

Liver Fibrosis, Liver Cancer, and Advances in Therapeutic Approaches


{kind=link}
{kind=link}
Abstract
1. Liver Fibrosis
1.1. Hepatic Stellate Cells
- Capillarization of LSECs;
- Apoptosis of hepatocytes.
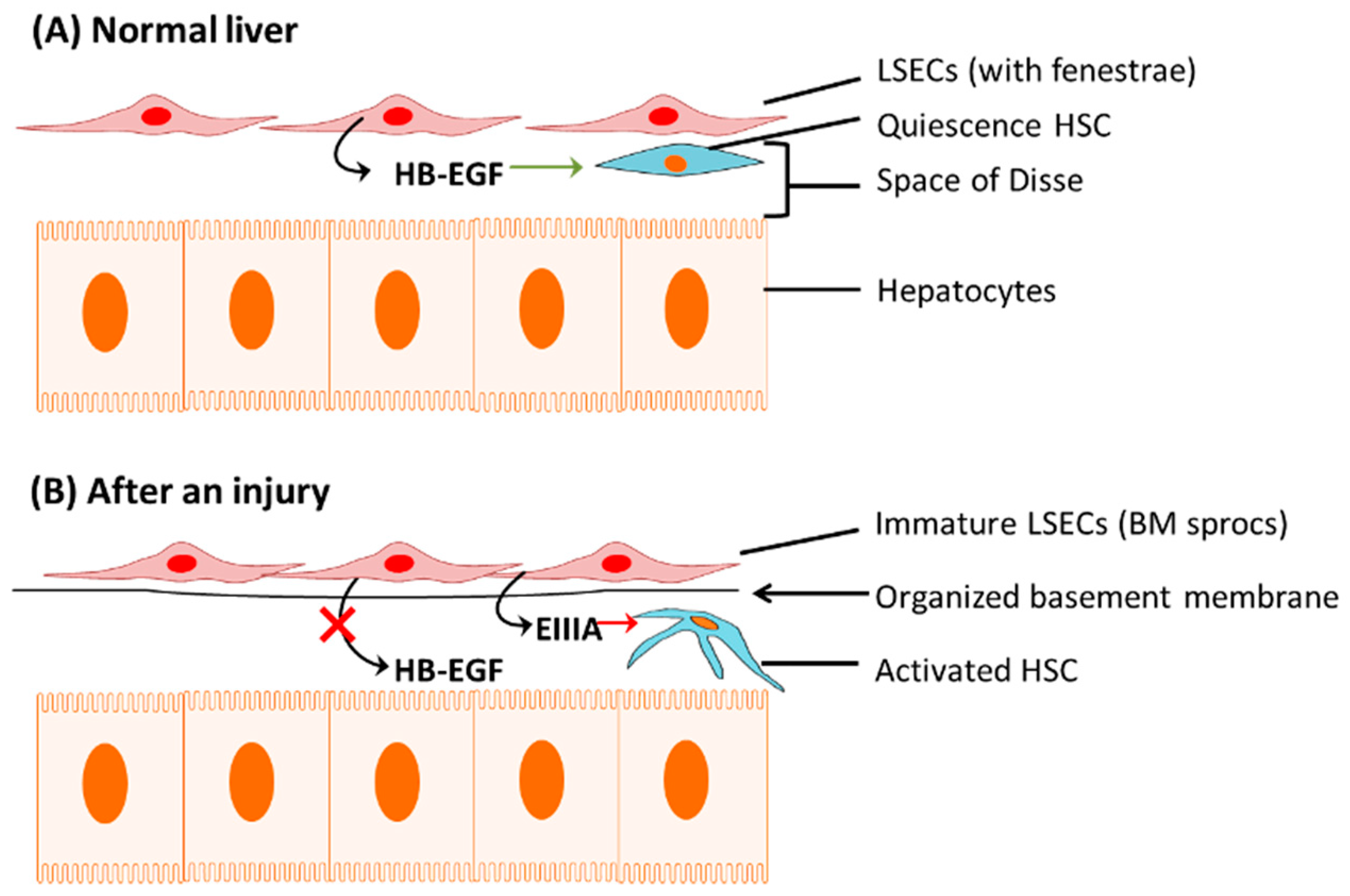
1.1.1. Capillarization of LSECs
1.1.2. Apoptosis of Hepatocytes
1.2. Cells That Contribute to Extracellular Matrix Synthesis (ECM) Other Than Hepatic Stellate Cells
- 1.
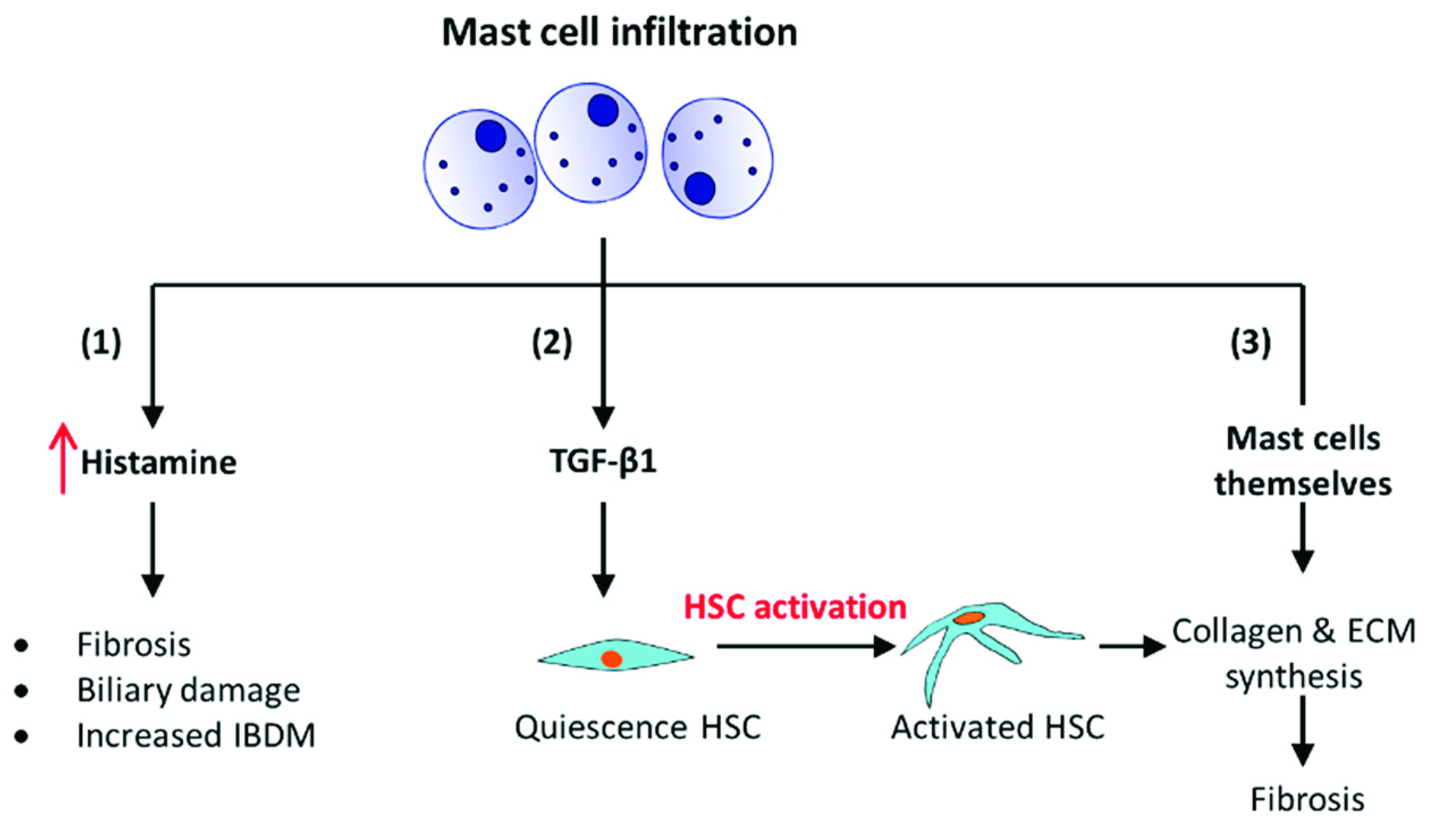
- It has been shown using a mouse model with progressive biliary fibrosis, Mdr2-KO; those mast cells infiltrate into the liver during the progression of biliary fibrosis. The presence of mast cells increases the local levels of histamine, which is a pro-fibrogenic and proliferative factor. It induces intrahepatic bile duct mass (IBDM) and proliferation leading to fibrosis [41].
- 2.
- 3.
1.3. Cirrhosis
2. Treatment for Liver Fibrosis
2.1. Non-Selective Beta-Blockers (NSBB)
2.2. Renin-Angiotensin System Inhibitors
2.3. Statins
2.4. Antifibrotic Therapies
3. Hepatocellular Carcinoma (HCC)
3.1. Liver Stem Cells and Mature Hepatocytes
3.2. Genetic Mutations and Oncogenic Genes
3.3. Immune Response
3.4. Treatments for Liver Cancer
3.4.1. Molecular Targeted Agents
3.4.2. Cytotoxic Chemotherapy
3.4.3. Immune Therapy
3.4.4. Nanomedical Approaches
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rajapaksha, I.; Gunarathne, L.; Angus, P.; Herath, C. Update on New Aspects of the Renin-Angiotensin System in Hepatic Fibrosis and Portal Hypertension: Implications for Novel Therapeutic Options. J. Clin. Med. 2021, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- Scorza, M.; Elce, A.; Zarrilli, F.; Liguori, R.; Amato, F.; Castaldo, G. Genetic Diseases That Predispose to Early Liver Cirrhosis. Int. J. Hepatol. 2014, 2014, 713754. [Google Scholar] [CrossRef]
- Minton, K. Extracellular matrix: Preconditioning the ECM for fibrosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 766–767. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Albanis, E.; Friedman, S.L. HEPATIC FIBROSIS: Pathogenesis and Principles of Therapy. Clin. Liver Dis. 2001, 5, 315–334. [Google Scholar] [CrossRef]
- Pinzani, M.; Rosselli, M.; Zuckermann, M. Liver cirrhosis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 281–290. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Benyon, R.C.; Iredale, J.P. Is liver fibrosis reversible? Gut 2000, 46, 443–446. [Google Scholar] [CrossRef]
- Arthur, M.J.P. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G245–G249. [Google Scholar]
- Tu, T.; Calabro, S.R.; Lee, A.; Maczurek, A.E.; Budzinska, M.A.; Warner, F.J.; McLennan, S.V.; Shackel, N.A. Hepatocytes in liver injury: Victim, bystander, or accomplice in progressive fibrosis? J. Gastroenterol. Hepatol. 2015, 30, 1696–1704. [Google Scholar] [CrossRef]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Zhangdi, H.-J.; Su, S.-B.; Wang, F.; Liang, Z.-Y.; Yan, Y.-D.; Qin, S.-Y.; Jiang, H.-X. Crosstalk network among multiple inflammatory mediators in liver fibrosis. World J. Gastroenterol. 2019, 25, 4835–4849. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Friedman, S.L. Chapter 6—Hepatic Fibrosis and Cirrhosis A2. In Zakim and Boyer’s Hepatology, 5th ed.; Boyer, T.D., Terrault, N.A., Zakim, D., Saunders, W.B., Eds.; Elsevier: Edinburgh, UK, 2006; pp. 87–109. [Google Scholar]
- Takahashi-Iwanaga, H.; Fujita, T. The three-dimensional fine structure of Ito cells and hepatocytes studied by a maceration method. In Biopathology of the Liver; Springer: Dordrecht, The Netherlands, 1988; pp. 59–68. [Google Scholar] [CrossRef]
- de Macêdo, S.M.; Guimarães, T.A.; Feltenberger, J.D.; Santos, S.H.S. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides 2014, 62, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef]
- Bataller, R.; Sancho-Bru, P.; Ginès, P.; Lora, J.M.; Al-Garawi, A.; Solé, M.; Colmenero, J.; Nicolás, J.M.; Jiménez, W.; Weich, N.; et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 2003, 125, 117–125. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Bataller, R.; Ginès, P.; Nicolás, J.M.; Görbig, M.; Garcia–Ramallo, E.; Gasull, X.; Bosch, J.; Arroyo, V.; Rodés, J. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology 2000, 118, 1149–1156. [Google Scholar] [CrossRef]
- Wei, H.S.; Lu, H.M.; Li, D.G.; Zhan, Y.T.; Wang, Z.R.; Huang, X.; Cheng, J.L.; Xu, Q.F. The regulatory role of AT1 receptor on activated HSCs in hepatic fibrogenesis: Effects of RAS inhibitors on hepatic fibrosis induced by CCl4. World J. Gastroenterol. 2000, 6, 824–828. [Google Scholar] [CrossRef]
- Friedman, S.L. The answer: Angiotensin II. The question: What do inflammation, oxidant stress and fibrogenesis have in common? J. Hepatol. 2004, 40, 1050–1052. [Google Scholar] [CrossRef]
- Paizis, G.; Cooper, M.E.; Schembri, J.M.; Tikellis, C.; Burrell, L.M.; Angus, P.W. Up-regulation of components of the renin-angiotensin system in the bile duct–ligated rat liver. Gastroenterology 2002, 123, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef]
- Verrecchia, F.; Chu, M.-L.; Mauviel, A. Identification of Novel TGF-β/Smad Gene Targets in Dermal Fibroblasts using a Combined cDNA Microarray/Promoter Transactivation Approach. J. Biol. Chem. 2001, 276, 17058–17062. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar]
- Dadlani, H.; Ballinger, M.L.; Osman, N.; Getachew, R.; Little, P.J. Smad and p38 MAP Kinase-mediated Signaling of Proteoglycan Synthesis in Vascular Smooth Muscle. J. Biol. Chem. 2008, 283, 7844–7852. [Google Scholar] [CrossRef]
- Margadant, C.; Sonnenberg, A. Integrin–TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [PubMed]
- Hen, Y.; Blom, I.E.; Sa, S.; Goldschmeding, R.; Abraham, D.J.; Leask, A. CTGF expression in mesangial cells: Involvement of SMADs, MAP kinase, and PKC. Kidney Int. 2002, 62, 1149–1159. [Google Scholar]
- Yuan, W.; Varga, J. Transforming Growth Factor-β Repression of Matrix Metalloproteinase-1 in Dermal Fibroblasts Involves Smad3. J. Biol. Chem. 2001, 276, 38502–38510. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Dewidar, B.; Soukupova, J.; Fabregat, I.; Dooley, S. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis: Updated. Curr. Pathobiol. Rep. 2015, 3, 291–305. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2014, 61, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Maretti-Mira, A.C.; Wang, X.; Wang, L.; DeLeve, L.D. 1667 Role of incomplete stem cell maturation in hepatic fibrosis. Hepatology 2016, 64, 811–1050. [Google Scholar] [CrossRef]
- Jarnagin, W.R.; Rockey, D.C.; Koteliansky, V.E.; Wang, S.S.; Bissell, D.M. Expression of variant fibronectins in wound healing: Cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J. Cell Biol. 1994, 127, 2037–2048. [Google Scholar] [CrossRef]
- Jiang, J.X.; Mikami, K.; Venugopal, S.; Li, Y.; Török, N.J. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-κB-dependent pathways. J. Hepatol. 2009, 51, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J.G. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Kinnman, N.; Housset, C. Peribiliary myofibroblasts in biliary type liver fibrosis. Front. Biosci.-Landmark 2002, 7, 496–503. [Google Scholar] [CrossRef]
- Rioux, K.P.; Sharkey, K.A.; Wallace, J.L.; Swain, M.G. Hepatic mucosal mast cell hyperplasia in rats with secondary biliary cirrhosis. Hepatology 1996, 23, 888–895. [Google Scholar] [CrossRef]
- Jennifer, D.; Hargrove, L.; Kennedy, L.; Jarido, V.; Francis, H.L. 181 Knockout of the HDC/histamine axis and reduction of mast cell number/function rescues Mdr2-KO mice from PSC-related biliary proliferation and fibrosis. Hepatology 2016, 64, 1–136. [Google Scholar] [CrossRef]
- Grizzi, F.; Di Caro, G.; Laghi, L.; Hermonat, P.; Mazzola, P.; Nguyen, D.D.; Radhi, S.; A Figueroa, J.; Cobos, E.; Annoni, G.; et al. Mast cells and the liver aging process. Immun. Ageing 2013, 10, 9–10. [Google Scholar] [CrossRef]
- Paizis, G.; E Gilbert, R.; E Cooper, M.; Murthi, P.; Schembri, J.M.; Wu, L.L.; Rumble, J.R.; Kelly, D.J.; Tikellis, C.; Cox, A.; et al. Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J. Hepatol. 2001, 35, 376–385. [Google Scholar] [CrossRef]
- Thompson, H.L.; Burbelo, P.D.; Gabriel, G.; Yamada, Y.; Metcalfe, D.D. Murine mast cells synthesize basement membrane components. A potential role in early fibrosis. J. Clin. Investig. 1991, 87, 619–623. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rajapaksha, I.G.; Angus, P.W.; Herath, C.B. Current therapies and novel approaches for biliary diseases. World J. Gastrointest. Pathophysiol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D. Liver fibrosis: Common mechanisms and antifibrotic therapies. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S51–S59. [Google Scholar] [CrossRef] [PubMed]
- Deleve, L.D. The Hepatic Sinusoidal Endothelial Cell: Morphology, Function, and Pathobiology. Liver Biol. Pathobiol. 2009, 371–388. [Google Scholar] [CrossRef]
- Fernández, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef]
- García-Pagán, J.-C.; Gracia-Sancho, J.; Bosch, J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J. Hepatol. 2012, 57, 458–461. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Herath, C.B.; A Grace, J.; Angus, P.W. Therapeutic potential of targeting the renin angiotensin system in portal hypertension. World J. Gastrointest. Pathophysiol. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Bosch, J.; Berzigotti, A.; Garcia-Pagan, J.C.; Gonzalez-Abraldes, J. The management of portal hypertension: Rational basis, available treatments and future options. J. Hepatol. 2008, 48, S68–S92. [Google Scholar] [CrossRef]
- Bosch, J.; Abraldes, J.G.; Fernández, M.; García-Pagán, J.C. Hepatic endothelial dysfunction and abnormal angiogenesis: New targets in the treatment of portal hypertension. J. Hepatol. 2010, 53, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G. Ascites. In Textbook of Clinical Gastroenterology and Hepatology, 2nd ed.; Blackwell Publishing: Hoboken, NJ, USA, 2012; pp. 103–106. [Google Scholar]
- Garcia-Tsao, G.; Veterans Affairs Hepatitis C Resource Center Program; Lim, J.K. Management and Treatment of Patients with Cirrhosis and Portal Hypertension: Recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am. J. Gastroenterol. 2009, 104, 1802–1829. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Torres, F.; Sarin, S.K.; Shah, H.A.; Tripathi, D.; Brujats, A.; Rodrigues, S.G.; Bhardwaj, A.; Azam, Z.; Hayes, P.C.; et al. Carvedilol reduces the risk of decompensation and mortality in patients with compensated cirrhosis in a competing-risk meta-analysis. J. Hepatol. 2022, 77, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Herath, C.B.; Grace, J.A.; Angus, P.W. Baveno VII—Renewing consensus in portal hypertension. J. Hepatol. 2022, 76, 959–974. [Google Scholar]
- Wong, S.Y.; Lee, J.; Anile Sule, A. Is carvedilol better than propranolol in portal hypertension? AME Med. J. 2017, 2, 1–3. [Google Scholar] [CrossRef]
- Turnes, J.; Garcia-Pagan, J.C.; Abraldes, J.G.; Hernandez-Guerra, M.; Dell’Era, A.; Bosch, J. Pharmacological Reduction of Portal Pressure and Long-Term Risk of First Variceal Bleeding in Patients with Cirrhosis. Am. J. Gastroenterol. 2006, 101, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.R.; Olson, H.G.; Amsterdam, E.A.; Mason, D.T. Propranolol-Withdrawal Rebound Phenomenon. N. Engl. J. Med. 1975, 293, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.S.; Runyon, B.A. The changing role of beta-blocker therapy in patients with cirrhosis. J. Hepatol. 2014, 60, 643–653. [Google Scholar] [CrossRef]
- Popp, D.A.; Tse, T.F.; Shah, S.D.; Clutter, W.E.; Cryer, P.E. Oral Propranolol and Metoprolol Both Impair Glucose Recovery from Insulin-induced Hypoglycemia in Insulin-dependent Diabetes Mellitus. Diabetes Care 1984, 7, 243–247. [Google Scholar] [CrossRef]
- Grace, J.A.; Herath, C.B.; Mak, K.Y.; Burrell, L.M.; Angus, P.W. Update on new aspects of the renin–angiotensin system in liver disease: Clinical implications and new therapeutic options. Clin. Sci. 2012, 123, 225–239. [Google Scholar] [CrossRef]
- Lubel, J.S.; Herath, C.B.; Burrell, L.M.; Angus, P.W. Liver disease and the renin-angiotensin system: Recent discoveries and clinical implications. J. Gastroenterol. Hepatol. 2008, 23, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Rodríguez-Vilarrupla, A.; Graupera, M.; Zafra, C.; García-Calderó, H.; García-Pagán, J.C.; Bosch, J. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J. Hepatol. 2007, 46, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Zafra, C.; Abraldes, J.G.; Turnes, J.; Berzigotti, A.; Fernández, M.; García-Pagán, J.C.; Rodés, J.; Bosch, J. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology 2004, 126, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-W.; Lee, C.-L.; Yang, S.-S.; Fu, S.-C.; Chen, Y.-Y.; Wang, T.-C.; Hu, J.-T.; Chen, D.-S. Statins Reduce the Risk of Cirrhosis and Its Decompensation in Chronic Hepatitis B Patients: A Nationwide Cohort Study. Am. J. Gastroenterol. 2016, 111, 976–985. [Google Scholar] [CrossRef]
- Ge, P.S.; Runyon, B.A. Treatment of Patients with Cirrhosis. N. Engl. J. Med. 2016, 375, 767–777. [Google Scholar] [CrossRef]
- Oseini, A.M.; Sanyal, A.J. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. 2017, 37 (Suppl. 1), 97–103. [Google Scholar] [CrossRef]
- Mak, K.Y.; Chin, R.; Cunningham, S.C.; Habib, M.R.; Torresi, J.; Sharland, A.F.; E Alexander, I.; Angus, P.W.; Herath, C.B. ACE2 Therapy Using Adeno-associated Viral Vector Inhibits Liver Fibrosis in Mice. Mol. Ther. 2015, 23, 1434–1443. [Google Scholar] [CrossRef]
- Rajapaksha, I.G.; Gunarathne, L.S.; Asadi, K.; Cunningham, S.C.; Sharland, A.; Alexander, I.E.; Angus, P.W.; Herath, C.B. Liver-Targeted Angiotensin Converting Enzyme 2 Therapy Inhibits Chronic Biliary Fibrosis in Multiple Drug-Resistant Gene 2-Knockout Mice. Hepatol. Commun. 2019, 3, 1656–1673. [Google Scholar] [CrossRef]
- Paradis, V. Histopathology of Hepatocellular Carcinoma. World J. Gastroenterol. 2012, 190, 21–32. [Google Scholar] [CrossRef]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef]
- Global Health Estimates 2014 Summary Tables: Deaths by Cause, Age and Sex, 2000–2012; WHO: Geneva, Switzerland, 2014.
- Choti, M.A.; Sitzmann, J.V.; Tiburi, M.F.; Sumetchotimetha, W.; Rangsin, R.; Schulick, R.D.; Lillemoe, K.D.; Yeo, C.J.; Cameron, J.L. Trends in Long-Term Survival Following Liver Resection for Hepatic Colorectal Metastases. Ann. Surg. 2002, 235, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Meloche, M.L.S. YES, a novel therapeutic target in hepatocellular carcinoma. Mol. Cell. Oncol. 2022, 9, 2069993. [Google Scholar]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Roskams, T.A.; Libbrecht, L.; Desmet, V.J. Progenitor Cells in Diseased Human Liver. Semin. Liver Dis. 2003, 23, 385–396. [Google Scholar] [CrossRef]
- Mu, X.; Español-Suñer, R.; Mederacke, I.; Affò, S.; Manco, R.; Sempoux, C.; Lemaigre, F.P.; Adili, A.; Yuan, D.; Weber, A.; et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J. Clin. Investig. 2015, 125, 3891–3903. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef]
- Benhamouche, S.; Curto, M.; Saotome, I.; Gladden, A.B.; Liu, C.H.; Giovannini, M.; McClatchey, A.I. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 2010, 24, 1718–1730. [Google Scholar] [CrossRef]
- Lee, K.P.; Lee, J.H.; Kim, T.S.; Kim, T.H.; Park, H.D.; Byun, J.S.; Kim, M.C.; Jeong, W.I.; Calvisi, D.F.; Kim, J.M.; et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 8248–8253. [Google Scholar] [CrossRef]
- Shin, S.; Wangensteen, K.J.; Teta-Bissett, M.; Wang, Y.J.; Mosleh-Shirazi, E.; Buza, E.L.; Greenbaum, L.E.; Kaestner, K.H. Genetic lineage tracing analysis of the cell of origin of hepatotoxin-induced liver tumors in mice. Hepatology 2016, 64, 1163–1177. [Google Scholar] [CrossRef]
- Lee, J.-S.; Heo, J.; Libbrecht, L.; Chu, I.-S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef]
- Roskams, T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 2006, 25, 3818–3822. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Paterlini-Bréchot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Bréchot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef]
- Perra, A.; Kowalik, M.A.; Ghiso, E.; Ledda-Columbano, G.M.; Di Tommaso, L.; Angioni, M.M.; Raschioni, C.; Testore, E.; Roncalli, M.; Giordano, S.; et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J. Hepatol. 2014, 61, 1088–1096. [Google Scholar] [CrossRef]
- Manmadhan, S.; Ehmer, U. Hippo Signaling in the Liver—A Long and Ever-Expanding Story. Front. Cell Dev. Biol. 2019, 7, 33. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; de Moura, M.C.; Putra, J.; Campreciós, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Wada, Y.; Nakashima, O.; Kutami, R.; Yamamoto, O.; Kojiro, M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology 1998, 27, 407–414. [Google Scholar] [CrossRef]
- Seehawer, M.; Heinzmann, F.; D’Artista, L.; Harbig, J.; Roux, P.-F.; Hoenicke, L.; Dang, H.; Klotz, S.; Robinson, L.; Doré, G.; et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018, 562, 69–75. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Chen, K.F.; Chen, P.J. Treatment of Liver Cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a021535. [Google Scholar] [PubMed]
- Ruman, U.; Fakurazi, S.; Masarudin, M.J.; Hussein, M.Z. Nanocarrier-Based Therapeutics and Theranostics Drug Delivery Systems for Next Generation of Liver Cancer Nanodrug Modalities. Int. J. Nanomed. 2020, 15, 1437–1456. [Google Scholar] [CrossRef]
- Verslype, C.; Van Cutsem, E.; Dicato, M.; Arber, N.; Berlin, J.D.; Cunningham, D.; De Gramont, A.; Diaz-Rubio, E.; Ducreux, M.; Gruenberger, T.; et al. The management of hepatocellular carcinoma. Current expert opinion and recommendations derived from the 10th World Congress on Gastrointestinal Cancer, Barcelona, 2008. Ann. Oncol. 2009, 20, vii1–vii6. [Google Scholar] [CrossRef]
- Fan, Y.; Xue, H.; Zheng, H. Systemic Therapy for Hepatocellular Carcinoma: Current Updates and Outlook. J. Hepatocell. Carcinoma 2022, 9, 233–263. [Google Scholar] [CrossRef]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib Suppresses Tumor Growth and Metastasis in Hepatocellular Carcinoma by a Dual Blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef]
- Syed, Y.Y. Ramucirumab: A Review in Hepatocellular Carcinoma. Drugs 2020, 80, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Johnson, P.J. Are there indications for chemotherapy in hepatocellular carcinoma? Surg. Oncol. Clin. N. Am. 2003, 12, 127–134. [Google Scholar] [CrossRef]
- Breous, E.; Thimme, R. Potential of immunotherapy for hepatocellular carcinoma. J. Hepatol. 2011, 54, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.E.A. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased Regulatory T Cells Correlate with CD8 T-Cell Impairment and Poor Survival in Hepatocellular Carcinoma Patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef]
- Neubert, R.H. Potentials of new nanocarriers for dermal and transdermal drug delivery. Eur. J. Pharm. Biopharm. 2011, 77, 1–2. [Google Scholar] [CrossRef]
- Kaushik, N.; Borkar, S.B.; Nandanwar, S.K.; Panda, P.K.; Choi, E.H.; Kaushik, N.K. Nanocarrier cancer therapeutics with functional stimuli-responsive mechanisms. J. Nanobiotechnol. 2022, 20, 152. [Google Scholar]
- Zhang, N.-N.; Yu, R.-S.; Xu, M.; Cheng, X.-Y.; Chen, C.-M.; Xu, X.-L.; Lu, C.-Y.; Lu, K.-J.; Chen, M.-J.; Zhu, M.-L.; et al. Visual targeted therapy of hepatic cancer using homing peptide modified calcium phosphate nanoparticles loading doxorubicin guided by T1 weighted MRI. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2167–2178. [Google Scholar] [CrossRef]
- Garcia–Tsao, G. Current management of the complications of cirrhosis and portal hypertension: Variceal hemorrhage, ascites, and spontaneous bacterial peritonitis. Gastroenterology 2001, 120, 726–748. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajapaksha, I. Liver Fibrosis, Liver Cancer, and Advances in Therapeutic Approaches. Livers 2022, 2, 372-386. https://doi.org/10.3390/livers2040028
Rajapaksha I. Liver Fibrosis, Liver Cancer, and Advances in Therapeutic Approaches. Livers. 2022; 2(4):372-386. https://doi.org/10.3390/livers2040028
Chicago/Turabian StyleRajapaksha, Indu. 2022. "Liver Fibrosis, Liver Cancer, and Advances in Therapeutic Approaches" Livers 2, no. 4: 372-386. https://doi.org/10.3390/livers2040028
APA StyleRajapaksha, I. (2022). Liver Fibrosis, Liver Cancer, and Advances in Therapeutic Approaches. Livers, 2(4), 372-386. https://doi.org/10.3390/livers2040028