Xie, J.; Wang, S.; Lu, L.; Cui, D. Role of Th22 Cells in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 2021, 12, 688066. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ota, N.; Manzanillo, P.; Kates, L.; Zavala-Solorio, J.; Eidenschenk, C.; Zhang, J.; Lesch, J.; Lee, W.P.; Ross, J.; et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 2014, 514, 237–241. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Wang, L.; Fan, F.; Zhu, L.; Li, Z.; Ruan, X.; Huang, H.; Wang, Z.; Huang, Z.; et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J. Hepatol. 2010, 53, 339–347. [Google Scholar] [CrossRef]
- Jiang, R.; Tan, Z.; Deng, L.; Chen, Y.; Xia, Y.; Gao, Y.; Wang, X.; Sun, B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011, 54, 900–909. [Google Scholar] [CrossRef]
- Barron, L.; Wynn, T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 723–728. [Google Scholar] [CrossRef]
- Fabre, T.; Molina, M.F.; Soucy, G.; Goulet, J.P.; Willems, B.; Villeneuve, J.P.; Bilodeau, M.; Shoukry, N.H. Type 3 cytokines IL-17A and IL-22 drive TGF-β-dependent liver fibrosis. Sci. Immunol. 2018, 3, 7754. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, D.; Weng, J.; Huang, Y.; Liu, S.; Zhang, Q.; Li, N.; Wen, M.; Zhu, G.; Lin, F.; et al. Neutralization of Interleukin-17 Attenuates Cholestatic Liver Fibrosis in Mice. Scand. J. Immunol. 2016, 83, 102–108. [Google Scholar] [CrossRef]
- Hara, M.; Kono, H.; Furuya, S.; Hirayama, K.; Tsuchiya, M.; Fujii, H. Interleukin-17A plays a pivotal role in cholestatic liver fibrosis in mice. J. Surg. Res. 2013, 183, 574–582. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Inzaugarat, M.E.; Eguchi, A.; Schuster, S.; Johnson, C.D.; Peña, C.A.; Geisler, L.J.; Papouchado, B.G.; Hoffman, H.M.; et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 2018, 67, 736–749. [Google Scholar] [CrossRef]
- Gieseck, R.L., 3rd; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Homer, R.J.; Zhu, Z.; Lanone, S.; Wang, X.; Koteliansky, V.; Shipley, J.M.; Gotwals, P.; Noble, P.; Chen, Q.; et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J. Exp. Med. 2001, 194, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Kaviratne, M.; Hesse, M.; Leusink, M.; Cheever, A.W.; Davies, S.J.; McKerrow, J.H.; Wakefield, L.M.; Letterio, J.J.; Wynn, T.A. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J. Immunol. 2004, 173, 4020–4029. [Google Scholar] [CrossRef]
- Gieseck, R.L., 3rd; Ramalingam, T.R.; Hart, K.M.; Vannella, K.M.; Cantu, D.A.; Lu, W.Y.; Ferreira-González, S.; Forbes, S.J.; Vallier, L.; Wynn, T.A. Interleukin-13 Activates Distinct Cellular Pathways Leading to Ductular Reaction, Steatosis, and Fibrosis. Immunity 2016, 45, 145–158. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, W.; Wei, H.; Sun, R.; Tian, Z.; Chen, Y. Hepatic NK Cells Attenuate Fibrosis Progression of Non-Alcoholic Steatohepatitis in Dependent of CXCL10-Mediated Recruitment. Liver Int. 2020, 40, 598–608. [Google Scholar] [CrossRef]
- Hart, K.M.; Fabre, T.; Sciurba, J.C.; Gieseck, R.L., 3rd; Borthwick, L.A.; Vannella, K.M.; Acciani, T.H.; de Queiroz Prado, R.; Thompson, R.W.; White, S.; et al. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-β. Sci. Transl. Med. 2017, 9, 3694. [Google Scholar]
- Tosello-Trampont, A.C.; Krueger, P.; Narayanan, S.; Landes, S.G.; Leitinger, N.; Hahn, Y.S. NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology 2016, 63, 799–812. [Google Scholar] [CrossRef]
- Zhang, H.; Meadows, G.G. Chronic alcohol consumption in mice increases the proportion of peripheral memory T cells by homeostatic proliferation. J. Leukoc. Biol. 2005, 78, 1070–1080. [Google Scholar] [CrossRef]
- Zuluaga, P.; Sanvisens, A.; Teniente-Serra, A.; El Ars, O.; Fuster, D.; Quirant-Sánchez, B.; Martínez-Cáceres, E.; Muga, R. Loss of naive T lymphocytes is associated with advanced liver fibrosis in alcohol use disorder. Drug Alcohol Depend. 2020, 213, 108046. [Google Scholar] [CrossRef]
- Kita, H.; Matsumura, S.; He, X.S.; Ansari, A.A.; Lian, Z.X.; Van de Water, J.; Coppel, R.L.; Kaplan, M.M.; Gershwin, M.E. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J. Clin. Investig. 2002, 109, 1231–1240. [Google Scholar] [CrossRef]
- Zhang, W.; Ono, Y.; Miyamura, Y.; Bowlus, C.L.; Gershwin, M.E.; Maverakis, E. T cell clonal expansions detected in patients with primary biliary cirrhosis express CX3CR1. J. Autoimmun. 2011, 37, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Immune tolerance in liver disease. Hepatology 2014, 60, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529. [Google Scholar] [CrossRef]
- He, B.; Wu, L.; Xie, W.; Shao, Y.; Jiang, J.; Zhao, Z.; Yan, M.; Chen, Z.; Cui, D. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol. 2017, 18, 33. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, Fat Mass and Immune System: Role for Leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef]
- Dywicki, J.; Buitrago-Molina, L.E.; Noyan, F.; Davalos-Misslitz, A.C.; Hupa-Breier, K.L.; Lieber, M.; Hapke, M.; Schlue, J.; Falk, C.S.; Raha, S.; et al. The Detrimental Role of Regulatory T Cells in Nonalcoholic Steatohepatitis. Hepatol. Commun. 2022, 6, 320–333. [Google Scholar] [CrossRef]
- Katz, S.C.; Ryan, K.; Ahmed, N.; Plitas, G.; Chaudhry, U.I.; Kingham, T.P.; Naheed, S.; Nguyen, C.; Somasundar, P.; Espat, N.J.; et al. Obstructive jaundice expands intrahepatic regulatory T cells, which impair liver T lymphocyte function but modulate liver cholestasis and fibrosis. J. Immunol. 2011, 187, 1150–1156. [Google Scholar] [CrossRef]
- Claassen, M.A.; de Knegt, R.J.; Tilanus, H.W.; Janssen, H.L.; Boonstra, A. Abundant numbers of regulatory T cells localize to the liver of chronic hepatitis C infected patients and limit the extent of fibrosis. J. Hepatol. 2010, 52, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Langhans, B.; Krämer, B.; Louis, M.; Nischalke, H.D.; Hüneburg, R.; Staratschek-Jox, A.; Odenthal, M.; Manekeller, S.; Schepke, M.; Kalff, J.; et al. Intrahepatic IL-8 producing Foxp3⁺CD4⁺ regulatory T cells and fibrogenesis in chronic hepatitis C. J. Hepatol. 2013, 59, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qiu, S.J.; She, W.M.; Wang, F.P.; Gao, H.; Li, L.; Tu, C.T.; Wang, J.Y.; Shen, X.Z.; Jiang, W. Significance of the balance between regulatory T (Treg) and T helper 17 (Th17) cells during hepatitis B virus related liver fibrosis. PLoS ONE 2012, 7, 39307. [Google Scholar] [CrossRef]
- Liu, C.; Zeng, X.; Yu, S.; Ren, L.; Sun, X.; Long, Y.; Wang, X.; Lu, S.; Song, Y.; Sun, X.H.; et al. Up-regulated DNA-binding inhibitor Id3 promotes differentiation of regulatory T cell to influence antiviral immunity in chronic hepatitis B virus infection. Life Sci. 2021, 285, 119991. [Google Scholar] [CrossRef]
- Trehanpati, N.; Vyas, A.K. Immune Regulation by T Regulatory Cells in Hepatitis B Virus-Related Inflammation and Cancer. Scand. J. Immunol. 2017, 85, 175–181. [Google Scholar] [CrossRef]
- Zeng, X.; Bahabayi, A.; Tuerhanbayi, B.; Zheng, M.; Liu, T.; Xu, L.; Long, Y.; Xia, C.; Lu, S.; Song, Y.; et al. The altered HLA-DQ expression in peripheral blood T cells of chronic hepatitis B patients characterizes the function of T cells. J. Viral Hepat. 2022, 29, 340–351. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, S.; Wong, D.K.; Huang, F.Y.; Cheung, K.S.; Mak, L.Y.; Fung, J.; Yuen, M.F.; Seto, W.K. Phenotypic Changes of PD-1 and GITR in T Cells Are Associated With Hepatitis B Surface Antigen Seroclearance. J. Clin. Gastroenterol. 2022, 56, e31–e37. [Google Scholar] [CrossRef]
- Wang, X.; Dong, Q.; Li, Q.; Li, Y.; Zhao, D.; Sun, J.; Fu, J.; Meng, F.; Lin, H.; Luan, J.; et al. Dysregulated Response of Follicular Helper T Cells to Hepatitis B Surface Antigen Promotes HBV Persistence in Mice and Associates With Outcomes of Patients. Gastroenterology 2018, 154, 2222–2236. [Google Scholar] [CrossRef]
- Tangye, S.G.; Ma, C.S. Regulation of the germinal center and humoral immunity by interleukin-21. J. Exp. Med. 2020, 217, e20191638. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, L.S.; Wu, S.D.; Wang, S.Q.; Li, L.; She, W.M.; Li, J.; Wang, J.Y.; Jiang, W. IL-10-producing regulatory B-cells suppressed effector T-cells but enhanced regulatory T-cells in chronic HBV infection. Clin. Sci. 2016, 130, 907–919. [Google Scholar] [CrossRef]
- Yang, L.; Shao, X.; Jia, S.; Zhang, Q.; Jin, Z. Interleukin-35 Dampens CD8+ T Cells Activity in Patients With Non-viral Hepatitis-Related Hepatocellular Carcinoma. Front. Immunol. 2019, 10, 1032. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, L.; Liu, S.; Zhang, M.; Jin, Z. Interleukin-35 Suppresses Interleukin-9-Secreting CD4+ T Cell Activity in Patients With Hepatitis B-Related Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 645835. [Google Scholar] [CrossRef]
- Tang, Y.; Ma, T.; Jia, S.; Zhang, Q.; Liu, S.; Qi, L.; Yang, L. The Mechanism of Interleukin-35 in Chronic Hepatitis B. Semin. Liver Dis. 2021, 41, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Peng, N.; Xiao, F.; Hu, D.; Wang, X.; Lu, L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. Int. J. Mol. Sci. 2020, 21, 5203. [Google Scholar] [CrossRef] [PubMed]
- Fillatreau, S. B cells and their cytokine activities implications in human diseases. Clin. Immunol. 2018, 186, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Novobrantseva, T.I.; Majeau, G.R.; Amatucci, A.; Kogan, S.; Brenner, I.; Casola, S.; Shlomchik, M.J.; Koteliansky, V.; Hochman, P.S.; Ibraghimov, A. Attenuated liver fibrosis in the absence of B cells. J. Clin. Investig. 2005, 115, 3072–3082. [Google Scholar] [CrossRef]
- Bruzzì, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic. Biol. Med. 2018, 124, 249–259. [Google Scholar] [CrossRef]
- Gong, Y.; Zhao, C.; Zhao, P.; Wang, M.; Zhou, G.; Han, F.; Cui, Y.; Qian, J.; Zhang, H.; Xiong, H.; et al. Role of IL-10-Producing Regulatory B Cells in Chronic Hepatitis B Virus Infection. Dig. Dis. Sci. 2015, 60, 1308–1314. [Google Scholar] [CrossRef]
- Jin, X.; Yan, Z.H.; Lu, L.; Lu, S.; Zhang, G.; Lin, W. Peripheral Immune Cells Exhaustion and Functional Impairment in Patients With Chronic Hepatitis B. Front. Med. 2021, 8, 759292. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, Y.; Zhu, T.; Jiang, M.; Tian, Z.; Tang, G.; Liang, X. Regulatory B Cells Dysregulated T Cell Function in an IL-35-Dependent Way in Patients With Chronic Hepatitis B. Front. Immunol. 2021, 12, 653198. [Google Scholar] [CrossRef]
- Zheng, P.; Dou, Y.; Wang, Q. Immune response and treatment targets of chronic hepatitis B virus infection: Innate and adaptive immunity. Front. Cell Infect. Microbiol. 2023, 13, 1206720. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Chen, J.; Zhang, X.; Bi, K.; Zhou, H.; Liu, T.; Xu, J.; Diao, H. IL-17A produced by invariant natural killer T cells and CD3+ CD56+ αGalcer-CD1d tetramer- T cells promote liver fibrosis in patients with primary biliary cholangitis. J. Leukoc. Biol. 2022, 112, 1079–1087. [Google Scholar] [CrossRef]
- Hammerich, L.; Tacke, F. Role of gamma-delta T cells in liver inflammation and fibrosis. World J. Gastrointest. Pathophysiol. 2014, 5, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Bartish, M.; Del Rincón, S.V.; Rudd, C.E.; Saragovi, H.U. Aiming for the Sweet Spot: Glyco-Immune Checkpoints and γδ T Cells in Targeted Immunotherapy. Front. Immunol. 2020, 11, 564499. [Google Scholar] [CrossRef]
- Pellicci, D.G.; Koay, H.F.; Berzins, S.P. Thymic development of unconventional T cells: How NKT cells, MAIT cells and γδ T cells emerge. Nat. Rev. Immunol. 2020, 20, 756–770. [Google Scholar] [CrossRef]
- Kurioka, A.; Walker, L.J.; Klenerman, P.; Willberg, C.B. MAIT cells: New guardians of the liver. Clin. Transl. Immunology. 2016, 5, 98. [Google Scholar] [CrossRef]
- Bandyopadhyay, K.; Marrero, I.; Kumar, V. NKT cell subsets as key participants in liver physiology and pathology. Cell Mol. Immunol. 2016, 13, 337–346. [Google Scholar] [CrossRef]
- Ibidapo-Obe, O.; Bruns, T. Tissue-resident and innate-like T cells in patients with advanced chronic liver disease. JHEP Rep. 2023, 5, 100812. [Google Scholar] [CrossRef]
- Wei, X.; Qian, J.; Yao, W.; Chen, L.; Guan, H.; Chen, Y.; Xie, Y.; Lu, H.; Zhang, Z.; Shi, L.; et al. Hyperactivated peripheral invariant natural killer T cells correlate with the progression of HBV-relative liver cirrhosis. Scand. J. Immunol. 2019, 90, 12775. [Google Scholar] [CrossRef]
- Tang, W.; Zhou, J.; Yang, W.; Feng, Y.; Wu, H.; Mok, M.T.S.; Zhang, L.; Liang, Z.; Liu, X.; Xiong, Z.; et al. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol. Immunol. 2022, 19, 834–847. [Google Scholar] [CrossRef]
- Zheng, S.; Yang, W.; Yao, D.; Tang, S.; Hou, J.; Chang, X. A comparative study on roles of natural killer T cells in two diet-induced non-alcoholic steatohepatitis-related fibrosis in mice. Ann. Med. 2022, 54, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.W.; Chen, H.W.; Wang, Y.W.; Lin, C.I.; Chuang, Y.H. IL-21, not IL-17A, exacerbates murine primary biliary cholangitis. Clin. Exp. Immunol. 2024, 215, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tian, Z. Roles of Hepatic Innate and Innate-Like Lymphocytes in Nonalcoholic Steatohepatitis. Front. Immunol. 2020, 11, 1500. [Google Scholar] [CrossRef]
- Torres-Hernandez, A.; Wang, W.; Nikiforov, Y.; Tejada, K.; Torres, L.; Kalabin, A.; Adam, S.; Wu, J.; Lu, L.; Chen, R.; et al. γδ T Cells Promote Steatohepatitis by Orchestrating Innate and Adaptive Immune Programming. Hepatology 2020, 71, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, B. γδT Cells and CD1d, Novel Immune Players in Alcoholic and Nonalcoholic Steatohepatitis? Hepatology 2020, 71, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hao, X.; Chen, Y.; Bai, L.; Gao, X.; Lian, Z.; Wei, H.; Sun, R.; Tian, Z. The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat. Commun. 2017, 7, 13839. [Google Scholar] [CrossRef]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014, 59, 630–642. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Wu, S.; Cheng, L.; Shen, Y.; Ma, W.; She, W.; Yang, C.; Wang, J.; Jiang, W. Type 3 innate lymphoid cell: A new player in liver fibrosis progression. Clin. Sci. 2018, 132, 2565–2582. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C. The Roles of Liver-Resident Lymphocytes in Liver Diseases. Front. Immunol. 2019, 10, 1582. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Nevzorova, Y.A.; Luedde, T.; Zimmermann, H.; Kroy, D.; Strnad, P.; Berres, M.L.; Bernhagen, J.; Tacke, F.; Nattermann, J.; et al. Liver Fibrosis-From Mechanisms of Injury to Modulation of Disease. Front. Med. 2022, 8, 814496. [Google Scholar] [CrossRef] [PubMed]
- Seillet, C.; Brossay, L.; Vivier, E. Natural killers or ILC1s? That is the question. Curr. Opin. Immunol. 2021, 68, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Ebbo, M.; Crinier, A.; Vély, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Forkel, M.; Berglin, L.; Kekäläinen, E.; Carlsson, A.; Svedin, E.; Michaëlsson, J.; Nagasawa, M.; Erjefält, J.S.; Mori, M.; Flodström-Tullberg, M.; et al. Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur. J. Immunol. 2017, 47, 1280–1294. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Koay, H.F.; McCluskey, J.; Gherardin, N.A. The biology and functional importance of MAIT cells. Nat. Immunol. 2019, 20, 1110–1128. [Google Scholar] [CrossRef]
- Hegde, P.; Weiss, E.; Paradis, V.; Wan, J.; Mabire, M.; Sukriti, S.; Rautou, P.E.; Albuquerque, M.; Picq, O.; Gupta, A.C.; et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat. Commun. 2018, 9, 2146. [Google Scholar] [CrossRef]
- Li, Y.; Huang, B.; Jiang, X.; Chen, W.; Zhang, J.; Wei, Y.; Chen, Y.; Lian, M.; Bian, Z.; Miao, Q.; et al. Mucosal-Associated Invariant T Cells Improve Nonalcoholic Fatty Liver Disease Through Regulating Macrophage Polarization. Front. Immunol. 2018, 9, 1994. [Google Scholar] [CrossRef]
- Guan, H.; Zhang, X.; Kuang, M.; Yu, J. The gut-liver axis in immune remodeling of hepatic cirrhosis. Front. Immunol. 2022, 13, 946628. [Google Scholar] [CrossRef]
- Tranah, T.H.; Edwards, L.A.; Schnabl, B.; Shawcross, D.L. Targeting the gut-liver-immune axis to treat cirrhosis. Gut 2021, 70, 982–994. [Google Scholar] [CrossRef]
- Koda, Y.; Teratani, T.; Chu, P.S.; Hagihara, Y.; Mikami, Y.; Harada, Y.; Tsujikawa, H.; Miyamoto, K.; Suzuki, T.; Taniki, N.; et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat. Commun. 2021, 12, 4474. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.G.; Kounatidis, D.; Psallida, S.; Vythoulkas-Biotis, N.; Adamou, A.; Zachariadou, T.; Kargioti, S.; Karampela, I.; Dalamaga, M. NAFLD/MASLD and the Gut-Liver Axis: From Pathogenesis to Treatment Options. Metabolites 2024, 14, 366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lu, S.; Li, Z. Extrahepatic factors in hepatic immune regulation. Front. Immunol. 2022, 13, 941721. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. Gammadelta T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef]
- Yang, F.; Li, H.; Li, Y.; Hao, Y.; Wang, C.; Jia, P.; Chen, X.; Ma, S.; Xiao, Z. Crosstalk between hepatic stellate cells and surrounding cells in hepatic fibrosis. Int. Immunopharmacol. 2021, 99, 108051. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, R.; Yang, A.G.; Zheng, G. Diversity of immune checkpoints in cancer immunotherapy. Front. Immunol. 2023, 14, 1121285. [Google Scholar] [CrossRef]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Zhong, Z.; Vong, C.T.; Chen, F.; Tan, H.; Zhang, C.; Wang, N.; Cui, L.; Wang, Y.; Feng, Y. Immunomodulatory potential of natural products from herbal medicines as immune checkpoints inhibitors: Helping to fight against cancer via multiple targets. Med. Res. Rev. 2022, 42, 1246–1279. [Google Scholar] [CrossRef]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef]
- Zhao, Y.; Shao, Q.; Peng, G. Exhaustion and senescence: Two crucial dysfunctional states of T cells in the tumor microenvironment. Cell Mol. Immunol. 2020, 17, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Zhang, X.; Fedorov, A.A.; Nathenson, S.G.; Almo, S.C. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 2001, 410, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Bradshaw, J.; Greene, J.; Peach, R.; Bennett, K.L.; Mittler, R.S. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 1996, 4, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Ruff, S.M.; Pawlik, T.M. Emerging data on immune checkpoint inhibitors in the neoadjuvant and adjuvant setting for patients with hepatocellular carcinoma. Hepatoma Res. 2024, 10, 22. [Google Scholar] [CrossRef]
- Baldanzi, G. Immune Checkpoint Receptors Signaling in T Cells. Int. J. Mol. Sci. 2022, 23, 3529. [Google Scholar] [CrossRef]
- Cai, J.; Qi, Q.; Qian, X.; Han, J.; Zhu, X.; Zhang, Q.; Xia, R. The role of PD-1/PD-L1 axis and macrophage in the progression and treatment of cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1377–1385. [Google Scholar] [CrossRef]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun. Rev. 2013, 12, 1091–1100. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebemberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.J.; et al. TGFβ1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fourcade, J.; Pagliano, O.; Chauvin, J.M.; Sander, C.; Kirkwood, J.M.; Zarour, H.M. IL10 and PD-1 Cooperate to Limit the Activity of Tumor-Specific CD8+ T Cells. Cancer Res. 2015, 75, 1635–1644. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef]
- Li, K.; Yuan, Z.; Lyu, J.; Ahn, E.; Davis, S.J.; Ahmed, R.; Zhu, C. PD-1 suppresses TCR-CD8 cooperativity during T-cell antigen recognition. Nat. Commun. 2021, 12, 2746. [Google Scholar] [CrossRef]
- Apol, Á.D.; Winckelmann, A.A.; Duus, R.B.; Bukh, J.; Weis, N. The Role of CTLA-4 in T Cell Exhaustion in Chronic Hepatitis B Virus Infection. Viruses 2023, 15, 1141. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Qorraj, M.; Bruns, H.; Böttcher, M.; Weigand, L.; Saul, D.; Mackensen, A.; Jitschin, R.; Mougiakakos, D. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia 2017, 31, 470–478. [Google Scholar] [CrossRef]
- Liu, J.; Xu, X.; Zhong, H.; Yu, M.; Abuduaini, N.; Zhang, S.; Yang, X.; Feng, B. Glycosylation and Its Role in Immune Checkpoint Proteins: From Molecular Mechanisms to Clinical Implications. Biomedicines 2024, 12, 1446. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.M.; Xia, W.; Hsu, Y.H.; Chan, L.C.; Yu, W.H.; Cha, J.H.; Chen, C.T.; Liao, H.W.; Kuo, C.W.; Khoo, K.H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qu, Y.; Hao, C.; Yao, W. PD-1/PD-L1 axis in organ fibrosis. Front. Immunol. 2023, 14, 1145682. [Google Scholar] [CrossRef]
- Aoki, T.; Nishida, N.; Kudo, M. Current Perspectives on the Immunosuppressive Niche and Role of Fibrosis in Hepatocellular Carcinoma and the Development of Antitumor Immunity. J. Histochem. Cytochem. 2022, 70, 53–81. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Zhang, Y.T.; Wang, Y.; Zhao, Y.; He, L.J. What role does PDL1 play in EMT changes in tumors and fibrosis? Front Immunol. 2023, 14, 1226038. [Google Scholar] [CrossRef]
- Ke, M.Y.; Xu, T.; Fang, Y.; Ye, Y.P.; Li, Z.J.; Ren, F.G.; Lu, S.Y.; Zhang, X.F.; Wu, R.Q.; Lv, Y.; et al. Liver fibrosis promotes immune escape in hepatocellular carcinoma via GOLM1-mediated PD-L1 upregulation. Cancer Lett. 2021, 513, 14–25. [Google Scholar] [CrossRef]
- Xiang, M.; Liu, T.; Tian, C.; Ma, K.; Gou, J.; Huang, R.; Li, S.; Li, Q.; Xu, C.; Li, L.; et al. Kinsenoside attenuates liver fibro-inflammation by suppressing dendritic cells via the PI3K-AKT-FoxO1 pathway. Pharmacol. Res. 2022, 177, 106092. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4+ T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 2019, 20, 27. [Google Scholar] [CrossRef]
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694. [Google Scholar] [CrossRef]
- Cho, H.; Kang, H.; Lee, H.H.; Kim, C.W. Programmed Cell Death 1 (PD-1) and Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4) in Viral Hepatitis. Int. J. Mol. Sci. 2017, 18, 1517. [Google Scholar] [CrossRef]
- Huang, D.; Yan, W.; Han, M.; Yuan, W.; Wang, P.; Chen, Y.; Wan, X.; Luo, X.; Wu, D.; Ning, Q. Insufficient immunity led to virologic breakthrough in NAs-treated chronic hepatitis B patients switching to Peg-IFN-ɑ. Antiviral Res. 2022, 197, 105220. [Google Scholar] [CrossRef] [PubMed]
- Ferrando-Martinez, S.; Huang, K.; Bennett, A.S.; Sterba, P.; Yu, L.; Suzich, J.A.; Janssen, H.L.A.; Robbins, S.H. HBeAg seroconversion is associated with a more effective PD-L1 blockade during chronic hepatitis B infection. JHEP Rep. 2019, 1, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Jiang, D.; Yan, C.; Liu, X.; Lv, Y.; Xie, J.; Chen, Y. Immune Checkpoint Molecules Expressed on CD4+ T Cell Subsets in Chronic Asymptomatic Hepatitis B Virus Carriers With Hepatitis B e Antigen-Negative. Front. Microbiol. 2022, 13, 887408. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; El-Badawy, A. Programmed death-1/programmed death-L1 signaling pathway and its blockade in hepatitis C virus immunotherapy. World J. Hepatol. 2015, 7, 2449–2458. [Google Scholar] [CrossRef]
- Park, S.J.; Hahn, Y.S. Hepatocytes infected with hepatitis C virus change immunological features in the liver microenvironment. Clin. Mol. Hepatol. 2023, 29, 65–76. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, J.; Wu, H.; Chen, S.; Zhang, L.; Tang, W.; Duan, L.; Wang, Y.; McCabe, E.; Hu, M.; et al. Fibrotic immune microenvironment remodeling mediates superior anti-tumor efficacy of a nano-PD-L1 trap in hepatocellular carcinoma. Mol. Ther. 2023, 31, 119–133. [Google Scholar] [CrossRef]
- Zhou, L.; Li, X.; Huang, X.; Chen, L.; Gu, L.; Huang, Y. Soluble programmed death-1 is a useful indicator for inflammatory and fibrosis severity in chronic hepatitis B. J. Viral Hepat. 2019, 26, 795–802. [Google Scholar] [CrossRef]
- Huang, N.; Zhou, R.; Chen, H.; Zhang, S.; Li, J.; Wei, W.; Sun, J.; Ren, S.; Li, B.; Deng, H.; et al. Splenic CD4+ and CD8+ T-cells highly expressed PD-1 and Tim-3 in cirrhotic patients with HCV infection and portal hypertension. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211061051. [Google Scholar] [CrossRef]
- Jiang, A.; Liu, N.; Wang, J.; Zheng, X.; Ren, M.; Zhang, W.; Yao, Y. The role of PD-1/PD-L1 axis in idiopathic pulmonary fibrosis: Friend or foe? Front. Immunol. 2022, 13, 1022228. [Google Scholar] [CrossRef]
- Lu, Y.; Zhong, W.; Liu, Y.; Chen, W.; Zhang, J.; Zeng, Z.; Huang, H.; Qiao, Y.; Wan, X.; Meng, X.; et al. Anti-PD-L1 antibody alleviates pulmonary fibrosis by inducing autophagy via inhibition of the PI3K/Akt/mTOR pathway. Int. Immunopharmacol. 2022, 104, 108504. [Google Scholar] [CrossRef]
- Paskeh, M.D.A.; Ghadyani, F.; Hashemi, M.; Abbaspour, A.; Zabolian, A.; Javanshir, S.; Razzazan, M.; Mirzaei, S.; Entezari, M.; Goharrizi, M.A.S.B.; et al. Biological impact and therapeutic perspective of targeting PI3K/Akt signaling in hepatocellular carcinoma: Promises and Challenges. Pharmacol. Res. 2023, 187, 106553. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Chen, T.Y.; Zhao, X.J.; Xu, Q.; Jiao, R.Q.; Li, J.M.; Kong, L.D. Pterostilbene prevents hepatocyte epithelial-mesenchymal transition in fructose-induced liver fibrosis through suppressing miR-34a/Sirt1/p53 and TGF-β1/Smads signalling. Br. J. Pharmacol. 2019, 176, 1619–1634. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Z.; Zhao, X.J.; Xu, H.J.; Wang, S.C.; Pan, Y.; Wang, S.J.; Xu, Q.; Jiao, R.Q.; Gu, H.M.; Kong, L.D. Magnesium isoglycyrrhizinate ameliorates high fructose-induced liver fibrosis in rat by increasing miR-375-3p to suppress JAK2/STAT3 pathway and TGF-β1/Smad signaling. Acta Pharmacol. Sin. 2019, 40, 879–894. [Google Scholar] [CrossRef]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef]
- Ye, Y.; Xu, Y.; Lai, Y.; He, W.; Li, Y.; Wang, R.; Luo, X.; Chen, R.; Chen, T. Long non-coding RNA cox-2 prevents immune evasion and metastasis of hepatocellular carcinoma by altering M1/M2 macrophage polarization. J. Cell Biochem. 2018, 119, 2951–2963. [Google Scholar] [CrossRef]
- Li, Q.; Deng, M.S.; Wang, R.T.; Luo, H.; Luo, Y.Y.; Zhang, D.D.; Chen, K.J.; Cao, X.F.; Yang, G.M.; Zhao, T.M.; et al. PD-L1 upregulation promotes drug-induced pulmonary fibrosis by inhibiting vimentin degradation. Pharmacol. Res. 2023, 187, 106636. [Google Scholar] [CrossRef]
- Kong, X.; Peng, H.; Liu, P.; Fu, X.; Wang, N.; Zhang, D. Programmed death ligand 1 regulates epithelial-mesenchymal transition and cancer stem cell phenotypes in hepatocellular carcinoma through the serum and glucocorticoid kinase 2/β-catenin signaling pathway. Cancer Sci. 2023, 114, 2265–2276. [Google Scholar] [CrossRef]
- Nakamoto, N.; Cho, H.; Shaked, A.; Olthoff, K.; Valiga, M.E.; Kaminski, M.; Gostick, E.; Price, D.A.; Freeman, G.J.; Wherry, E.J.; et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. 2009, 5, 1000313. [Google Scholar] [CrossRef]
- Hazrati, A.; Malekpour, K.; Khorramdelazad, H.; Rajaei, S.; Hashemi, S.M. Therapeutic and immunomodulatory potentials of mesenchymal stromal/stem cells and immune checkpoints related molecules. Biomark. Res. 2024, 12, 35. [Google Scholar] [CrossRef]
- Bally, A.P.; Lu, P.; Tang, Y.; Austin, J.W.; Scharer, C.D.; Ahmed, R.; Boss, J.M. NF-κB regulates PD-1 expression in macrophages. J. Immunol. 2015, 194, 4545–4554. [Google Scholar] [CrossRef]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef]
- Huang, X.; Venet, F.; Wang, Y.L.; Lepape, A.; Yuan, Z.; Chen, Y.; Swan, R.; Kherouf, H.; Monneret, G.; Chung, C.S.; et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 6303–6308. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Gao, Y.; Liu, Y.; Zhang, B.; Liu, Q.; Wu, J.; Fan, L.; Ou, Q.; Zhang, W.; Shao, L. PD-1/PD-L pathway inhibits M.tb-specific CD4+ T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci. Rep. 2016, 6, 38362. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.P.; Chow, L.; Ammons, D.T.; Wheat, W.H.; Dow, S.W. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol. Res. 2018, 6, 1260–1273. [Google Scholar] [CrossRef] [PubMed]
- Kassel, R.; Cruise, M.W.; Iezzoni, J.C.; Taylor, N.A.; Pruett, T.L.; Hahn, Y.S. Chronically inflamed livers up-regulate expression of inhibitory B7 family members. Hepatology 2009, 50, 1625–1637. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Gudd, C.L.; Mawhin, M.A.; Husbyn, H.C.; Trovato, F.M.; Siggins, M.K.; O’Connor, T.; Kudo, H.; Mukherjee, S.K.; Wendon, J.A.; et al. PD-1 blockade improves Kupffer cell bacterial clearance in acute liver injury. J. Clin. Investig. 2021, 131, 140196. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Woollard, K.J.; McPhail, M.J.W.; Antoniades, C.G.; Possamai, L.A. The Role of Monocytes and Macrophages in Acute and Acute-on-Chronic Liver Failure. Front. Immunol. 2018, 9, 2948. [Google Scholar] [CrossRef]
- Charles, R.; Chou, H.S.; Wang, L.; Fung, J.J.; Lu, L.; Qian, S. Human hepatic stellate cells inhibit T-cell response through B7-H1 pathway. Transplantation 2013, 96, 17–24. [Google Scholar] [CrossRef]
- Onorati, A.; Havas, A.P.; Lin, B.; Rajagopal, J.; Sen, P.; Adams, P.D.; Dou, Z. Upregulation of PD-L1 in Senescence and Aging. Mol. Cell Biol. 2022, 42, 0017122. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A. Inhibitory immune checkpoints suppress the surveillance of senescent cells promoting their accumulation with aging and in age-related diseases. Biogerontology 2024, 25, 749–773. [Google Scholar] [CrossRef]
- Salminen, A. The role of the immunosuppressive PD-1/PD-L1 checkpoint pathway in the aging process and age-related diseases. J. Mol. Med. 2024, 102, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Y.; Wang, X.; Navarro-Corcuera, A.; Ilyas, S.; Jalan-Sakrikar, N.; Gan, C.; Tu, X.; Shi, Y.; Tu, K.; et al. PD-L1 promotes myofibroblastic activation of hepatic stellate cells by distinct mechanisms selective for TGF-β receptor I versus II. Cell Rep. 2022, 38, 110349. [Google Scholar] [CrossRef] [PubMed]
- Kontos, F.; Michelakos, T.; Kurokawa, T.; Sadagopan, A.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. B7-H3: An Attractive Target for Antibody-based Immunotherapy. Clin. Cancer Res. 2021, 27, 1227–1235. [Google Scholar] [CrossRef]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef]
- Podojil, J.R.; Miller, S.D. Potential targeting of B7-H4 for the treatment of cancer. Immunol. Rev. 2017, 276, 40–51. [Google Scholar] [CrossRef]
- Sica, G.L.; Choi, I.H.; Zhu, G.; Tamada, K.; Wang, S.D.; Tamura, H.; Chapoval, A.I.; Flies, D.B.; Bajorath, J.; Chen, L. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003, 18, 849–861. [Google Scholar] [CrossRef]
- Jiang, D.; Chen, C.; Yan, D.; Zhang, X.; Liu, X.; Yan, D.; Cui, D.; Yang, S. Exhausted phenotype of circulating CD8+ T cell subsets in hepatitis B virus carriers. BMC Immunol. 2022, 23, 18. [Google Scholar] [CrossRef]
- Mohammadizad, H.; Shahbazi, M.; Hasanjani Roushan, M.R.; Soltanzadeh-Yamchi, M.; Mohammadnia-Afrouzi, M. TIM-3 as a marker of exhaustion in CD8+ T cells of active chronic hepatitis B patients. Microb. Pathog. 2019, 128, 323–328. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Papadakos, S.P.; Vakadaris, G.; Chatzikalil, E.; Stergiou, I.E.; Kalopitas, G.; Theocharis, S.; Germanidis, G. Shedding light on the role of LAG-3 in hepatocellular carcinoma: Unraveling immunomodulatory pathways. Hepatoma Res. 2024, 10, 21. [Google Scholar] [CrossRef]
- Heim, K.; Neumann-Haefelin, C.; Thimme, R.; Hofmann, M. Heterogeneity of HBV-Specific CD8+ T-Cell Failure: Implications for Immunotherapy. Front. Immunol. 2019, 10, 2240. [Google Scholar] [CrossRef] [PubMed]
- Ogiso, H.; Ito, H.; Ando, T.; Arioka, Y.; Kanbe, A.; Ando, K.; Ishikawa, T.; Saito, K.; Hara, A.; Moriwaki, H.; et al. The Deficiency of Indoleamine 2,3-Dioxygenase Aggravates the CCl4-Induced Liver Fibrosis in Mice. PLoS ONE 2016, 11, 0162183. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Osawa, Y.; Nakamoto, K.; Morita, N.; Yamamoto, Y.; Ando, T.; Tashita, C.; Nabeshima, T.; Saito, K. Kynurenine produced by indoleamine 2,3-dioxygenase 2 exacerbates acute liver injury by carbon tetrachloride in mice. Toxicology 2020, 438, 152458. [Google Scholar] [CrossRef]
- Nebbia, G.; Peppa, D.; Schurich, A.; Khanna, P.; Singh, H.D.; Cheng, Y.; Rosenberg, W.; Dusheiko, G.; Gilson, R.; ChinAleong, J.; et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE 2012, 7, e47648. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology 2010, 138, 682–693. [Google Scholar] [CrossRef]
- Wu, W.; Shi, Y.; Li, S.; Zhang, Y.; Liu, Y.; Wu, Y.; Chen, Z. Blockade of Tim-3 signaling restores the virus-specific CD8⁺ T-cell response in patients with chronic hepatitis B. Eur. J. Immunol. 2012, 42, 1180–1191. [Google Scholar] [CrossRef]
- Zong, L.; Peng, H.; Sun, C.; Li, F.; Zheng, M.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Breakdown of adaptive immunotolerance induces hepatocellular carcinoma in HBsAg-tg mice. Nat. Commun. 2019, 10, 221. [Google Scholar] [CrossRef]
- Li, Q.; Han, J.; Yang, Y.; Chen, Y. PD-1/PD-L1 checkpoint inhibitors in advanced hepatocellular carcinoma immunotherapy. Front. Immunol. 2022, 13, 1070961. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Gupta, T.; Jarpula, N.S. Hepatocellular carcinoma immune microenvironment and check point inhibitors-current status. World J. Hepatol. 2024, 16, 353–365. [Google Scholar] [CrossRef]
- Haber, P.K.; Puigvehí, M.; Castet, F.; Lourdusamy, V.; Montal, R.; Tabrizian, P.; Buckstein, M.; Kim, E.; Villanueva, A.; Schwartz, M.; et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002–2020). Gastroenterology 2021, 161, 879–898. [Google Scholar] [CrossRef]
- Li, X.; Gao, L.; Wang, B.; Hu, J.; Yu, Y.; Gu, B.; Xiang, L.; Li, X.; Li, H.; Zhang, T.; et al. FXa-mediated PAR-2 promotes the efficacy of immunotherapy for hepatocellular carcinoma through immune escape and anoikis resistance by inducing PD-L1 transcription. J. Immunother. Cancer 2024, 12, e009565. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Mainoli, B.; Duarte, G.S.; Machado, T.; Tinoco, R.G.; Esperança-Martins, M.; Ferreira, J.J.; Costa, J. Immune-related serious adverse events with immune checkpoint inhibitors: Systematic review and network meta-analysis. Eur. J. Clin. Pharmacol. 2024, 80, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xiao, H.; Li, H.; Chen, X.; Li, Y. Adverse events associated with immune checkpoint inhibitors in non-small cell lung cancer: A safety analysis of clinical trials and FDA pharmacovigilance system. Front. Immunol. 2024, 15, 1396752. [Google Scholar] [CrossRef] [PubMed]
- Godbert, B.; Petitpain, N.; Lopez, A.; Nisse, Y.E.; Gillet, P. Hepatitis B reactivation and immune check point inhibitors. Dig. Liver Dis. 2021, 53, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wang, H.; Deng, L.; Wang, W.; Wang, Y. Immune-related adverse events of immune checkpoint inhibitors combined with angiogenesis inhibitors: A real-world pharmacovigilance analysis of the FDA Adverse Event Reporting System (FAERS) database (2014–2022). Int. Immunopharmacol. 2024, 136, 112301. [Google Scholar] [CrossRef]

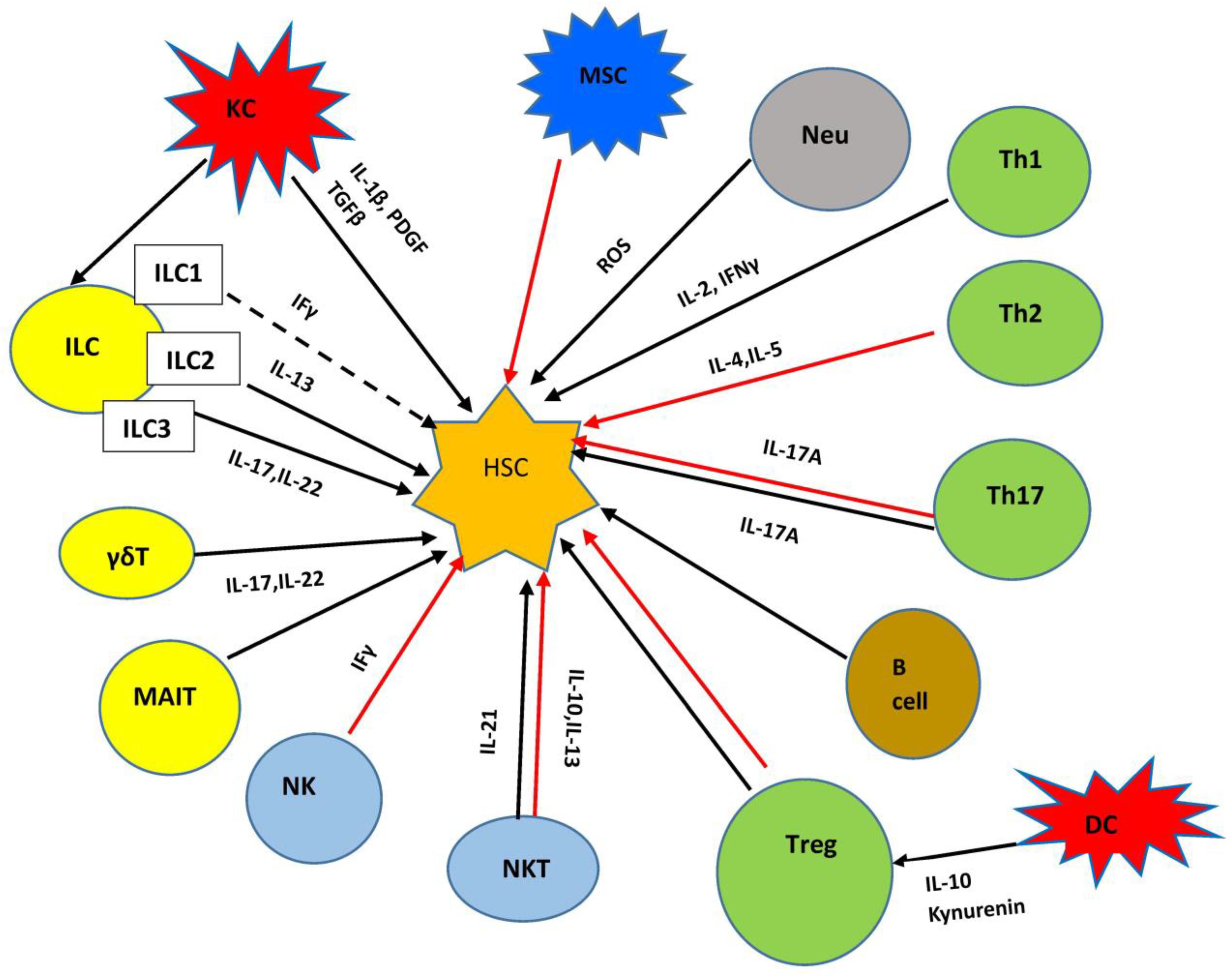

{kind=link}
{kind=link}
| Molecules | Functions | References |
|---|---|---|
| TGF-β | Primarily produced by macrophages. Enhances ECM production in HSCs through Smad-dependent pathways and Smad-independent pathways. | [131,132] |
| PDGF | Produced by macrophages. It contributes to fibrosis progression through HSC activation. | [133] |
| TNF-α | Upregulates TIMP-1 production, prevents HSC apoptosis. | [134,135] |
| IL-1β,IL-18 | Produced by pro-inflammatory macrophages through activation of the NLRP3 inflammasome. Activates HSCs, upregulates TIMP production. | [136,137] |
| IL-13, IL-4 | Produced by M2 macrophages. Promotes the activation of HSCs. | [138] |
| MCP1 | Activates CCR2 in Kupffer cells and HSCs. | [139,140] |
| CCL2 CCL5 | Produced by macrophages and HSCs. Increases macrophage infiltration and fibrotic phenotype of HSCs. | [141,142] |
| CXCL6 | Induces TGF β production by KCs and indirectly promotes fibrosis. | [143] |
| Cytokines | Functions | References |
|---|---|---|
| TNF-α | Inhibit HBV replication, provide antiviral immunity, induce inflammation. | [432,461] |
| TGF-β | Impair NK cell function, promote fibrosis and HCC. | [131,132,177] |
| IL-10 | Inhibit cytokine production, regulate T cell immunity, develop persistence of HBV infection. | [481,482,484] |
| IL-13 | Induce inflammation, liver fibrosis. and cirrhosis. | [471,472] |
| IL-6 | Produced by macrophages. Induce inflammation and fibrosis. Inhibit HBV replication; inhibits HBV entry. | [200,202,203] |
| IL-18, IL-1β | Pro-inflammatory. Activate HSCs. IL-1β induces the phosphorylation of Smad2/3 to promote the transformation of hepatocytes to EMT. IL-18 rs187238 GG genotype increases the risk of HCC in a healthy population and the risk of cirrhosis in CHB carriers. | [137,199,202] |
| IL-17 | Exacerbate inflammation, induce liver fibrosis and cirrhosis. | [436,450,451] |
| IL-21 | Produced by activated CD4+ T cells. Activate T and B cells. Maintenance of specific CD8+ T-cell functions and control of viremia. Increased levels may promote cirrhosis and exacerbate liver injury. | [424] |
| IL-22 | Inhibit liver inflammation and fibrosis. | [462,463] |
| IL-27 | Higher levels in patients with liver cirrhosis or hepatocellular carcinoma. Compensate the function of IL-21 by supporting Tfh-B cell function, required for protective antibody response. | [424] |
| IL-33 | Induce liver damage and fibrosis, activate Tfh cells, and enhance humoral immunity; suppress HBV replication. | [426,433,440] |
| IL-35 | Development of fibrosis, cirrhosis, and HCC. Inhibit HBV-specific CD8 T cells cytotoxicity. Inhibit cytokines and induce antiviral immunity. | [492,493,494] |
| IFN-γ | Antiviral immunity. Inhibit HBV replication; induce inflammation. | [432,434] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsomidis, I.; Voumvouraki, A.; Kouroumalis, E. Immune Checkpoints and the Immunology of Liver Fibrosis. Livers 2025, 5, 5. https://doi.org/10.3390/livers5010005
Tsomidis I, Voumvouraki A, Kouroumalis E. Immune Checkpoints and the Immunology of Liver Fibrosis. Livers. 2025; 5(1):5. https://doi.org/10.3390/livers5010005
Chicago/Turabian StyleTsomidis, Ioannis, Argyro Voumvouraki, and Elias Kouroumalis. 2025. "Immune Checkpoints and the Immunology of Liver Fibrosis" Livers 5, no. 1: 5. https://doi.org/10.3390/livers5010005
APA StyleTsomidis, I., Voumvouraki, A., & Kouroumalis, E. (2025). Immune Checkpoints and the Immunology of Liver Fibrosis. Livers, 5(1), 5. https://doi.org/10.3390/livers5010005