
The Pivotal Role of the Membrane-Bound O-Acyltransferase Domain Containing 7 in Non-Alcoholic Fatty Liver Disease



Abstract
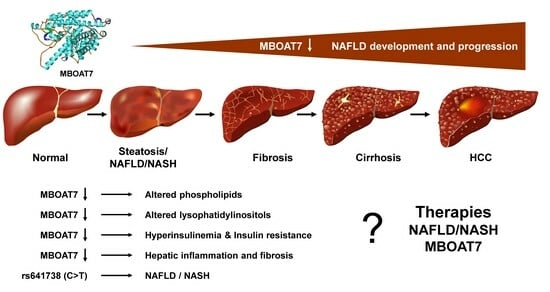
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. MBOAT7 Gene—An Overview
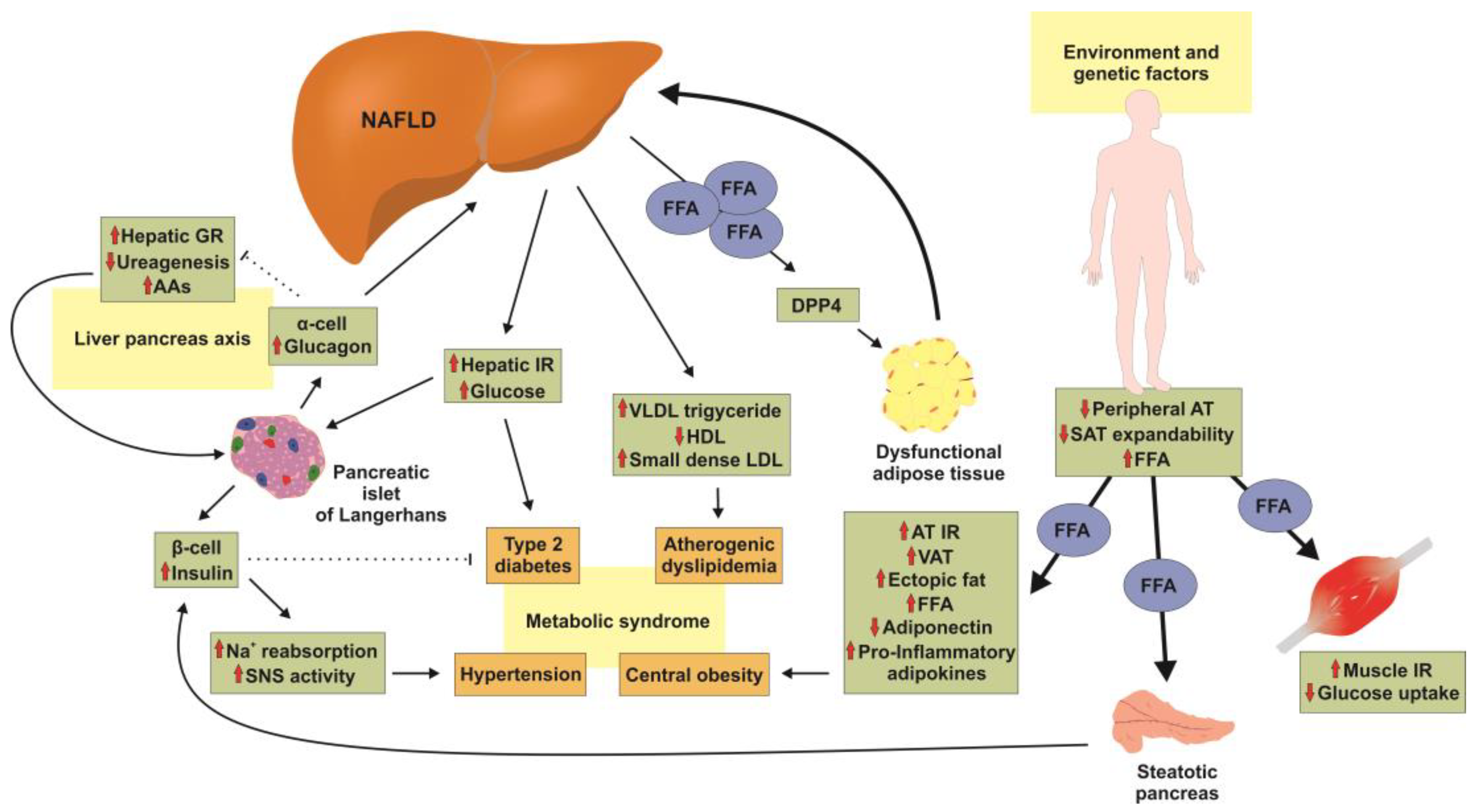
3. Pathogenesis of NAFLD
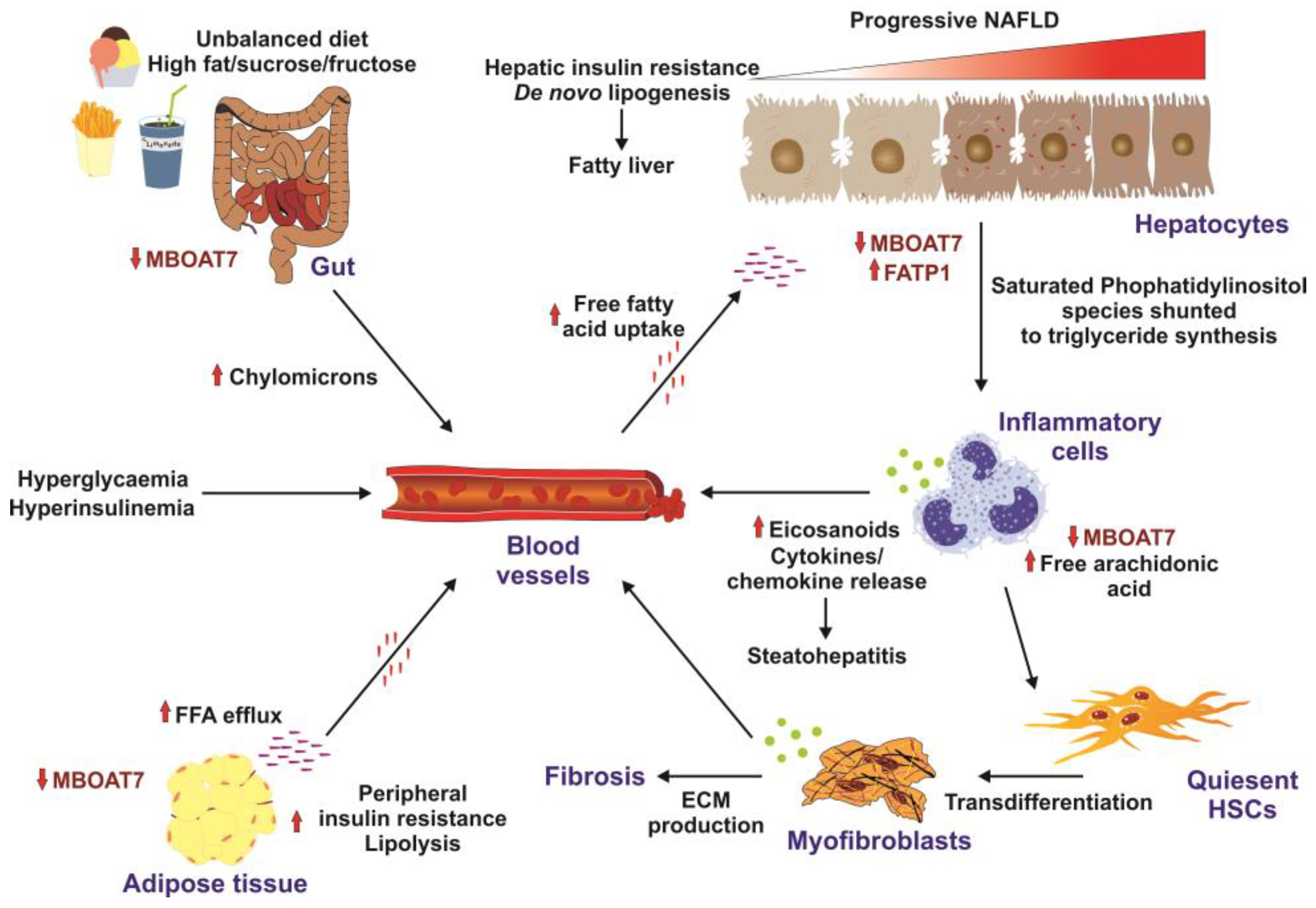
4. Mechanistic Link between MBOAT7 and NAFLD and Related In Vivo/In Vitro Studies
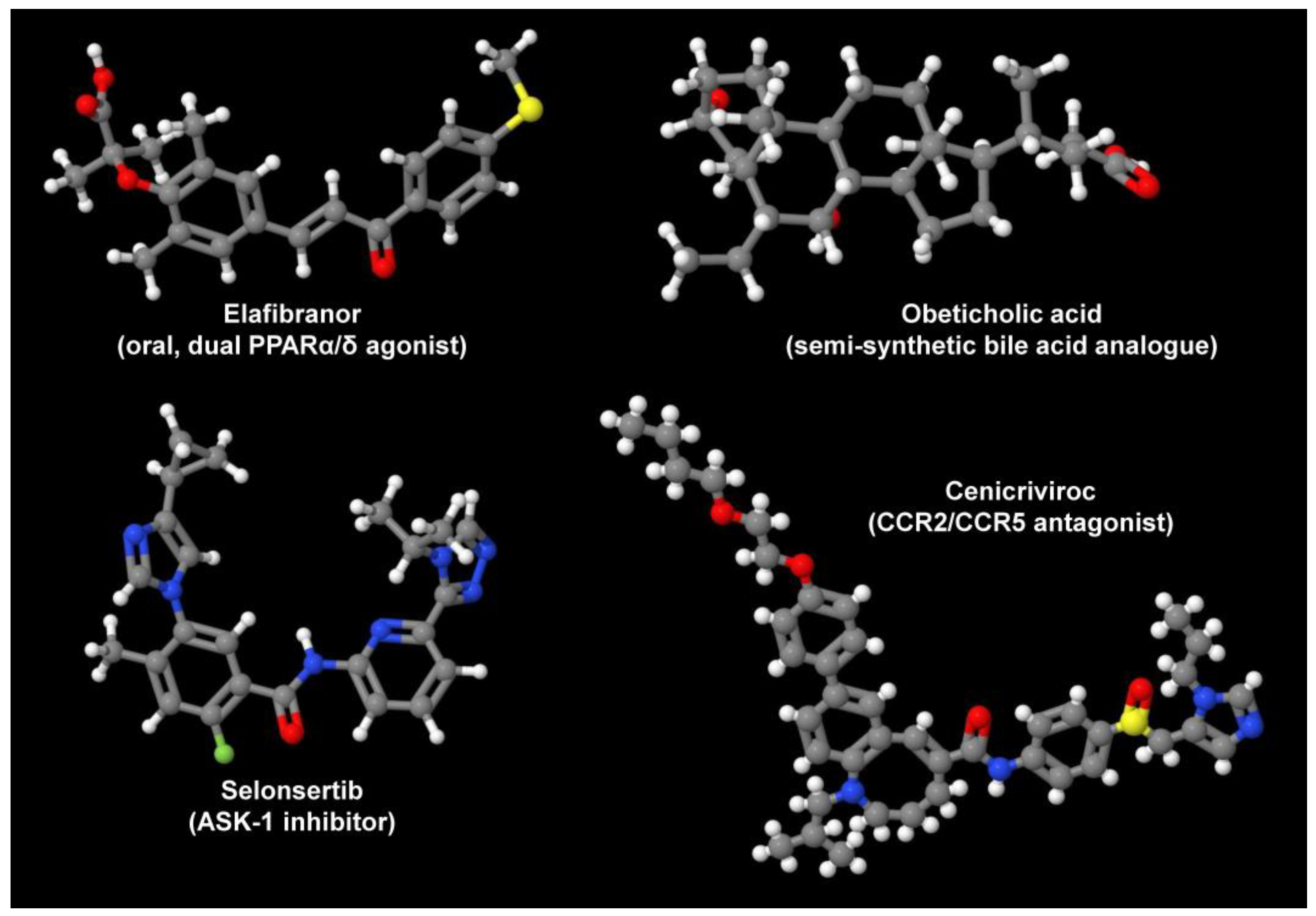
5. Challenges and Recent Progress in Treating Complex NAFLD
6. Future Directions in Management of NAFLD
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of non-alcoholic fatty liver disease. Qjm 2010, 103, 71–83. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Shaker, M.; Tabbaa, A.; Albeldawi, M.; Alkhouri, N. Liver transplantation for nonalcoholic fatty liver disease: New challenges and new opportunities. World J. Gastroenterol. 2014, 20, 5320–5330. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 2014, 59, 1174–1197. [Google Scholar] [CrossRef]
- Nagral, A.; Bangar, M.; Menezes, S.; Bhatia, S.; Butt, N.; Ghosh, J.; Manchanayake, J.H.; Mahtab, M.A.; Singh, S.P. Gender differences in nonalcoholic fatty liver disease. Euroasian J. Hepatogastroenterol. 2022, 12, S19–S25. [Google Scholar] [CrossRef]
- Caddeo, A.; Spagnuolo, R.; Maurotti, S. MBOAT7 in liver and extrahepatic diseases. Liver Int. 2023, 43, 2351–2364. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016, 150, 1219–1230.e1216. [Google Scholar] [CrossRef]
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K.; et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. eLife 2019, 8, e49882. [Google Scholar] [CrossRef]
- Tavaglione, F.; Kono, N.; Romeo, S. Understanding the underlying molecular pathways by which MBOAT7/Lpiat1 depletion induces hepatic steatosis. J. Lipid Res. 2021, 62, 100047. [Google Scholar] [CrossRef]
- Caddeo, A.; Jamialahmadi, O.; Solinas, G.; Pujia, A.; Mancina, R.M.; Pingitore, P.; Romeo, S. MBOAT7 is anchored to endomembranes by six transmembrane domains. J. Struct. Biol. 2019, 206, 349–360. [Google Scholar] [CrossRef]
- Lee, H.-C.; Inoue, T.; Sasaki, J.; Kubo, T.; Matsuda, S.; Nakasaki, Y.; Hattori, M.; Tanaka, F.; Udagawa, O.; Kono, N.; et al. LPIAT1 regulates arachidonic acid content in phosphatidylinositol and is required for cortical lamination in mice. Mol. Biol. Cell 2012, 23, 4689–4700. [Google Scholar] [CrossRef]
- Wang, K.; Lee, C.-W.; Sui, X.; Kim, S.; Wang, S.; Higgs, A.B.; Baublis, A.J.; Voth, G.A.; Liao, M.; Walther, T.C.; et al. The structure of phosphatidylinositol remodeling MBOAT7 reveals its catalytic mechanism and enables inhibitor identification. Nat. Commun. 2023, 14, 3533. [Google Scholar] [CrossRef]
- National Library of Medicine. Available online: https://www.ncbi.nlm.nih.gov/protein (accessed on 9 November 2023).
- Clustal Omega. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 9 November 2023).
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Byrne, C.D. Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proc. Nutr. Soc. 2013, 72, 412–419. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, X.-J.; Ji, Y.-X.; Zhang, P.; She, Z.-G.; Li, H. Nonalcoholic fatty liver disease pandemic fuels the upsurge in cardiovascular diseases. Circ. Res. 2020, 126, 679–704. [Google Scholar] [CrossRef]
- Pais, R.; Redheuil, A.; Cluzel, P.; Ratziu, V.; Giral, P. Relationship among fatty liver, specific and multiple-site atherosclerosis, and 10-year Framingham Score. Hepatology 2019, 69, 1453–1463. [Google Scholar] [CrossRef]
- Cheung, A.; Ahmed, A. Nonalcoholic fatty liver disease and chronic kidney disease: A review of links and risks. Clin. Exp. Gastroenterol. 2021, 14, 457–465. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Massey, W.J.; Varadharajan, V.; Banerjee, R.; Brown, A.L.; Horak, A.J.; Hohe, R.C.; Jung, B.M.; Qiu, Y.; Chan, E.R.; Pan, C.; et al. MBOAT7-driven lysophosphatidylinositol acylation in adipocytes contributes to systemic glucose homeostasis. J. Lipid Res. 2023, 64, 100349. [Google Scholar] [CrossRef]
- Xia, M.; Chandrasekaran, P.; Rong, S.; Fu, X.; Mitsche, M.A. Hepatic deletion of MBOAT7 (LPIAT1) causes activation of SREBP-1c and fatty liver. J. Lipid Res. 2021, 62, 100031. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Alharthi, J.; Bayoumi, A.; Thabet, K.; Pan, Z.; Gloss, B.S.; Latchoumanin, O.; Lundberg, M.; Twine, N.A.; McLeod, D.; Alenizi, S.; et al. A metabolic associated fatty liver disease risk variant in MBOAT7 regulates toll like receptor induced outcomes. Nat. Commun. 2022, 13, 7430. [Google Scholar] [CrossRef]
- Zahr, T.; Sun, K.; Qiang, L. The polarizable and reprogrammable identity of Kupffer cells in nonalcoholic steatohepatitis. Med. Rev. 2022, 2, 324–327. [Google Scholar] [CrossRef]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of inflammatory cytokines with non-alcoholic fatty liver disease. Front. Immunol. 2022, 13, 880298. [Google Scholar] [CrossRef]
- Zhu, B.; Chan, S.L.; Li, J.; Li, K.; Wu, H.; Cui, K.; Chen, H. Non-alcoholic steatohepatitis pathogenesis, diagnosis, and treatment. Front. Cardiovasc. Med. 2021, 8, 742382. [Google Scholar] [CrossRef]
- Mehal, W.Z. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 637–644. [Google Scholar] [CrossRef]
- Galatou, E.; Mourelatou, E.; Hatziantoniou, S.; Vizirianakis, I.S. Nonalcoholic steatohepatitis (NASH) and atherosclerosis: Explaining their pathophysiology, association and the role of incretin-based drugs. Antioxidants 2022, 11, 1060. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Varadharajan, V.; Massey, W.J.; Brown, J.M. Membrane-bound O-acyltransferase 7 (MBOAT7)-driven phosphatidylinositol remodeling in advanced liver disease. J. Lipid Res. 2022, 63, 100234. [Google Scholar] [CrossRef]
- Yuki, T.; Yuta, S.; Andrea, C.; Takuya, K.; Yanli, M.; Tetsuya, K.; Naoto, K.; Toshimasa, Y.; Rosellina Margherita, M.; Guido, B.; et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut 2021, 70, 180. [Google Scholar] [CrossRef]
- Lee, H.-C.; Inoue, T.; Imae, R.; Kono, N.; Shirae, S.; Matsuda, S.; Gengyo-Ando, K.; Mitani, S.; Arai, H. Caenorhabditis elegans mboa-7, a member of the MBOAT family, is required for selective incorporation of polyunsaturated fatty acids into phosphatidylinositol. Mol. Biol. Cell 2007, 19, 1174–1184. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Fracanzani, A.L.; Dongiovanni, P. MBOAT7 down-regulation by genetic and environmental factors predisposes to MAFLD. eBioMedicine 2020, 57, 102866. [Google Scholar] [CrossRef]
- Sharpe, M.C.; Pyles, K.D.; Hallcox, T.; Kamm, D.R.; Piechowski, M.; Fisk, B.; Albert, C.J.; Carpenter, D.H.; Ulmasov, B.; Ford, D.A.; et al. Enhancing hepatic MBOAT7 expression in mice with nonalcoholic steatohepatitis. Gastro Hep Adv. 2023, 2, 558–572. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-function genetic variants impact on NAFLD development and progression both in patients and in in vitro models. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 759–788. [Google Scholar] [CrossRef]
- Krawczyk, M.; Rau, M.; Schattenberg, J.M.; Bantel, H.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Lammert, F.; Geier, A. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: A multicenter biopsy-based study. J. Lipid Res. 2017, 58, 247–255. [Google Scholar] [CrossRef]
- Raja, A.M.; Ciociola, E.; Ahmad, I.N.; Dar, F.S.; Naqvi, S.M.S.; Moaeen-Ud-Din, M.; Kaukab Raja, G.; Romeo, S.; Mancina, R.M. Genetic susceptibility to chronic liver disease in individuals from Pakistan. Int. J. Mol. Sci. 2020, 21, 3558. [Google Scholar] [CrossRef]
- Sookoian, S.; Flichman, D.; Garaycoechea, M.E.; Gazzi, C.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Lack of evidence supporting a role of TMC4-rs641738 missense variant—MBOAT7- intergenic downstream variant—In the susceptibility to nonalcoholic fatty liver disease. Sci. Rep. 2018, 8, 5097. [Google Scholar] [CrossRef]
- Teo, K.; Abeysekera, K.W.M.; Adams, L.; Aigner, E.; Anstee, Q.M.; Banales, J.M.; Banerjee, R.; Basu, P.; Berg, T.; Bhatnagar, P.; et al. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J. Hepatol. 2021, 74, 20–30. [Google Scholar] [CrossRef]
- Sivell, C. Nonalcoholic fatty liver disease: A silent epidemic. Gastroenterol. Nurs. 2019, 42, 428–434. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and genetics in NAFLD: The perfect binomium. Int. J. Mol. Sci. 2020, 21, 2986. [Google Scholar] [CrossRef]
- Arab, J.P.; Díaz, L.A.; Dirchwolf, M.; Mark, H.E.; Lazarus, J.V.; Vaughan, E.; Méndez-Sánchez, N.; Oliveira, C.P.; Gadano, A.; Arrese, M. NAFLD: Challenges and opportunities to address the public health problem in Latin America. Ann. Hepatol. 2021, 24, 100359. [Google Scholar] [CrossRef]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef]
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic MBOAT7 leads to liver fibrosis. Gut 2021, 70, 940–950. [Google Scholar] [CrossRef]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of vitamin E for nonalcoholic steatohepatitis in patients with type 2 diabetes: A randomized controlled trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef]
- Iacono, A.; Raso, G.M.; Canani, R.B.; Calignano, A.; Meli, R. Probiotics as an emerging therapeutic strategy to treat NAFLD: Focus on molecular and biochemical mechanisms. J. Nutr. Biochem. 2011, 22, 699–711. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Sepe, V.; Zampella, A.; Distrutti, E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 623–632. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D with Features for Chemicals, Crystals, Materials and Biomolecules. Available online: https://jmol.sourceforge.net/ (accessed on 9 November 2023).
- PubChem. National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 9 November 2023).
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.F. PPAR-targeted therapies in the treatment of non-alcoholic fatty liver disease in diabetic patients. Int. J. Mol. Sci. 2022, 23, 4305. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Carr, R.M.; Reid, A.E. FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr. Atheroscler. Rep. 2015, 17, 500. [Google Scholar] [CrossRef]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist Cenicriviroc in animal models of liver and kidney fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef]
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. MBOAT7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. eBioMedicine 2020, 52, 102658. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375. [Google Scholar] [CrossRef] [PubMed]
- Kairuz, D.; Singh, P.; Smith, T.; Arbuthnot, P.; Ely, A.; Bloom, K. Synthetic mRNA gene therapies and hepatotropic non-viral vectors for the treatment of chronic HBV infections. In Messenger RNA Therapeutics, RNA Technologies; Jurga, S., Barciszewski, J., Eds.; Springer: Cham, Switzerland, 2022; Volume 13. [Google Scholar] [CrossRef]
- Khurana, A.; Sayed, N.; Singh, V.; Khurana, I.; Allawadhi, P.; Rawat, P.S.; Navik, U.; Pasumarthi, S.K.; Bharani, K.K.; Weiskirchen, R. A comprehensive overview of CRISPR/Cas 9 technology and application thereof in drug discovery. J. Cell. Biochem. 2022, 123, 1674–1698. [Google Scholar] [CrossRef]
- Burra, P.; Becchetti, C.; Germani, G. NAFLD and liver transplantation: Disease burden, current management and future challenges. JHEP Rep. 2020, 2, 100192. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekaran, P.; Weiskirchen, R. The Pivotal Role of the Membrane-Bound O-Acyltransferase Domain Containing 7 in Non-Alcoholic Fatty Liver Disease. Livers 2024, 4, 1-14. https://doi.org/10.3390/livers4010001
Chandrasekaran P, Weiskirchen R. The Pivotal Role of the Membrane-Bound O-Acyltransferase Domain Containing 7 in Non-Alcoholic Fatty Liver Disease. Livers. 2024; 4(1):1-14. https://doi.org/10.3390/livers4010001
Chicago/Turabian StyleChandrasekaran, Preethi, and Ralf Weiskirchen. 2024. "The Pivotal Role of the Membrane-Bound O-Acyltransferase Domain Containing 7 in Non-Alcoholic Fatty Liver Disease" Livers 4, no. 1: 1-14. https://doi.org/10.3390/livers4010001
APA StyleChandrasekaran, P., & Weiskirchen, R. (2024). The Pivotal Role of the Membrane-Bound O-Acyltransferase Domain Containing 7 in Non-Alcoholic Fatty Liver Disease. Livers, 4(1), 1-14. https://doi.org/10.3390/livers4010001