

Role of Oxidative Stress in Liver Disorders

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
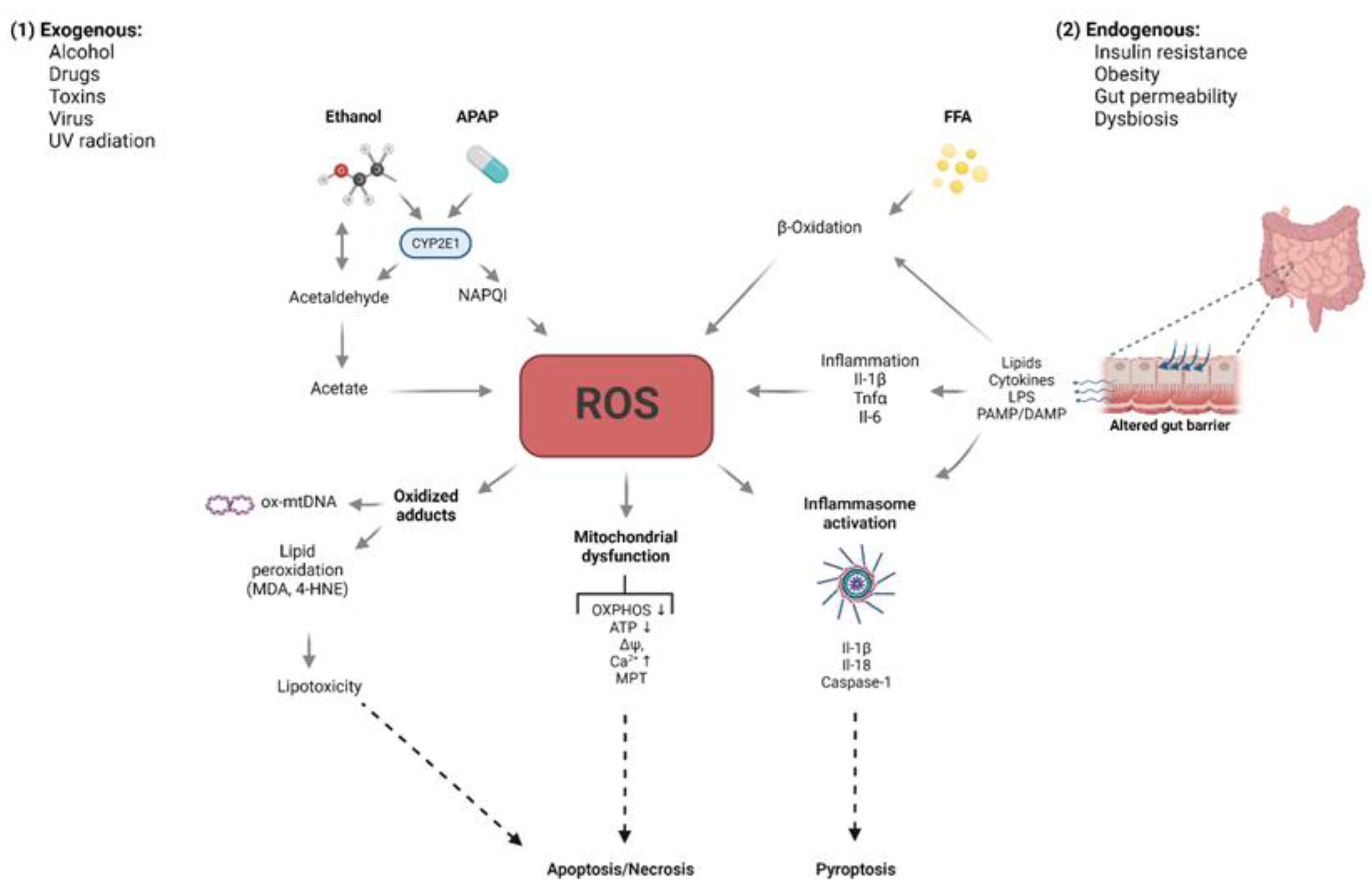
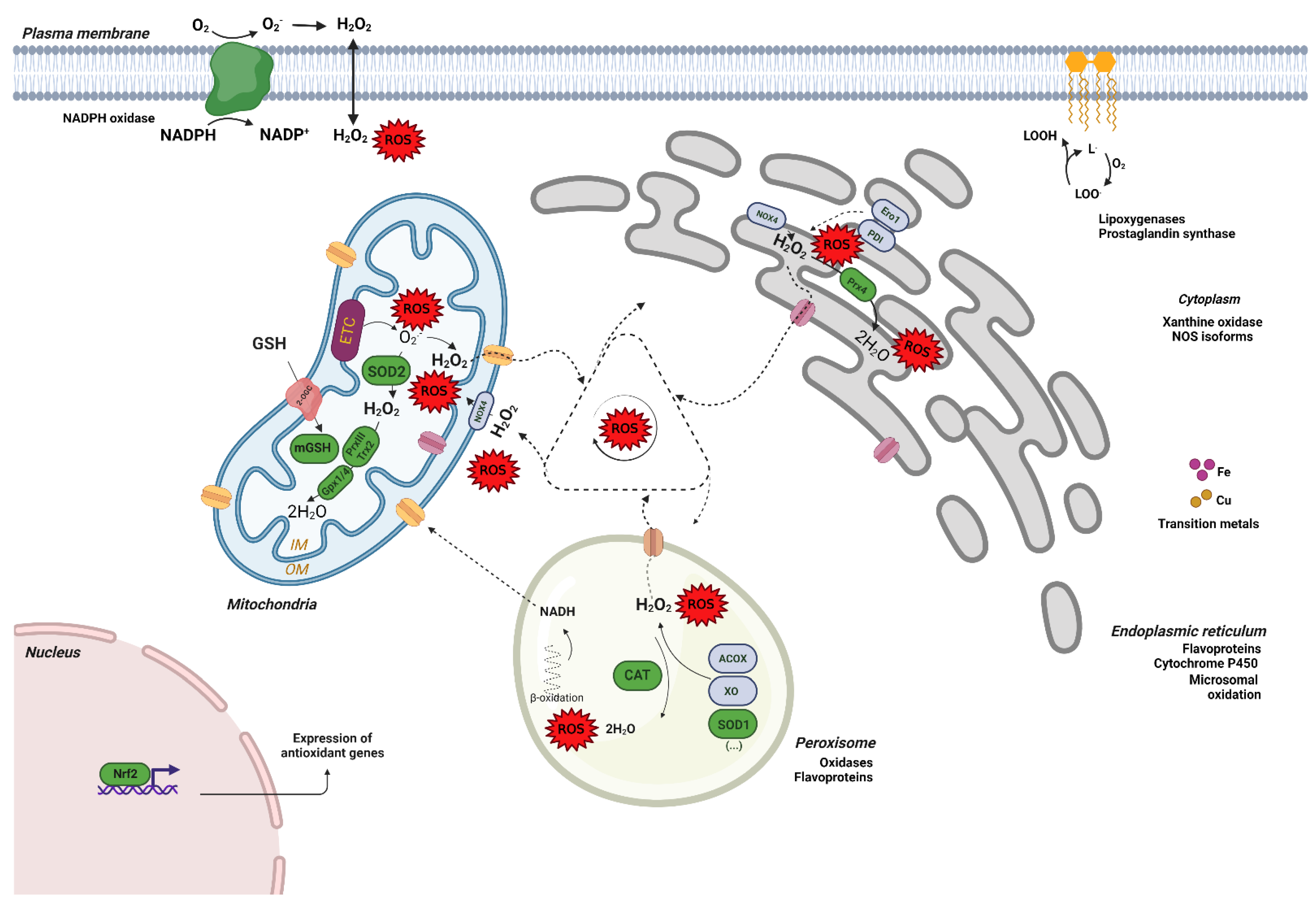
2. Free Radicals: Sources and Defense
3. Redox Control in the Liver
4. Cell Aging and Cell Death Regulation in the Liver
4.1. Cell Aging in the Liver
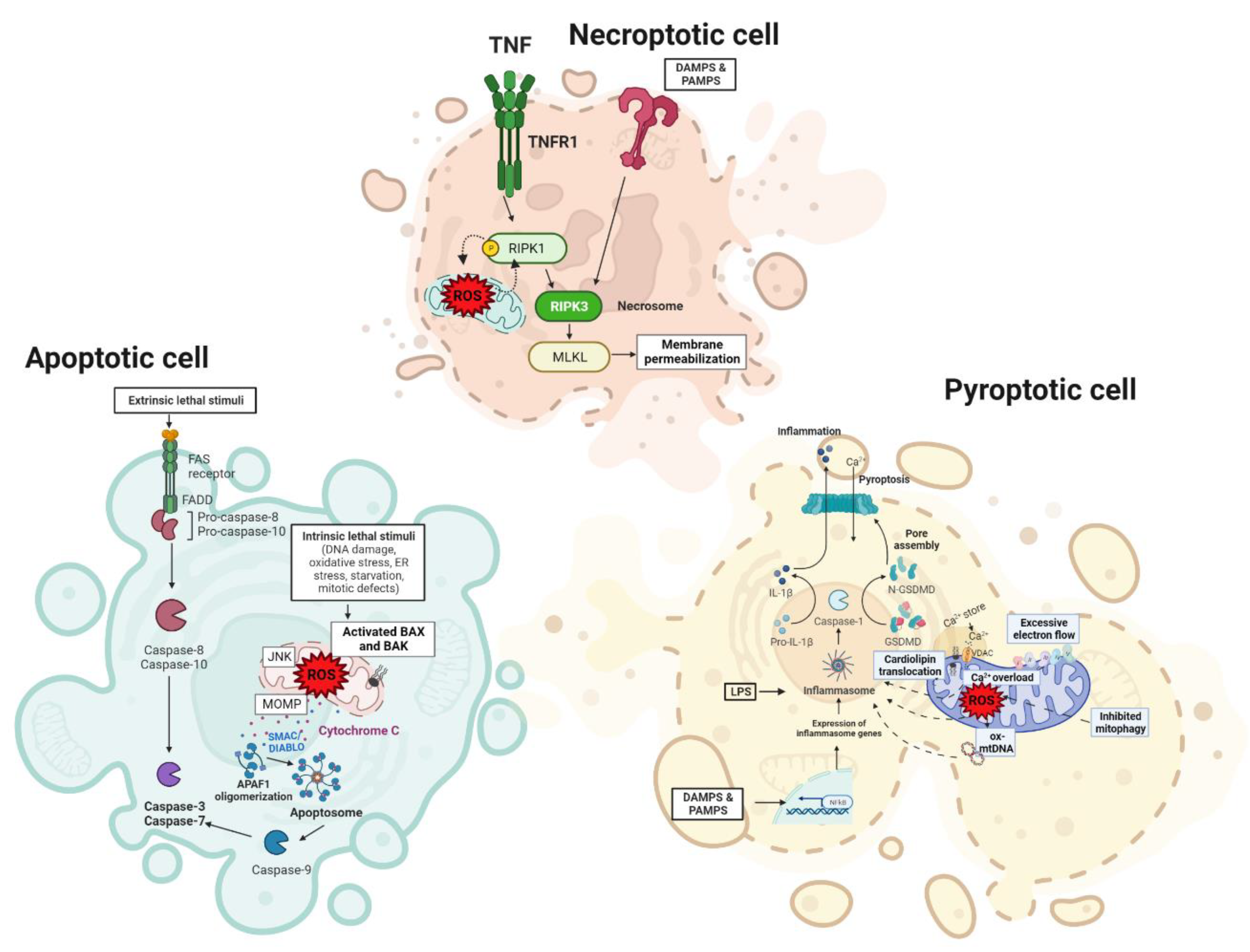
4.2. Cell Death Regulation in the Liver
4.2.1. Apoptosis
4.2.2. Necrosis
4.2.3. Pyroptosis
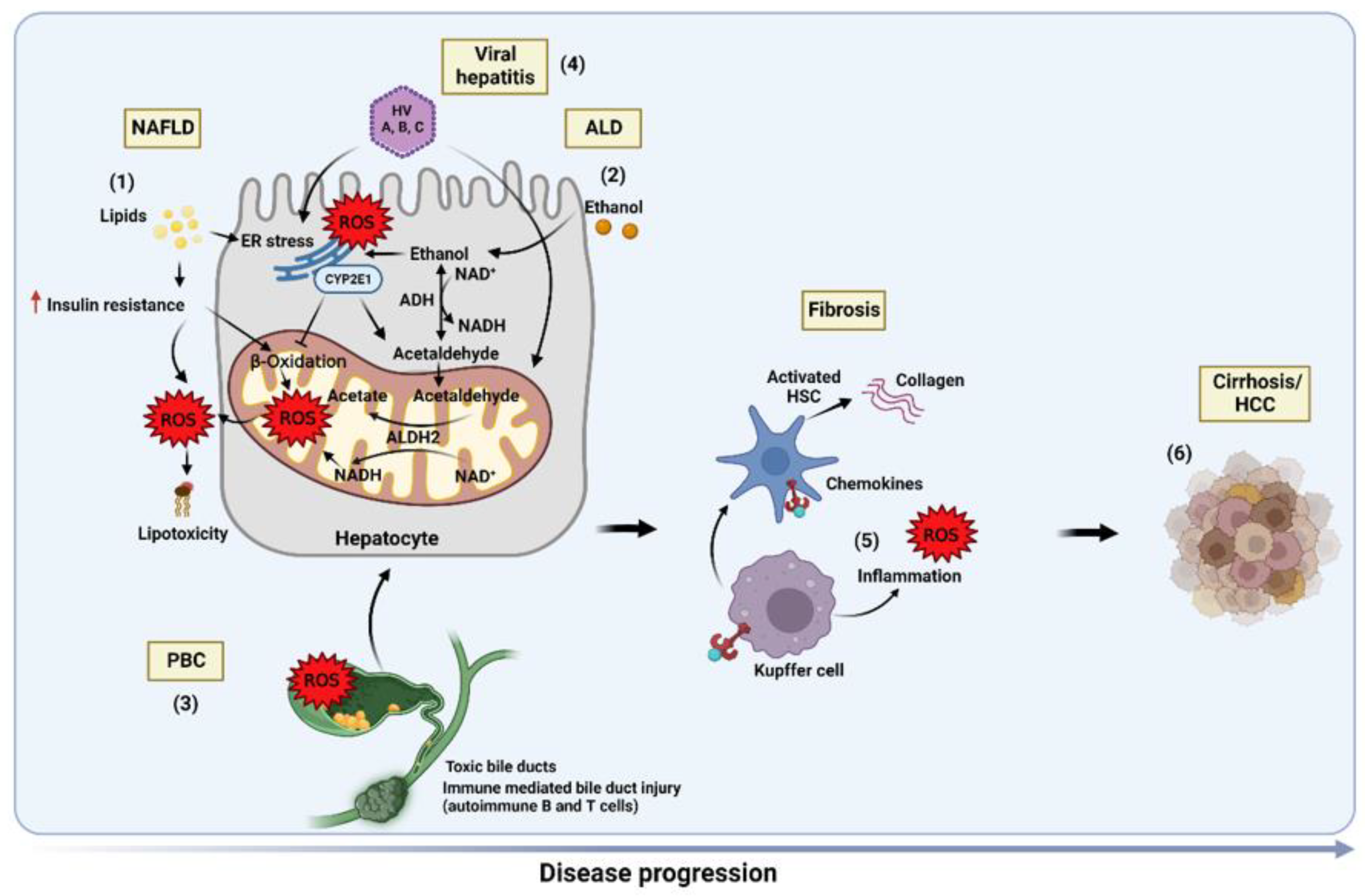
5. Role of Oxidative Stress in Liver Diseases
5.1. Non-Alcoholic Fatty Liver Disease (NAFLD)
5.2. Alcoholic Liver Disease (ALD)
5.3. Primary Biliary Cholangitis (PBC)
5.4. Viral Hepatitis
5.5. Liver Fibrosis
5.6. Hepatocellular Carcinoma (HCC)
6. Antioxidant Therapies
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADH | alcohol dehydrogenase |
| ALD | alcoholic liver disease |
| AMPK | 5’ adenosine monophosphate-activated protein kinase |
| ATP | adenosine triphosphate |
| ASC | apoptosis-associated speck-like protein containing a caspase recruit domain |
| ASH | alcoholic steatohepatitis |
| BDL | bile duct ligation |
| Ca2+ | calcium ion |
| DNA | deoxyribonucleic acid |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| GSH | glutathione |
| GSSG | oxidized glutathione |
| FADH2, | flavin adenine dinucleotide (reduced form) |
| FFA | free fatty acid |
| H2O2 | hydrogen peroxide |
| HCC | hepatocellular cancer |
| HFD | high fat diet |
| HSC | hepatic stellate cells |
| MAFLD | metabolic-dysfunction-associated fatty liver disease |
| MCD | methionine-choline deficient diet |
| MDA | malondialdehyde |
| MMOs | microsomal monooxygenases |
| MPT | mitochondrial permeability transition |
| mDNA | mitochondrial deoxyribonucleic acid |
| NAD+ | nicotinamide adenine dinucleotide phosphate, (oxidized form) |
| NADPH | nicotinamide adenine dinucleotide phosphate, (reduced form) |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NLRP3 | NOD-like receptor family pyrin domain containing 3 |
| NOX | NAPDH oxidases |
| Nrf2 | nuclear factor E2-related factor 2 |
| O2•− | superoxide anion |
| 1O2 | singlet oxygen |
| •OH | hydroxy radical |
| -OH | hydroxy group |
| ONOO− | peroxynitrite |
| OXPHOS | oxidative phosphorylation system |
| PBC | primary biliary cholangitis |
| Prx | peroxiredoxin |
| ROS | reactive oxygen species |
| SAMe | S-(5-Adenosyl)-L-methionine |
| SOD1 | superoxide dismutase 1 |
| SOD2 | superoxide dismutase 2 |
| StARD1 | steroidogenic acute regulatory protein |
| TNFα | tumor necrosis factor alpha |
References
- Chiang, J. Liver Physiology: MetaboLism and Detoxification. In Pathobiology of Human Disease; Academic Press: Oxford, UK, 2014; pp. 1770–1782. [Google Scholar]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S. Reactive Oxygen Species in Plant Biology; Springer: New Delhi, India, 2019; ISBN 978-81-322-3939-0. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-J.; Seo, J.-M.; Kim, J.-H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef] [PubMed]
- de Haan, J.B.; Cristiano, F.; Iannello, R.; Bladier, C.; Kelner, M.J.; Kola, I. Elevation in the Ratio of Cu/Zn-Superoxide Dismutase to Glutathione Peroxidase Activity Induces Features of Cellular Senescence and This Effect Is Mediated by Hydrogen Peroxide. Hum. Mol. Genet. 1996, 5, 283–292. [Google Scholar] [CrossRef]
- Pamplona, R.; Costantini, D. Molecular and structural antioxidant defenses against oxidative stress in animals. Am. J. Physiol. Integr. Comp. Physiol. 2011, 301, R843–R863. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.Y.; Elmasry, M.; Forootan, S.S.; Russomanno, G.; Bunday, T.M.; Zhang, F.; Brillant, N.; Starkey Lewis, P.J.; Aird, R.; Ricci, E.; et al. Pharmacological Activation of Nrf2 Enhances Functional Liver Regeneration. Hepatology 2021, 74, 973–986. [Google Scholar] [CrossRef]
- Lluis, J.M.; Colell, A.; García–Ruiz, C.; Kaplowitz, N.; Fernández–Checa, J.C. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology 2003, 124, 708–724. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Matias, N.; Fucho, R.; Ribas, V.; Von Montfort, C.; Nuño, N.; Baulies, A.; Martinez, L.; Tarrats, N.; Mari, M.; et al. ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J. Hepatol. 2013, 59, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Josekutty, J.; Iqbal, J.; Iwawaki, T.; Kohno, K.; Hussain, M.M. Microsomal Triglyceride Transfer Protein Inhibition Induces Endoplasmic Reticulum Stress and Increases Gene Transcription via Ire1α/cJun to Enhance Plasma ALT/AST. J. Biol. Chem. 2013, 288, 14372–14383. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Morales, A.; Ballesta, A.; Rodés, J.; Kaplowitz, N.; Fernández-Checa, J.C. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J. Clin. Investig. 1994, 94, 193–201. [Google Scholar] [CrossRef]
- Colell, A.; Garcia-Ruiz, C.; Morales, A.; Ballesta, A.; Ookhtens, M.; Rodes, J.; Kaplowitz, N.; Fernandez-Checa, J.C. Transport of reduced glutathione in hepatic mitochondria and mitoplasts from ethanol-treated rats: Effect of membrane physical properties and S-adenosyl-L-methionine. Hepatology 1997, 26, 699–708. [Google Scholar] [CrossRef]
- Colell, A.; García-Ruiz, C.; Miranda, M.; Ardite, E.; Marí, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernández-Checa, J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Marí, M.; Colell, A.; Morales, A.; Caballero, F.; Moles, A.; Fernández, A.; Terrones, O.; Basañez, G.; Antonsson, B.; García–Ruiz, C.; et al. Mechanism of Mitochondrial Glutathione-Dependent Hepatocellular Susceptibility to TNF Despite NF-κB Activation. Gastroenterology 2008, 134, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Ciriolo, M.R. Redox Control of Apoptosis. Antioxid. Redox Signal. 2005, 7, 432–435. [Google Scholar] [CrossRef]
- Mahmood, S.; Kawanaka, M.; Kamei, A.; Izumi, A.; Nakata, K.; Niiyama, G.; Ikeda, H.; Hanano, S.; Suehiro, M.; Togawa, K.; et al. Immunohistochemical Evaluation of Oxidative Stress Markers in Chronic Hepatitis C. Antioxid. Redox Signal. 2004, 6, 19–24. [Google Scholar] [CrossRef]
- Stiuso, P.; Scognamiglio, I.; Murolo, M.; Ferranti, P.; De Simone, C.; Rizzo, M.R.; Tuccillo, C.; Caraglia, M.; Loguercio, C.; Federico, A. Serum Oxidative Stress Markers and Lipidomic Profile to Detect NASH Patients Responsive to an Antioxidant Treatment: A Pilot Study. Oxid. Med. Cell. Longev. 2014, 2014, 169216. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.-L.; Nath, R.; Ocando, J.; Nishikawa, A.; Zhang, L. Deoxyguanosine adducts of t-4-Hydroxy-2-nonenal are endogenous DNA lesions in rodents and humans: Detection and potential sources. Cancer Res. 2000, 60, 1507–1511. [Google Scholar] [PubMed]
- Boyman, L.; Coleman, A.K.; Zhao, G.; Wescott, A.P.; Joca, H.C.; Greiser, B.M.; Karbowski, M.; Ward, C.W.; Lederer, W.J. Dynamics of the mitochondrial permeability transition pore: Transient and permanent opening events. Arch. Biochem. Biophys. 2019, 666, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Maciel, E.N.; Vercesi, A.E.; Castilho, R.F. Oxidative stress in Ca2+-induced membrane permeability transition in brain mitochondria. J. Neurochem. 2001, 79, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, D.L. Age-related changes in liver structure and function: Implications for disease? Exp. Gerontol. 2005, 40, 650–659. [Google Scholar] [CrossRef]
- Sheedfar, F.; Biase, S.D.; Koonen, D.; Vinciguerra, M. Liver diseases and aging: Friends or foes? Aging Cell 2013, 12, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Aravinthan, A.; Pietrosi, G.; Hoare, M.; Jupp, J.; Marshall, A.; Verrill, C.; Davies, S.; Bateman, A.; Sheron, N.; Allison, M.; et al. Hepatocyte Expression of the Senescence Marker p21 Is Linked to Fibrosis and an Adverse Liver-Related Outcome in Alcohol-Related Liver Disease. PLoS ONE 2013, 8, e72904. [Google Scholar] [CrossRef]
- Ramirez, T.; Li, Y.-M.; Yin, S.; Xu, M.-J.; Feng, D.; Zhou, Z.; Zang, M.; Mukhopadhyay, P.; Varga, Z.V.; Pacher, P.; et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J. Hepatol. 2017, 66, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; Svistounov, D.; Warren, A.; Zykova, S.; Melvin, R.G.; Solon-Biet, S.M.; O’Reilly, J.N.; McMahon, A.C.; Ballard, J.W.O.; De Cabo, R.; et al. Liver Aging and Pseudocapillarization in a Werner Syndrome Mouse Model. J. Gerontol. Ser. A 2014, 69, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; Warren, A.; Fraser, R.; Ngu, M.; McLean, A.J.; Le Couteur, D.G. Hepatic sinusoidal pseudocapillarization with aging in the non-human primate. Exp. Gerontol. 2003, 38, 1101–1107. [Google Scholar] [CrossRef]
- Mohamad, M.; Mitchell, S.J.; Wu, L.E.; White, M.Y.; Cordwell, S.J.; Mach, J.; Solon-Biet, S.M.; Boyer, D.; Nines, D.; Das, A.; et al. Ultrastructure of the liver microcirculation influences hepatic and systemic insulin activity and provides a mechanism for age-related insulin resistance. Aging Cell 2016, 15, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Simon-Santamaria, J.; Malovic, I.; Warren, A.; Oteiza, A.; Le Couteur, D.; Smedsrød, B.; McCourt, P.; Sørensen, K.K. Age-Related Changes in Scavenger Receptor–Mediated Endocytosis in Rat Liver Sinusoidal Endothelial Cells. J. Gerontol. Ser. A 2010, 65A, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Maeso-Díaz, R.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Hide, D.; Muñoz, L.; Hessheimer, A.J.; Vila, S.; Francés, R.; Fondevila, C.; Albillos, A.; et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell 2018, 17, e12829. [Google Scholar] [CrossRef]
- Wynne, H.A.; Cope, L.H.; Mutch, E.; Rawlins, M.D.; Woodhouse, K.W.; James, O.F.W. The effect of age upon liver volume and apparent liver blood flow in healthy man. Hepatology 1989, 9, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Zoli, M.; Magalotti, D.; Bianchi, G.; Gueli, C.; Orlandini, C.; Grimaldi, M.; Marchesini, G. Total and functional hepatic blood flow decrease in parallel with ageing. Age Ageing 1999, 28, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Sørensen, K.K.; Bethea, N.W.; Svistounov, D.; McCuskey, M.K.; Smedsrød, B.H.; McCuskey, R.S. Age-related changes in the hepatic microcirculation in mice. Exp. Gerontol. 2007, 42, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.; Cogger, V.C.; Fraser, R.; DeLeve, L.D.; McCuskey, R.S.; Le Couteur, D.G. The Effects of Old Age on Hepatic Stellate Cells. Curr. Gerontol. Geriatr. Res. 2011, 2011, 439835. [Google Scholar] [CrossRef]
- Stahl, E.; LoPresti, S.; Delgado, E.; Alencastro, F.; Wilkinson, P.; Duncan, A.W.; Brown, B.N. Age-induced Hepatic Steatosis and Inflammation of Murine Livers is Influenced by MCP-1. FASEB J. 2018, 32, 150.5. [Google Scholar] [CrossRef]
- Hunt, N.J.; Lockwood, G.P.; Kang, S.W.; Pulpitel, T.; Clark, X.; Mao, H.; McCourt, P.A.G.; Cooney, G.J.; Wali, J.A.; Le Couteur, F.H.; et al. The Effects of Metformin on Age-Related Changes in the Liver Sinusoidal Endothelial Cell. J. Gerontol. Ser. A 2020, 75, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.J.; Lockwood, G.P.; Warren, A.; Mao, H.; McCourt, P.A.G.; Le Couteur, D.G.; Cogger, V.C. Manipulating fenestrations in young and old liver sinusoidal endothelial cells. Am. J. Physiol. Liver Physiol. 2018, 316, G144–G154. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta-Proteins Proteom. 2005, 1703, 93–109. [Google Scholar] [CrossRef]
- Miki, H.; Funato, Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J. Biochem. 2012, 151, 255–261. [Google Scholar] [CrossRef]
- Hawkes, W.; Alkan, Z. Regulation of Redox Signaling by Selenoproteins. Biol. Trace Element Res. 2010, 134, 235–251. [Google Scholar] [CrossRef]
- Lee, J.-W.; Helmann, J.D. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature 2006, 440, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, T.; Heinemann, S.H. Regulation of cell function by methionine oxidation and reduction. J. Physiol. 2001, 531, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen Peroxide as a Cell-Survival Signaling Molecule. Antioxid. Redox Signal. 2009, 11, 2655–2671. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen Peroxide Sensing and Signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Conde de la Rosa, L.; Schoemaker, M.H.; Vrenken, T.E.; Buist-Homan, M.; Havinga, R.; Jansen, P.L.M.; Moshage, H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: Involvement of JNK and ERK MAP kinases. J. Hepatol. 2006, 44, 918–929. [Google Scholar] [CrossRef]
- Conde de la Rosa, L.; Vrenken, T.E.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Metformin protects primary rat hepatocytes against oxidative stress-induced apoptosis. Pharmacol. Res. Perspect. 2015, 3, e00125. [Google Scholar] [CrossRef]
- Carmody, R.J.; Cotter, T.G. Signalling apoptosis: A radical approach. Redox Rep. 2001, 6, 77–90. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Pelicano, H.; Feng, L.; Zhou, Y.; Carew, J.S.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Inhibition of mitochondrial respiration: A novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003, 278, 37832–37839. [Google Scholar] [CrossRef]
- Sastre, J.; Pallardó, F.V.; Viña, J. Mitochondrial Oxidative Stress Plays a Key Role in Aging and Apoptosis. IUBMB Life 2000, 49, 427–435. [Google Scholar] [CrossRef]
- Ventura, J.-J.; Hübner, A.; Zhang, C.; Flavell, R.A.; Shokat, K.M.; Davis, R.J. Chemical Genetic Analysis of the Time Course of Signal Transduction by JNK. Mol. Cell 2006, 21, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive Oxygen Species Promote TNFα-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gunarta, K.; Suzuki, R.; Jambaldorj, B.; Nakazato, R.; Yuliana, D.; Davaakhuu, G.; Tsendsuren, O.; Takamatsu, N.; Kobayashi, M.; et al. JLP-JNK signaling protects cancer cells from reactive oxygen species-induced cell death. Biochem. Biophys. Res. Commun. 2018, 501, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Putcha, G.V.; Le, S.; Frank, S.; Besirli, C.G.; Clark, K.; Chu, B.; Alix, S.; Youle, R.J.; LaMarche, A.; Maroney, A.C.; et al. JNK-Mediated BIM Phosphorylation Potentiates BAX-Dependent Apoptosis. Neuron 2003, 38, 899–914. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef]
- Deng, Y.; Ren, X.; Yang, L.; Lin, Y.; Wu, X. A JNK-Dependent Pathway Is Required for TNFα-Induced Apoptosis. Cell 2003, 115, 61–70. [Google Scholar] [CrossRef]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.-L.; Maeda, S.; Venuprasad, K.; Liu, Y.-C.; Karin, M. The E3 Ubiquitin Ligase Itch Couples JNK Activation to TNFα-induced Cell Death by Inducing c-FLIPL Turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef]
- Chaudhary, P.; Sharma, R.; Sharma, A.; Vatsyayan, R.; Yadav, S.; Singhal, S.S.; Rauniyar, N.; Prokai, L.; Awasthi, S.; Awasthi, Y.C. Mechanisms of 4-Hydroxy-2-nonenal Induced Pro- and Anti-Apoptotic Signaling. Biochemistry 2010, 49, 6263–6275. [Google Scholar] [CrossRef]
- Forman, H.J.; Dickinson, D.A.; Iles, K.E. HNE––signaling pathways leading to its elimination. Mol. Asp. Med. 2003, 24, 189–194. [Google Scholar] [CrossRef]
- McElhanon, K.; Bose, C.; Sharma, R.; Wu, L.; Awasthi, Y.; Singh, S. Gsta 4 Null Mouse Embryonic Fibroblasts Exhibit Enhanced Sensitivity to Oxidants: Role of 4-Hydroxynonenal in Oxidant Toxicity. Open J. Apoptosis 2013, 2, 1–11. [Google Scholar] [CrossRef]
- Zhong, H.; Xiao, M.; Zarkovic, K.; Zhu, M.; Sa, R.; Lu, J.; Tao, Y.; Chen, Q.; Xia, L.; Cheng, S.; et al. Mitochondrial control of apoptosis through modulation of cardiolipin oxidation in hepatocellular carcinoma: A novel link between oxidative stress and cancer. Free Radic. Biol. Med. 2017, 102, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Mari, M.; Colell, A.; Morales, A.; Basañez, G.; Garcia-Ruiz, C.; Fernández-Checa, J.C. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim. Biophys. Acta-Bioenerg. 2010, 1797, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 2022–2031. [Google Scholar] [CrossRef]
- Landeta, O.; Landajuela, A.; Gil-Carton, D.; Taneva, S.; Primo, C.; Sot, B.; Valle, M.; Frolov, V.; Basañez, G. Reconstitution of Proapoptotic BAK Function in Liposomes Reveals a Dual Role for Mitochondrial Lipids in the BAK-driven Membrane Permeabilization Process. J. Biol. Chem. 2011, 286, 8213–8230. [Google Scholar] [CrossRef]
- D’Alessio, M.; Cerella, C.; De Nicola, M.; Bergamaschi, A.; Magrini, A.; Gualandi, G.; Alfonsi, A.M.; Ghibelli, L. Apoptotic GSH Extrusion Is Associated with Free Radical Generation. Ann. N. Y. Acad. Sci. 2003, 1010, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Nicole, A.; Santiard-Baron, D.; Ceballos-Picot, I. Direct evidence for glutathione as mediator of apoptosis in neuronal cells. Biomed. Pharmacother. 1998, 52, 349–355. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell Death and Cell Death Responses in Liver Disease: Mechanisms and Clinical Relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: Role of short-form leptin receptors and osteopontin. Am. J. Physiol. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef]
- Malhi, H.; Gores, G.J. Cellular and Molecular Mechanisms of Liver Injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pacher, P.; De Lisle, R.C.; Huang, H.; Ding, W.-X. A Mechanistic Review of Cell Death in Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 2016, 40, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic Liver Disease: The Gut Microbiome and Liver Cross Talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Emilie, D.; Balian, A.; Grangeot-Keros, L.; Borotto, E.; Portier, A.; Giraud, V.; Capron, F.; Galanaud, P.; Chaput, J.-C. Plasma levels of soluble tumor necrosis factor receptors p55 and p75 in patients with alcoholic liver disease of increasing severity. J. Hepatol. 1998, 28, 778–784. [Google Scholar] [CrossRef]
- Natori, S.; Rust, C.; Stadheim, L.M.; Srinivasan, A.; Burgart, L.J.; Gores, G.J. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J. Hepatol. 2001, 34, 248–253. [Google Scholar] [CrossRef]
- Naveau, S.; Chollet-Martin, S.; Dharancy, S.; Mathurin, P.; Jouet, P.; Piquet, M.-A.; Davion, T.; Oberti, F.; Broët, P.; Emilie, D.; et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 2004, 39, 1390–1397. [Google Scholar] [CrossRef]
- Cazanave, S.; Gores, G. Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clin. Lipidol. 2010, 5, 71–85. [Google Scholar] [CrossRef]
- Ferreira, D.M.S.; Castro, R.E.; Machado, M.V.; Evangelista, T.; Silvestre, A.; Costa, A.; Coutinho, J.; Carepa, F.; Cortez-Pinto, H.; Rodrigues, C.M.P. Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease. Diabetologia 2011, 54, 1788–1798. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Wieckowska, A.; Lopez, A.R.; Liu, Y.-C.; Zein, N.N.; McCullough, A.J. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: A multicenter validation study. Hepatology 2009, 50, 1072–1078. [Google Scholar] [CrossRef]
- Wieckowska, A.; Zein, N.N.; Yerian, L.M.; Lopez, A.R.; McCullough, A.J.; Feldstein, A.E. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 2006, 44, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Akazawa, Y.; Cazanave, S.C.; Bronk, S.F.; Elmi, N.A.; Werneburg, N.W.; Billadeau, D.D.; Gores, G.J. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J. Hepatol. 2011, 54, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Le, B.H.A.; García-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Cazanave, S.; Mott, J.L.; Elmi, N.; Bronk, S.F.; Kohno, S.; Charlton, M.R.; Gores, G.J. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J. Hepatol. 2010, 52, 586–593. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Gores, G.J. Who pulls the trigger: JNK activation in liver lipotoxicity? J. Hepatol. 2012, 56, 17–19. [Google Scholar] [CrossRef][Green Version]
- Kakisaka, K.; Cazanave, S.C.; Fingas, C.D.; Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Gores, G.J. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am. J. Physiol. Liver Physiol. 2011, 302, G77–G84. [Google Scholar] [CrossRef]
- Kant, S.; Swat, W.; Zhang, S.; Zhang, Z.-Y.; Neel, B.G.; Flavell, R.A.; Davis, R.J. TNF-stimulated MAP kinase activation mediated by a Rho family GTPase signaling pathway. Genes Dev. 2011, 25, 2069–2078. [Google Scholar] [CrossRef]
- Sharma, M.; Gadang, V.; Jaeschke, A. Critical Role for Mixed-Lineage Kinase 3 in Acetaminophen-Induced Hepatotoxicity. Mol. Pharmacol. 2012, 82, 1001–1007. [Google Scholar] [CrossRef]
- Cazanave, S.C.; Wang, X.; Zhou, H.; Rahmani, M.; Grant, S.; Durrant, D.E.; Klaassen, C.D.; Yamamoto, M.; Sanyal, A.J. Degradation of Keap1 activates BH3-only proteins Bim and PUMA during hepatocyte lipoapoptosis. Cell Death Differ. 2014, 21, 1303–1312. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH Oxidase-Mediated Redox Signaling: Roles in Cellular Stress Response, Stress Tolerance, and Tissue Repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Li, Z.; Bronk, S.F.; Gores, G.J. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am. J. Physiol. Liver Physiol. 2006, 290, G1339–G1346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Y.; Zhao, L.-P.; Tian, X.-X.; Yan, C.-H.; Li, Y.; Liu, Y.-X.; Wang, P.-X.; Zhang, X.-J.; Han, Y.-L. The novel intracellular protein CREG inhibits hepatic steatosis, obesity, and insulin resistance. Hepatology 2017, 66, 834–854. [Google Scholar] [CrossRef] [PubMed]
- Pulli, B.; Ali, M.; Iwamoto, Y.; Zeller, M.; Schob, S.; Linnoila, J.; Chen, J. Myeloperoxidase–Hepatocyte–Stellate Cell Crosstalk Promotes Hepatocyte Injury and Fibrosis in Experimental NASH. Antioxid. Redox Signal. 2015, 23, 1255–1269. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Hatting, M.; Zhao, G.; Schumacher, F.; Sellge, G.; Al Masaoudi, M.; Gaβler, N.; Boekschoten, M.; Müller, M.; Liedtke, C.; Cubero, F.J.; et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology 2013, 57, 2189–2201. [Google Scholar] [CrossRef]
- Thapaliya, S.; Wree, A.; Povero, D.; Inzaugarat, M.E.; Berk, M.; Dixon, L.; Papouchado, B.G.; Feldstein, A.E. Caspase 3 Inactivation Protects Against Hepatic Cell Death and Ameliorates Fibrogenesis in a Diet-Induced NASH Model. Dig. Dis. Sci. 2014, 59, 1197–1206. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; Wong, K.I.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK–caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef]
- Roychowdhury, S.; McCullough, R.L.; Sanz-Garcia, C.; Saikia, P.; Alkhouri, N.; Matloob, A.; Pollard, K.A.; McMullen, M.R.; Croniger, C.M.; Nagy, L.E. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology 2016, 64, 1518–1533. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef]
- Ou-Yang, Q.; Xuan, C.; Wang, X.; Luo, H.; Liu, J.-E.; Wang, L.; Li, T.; Chen, Y.; Liu, J. 3-Acetyl-oleanolic acid ameliorates non-alcoholic fatty liver disease in high fat diet-treated rats by activating AMPK-related pathways. Acta Pharmacol. Sin. 2018, 39, 1284–1293. [Google Scholar] [CrossRef]
- Shi, T.; Yang, X.; Zhou, H.; Xi, J.; Sun, J.; Ke, Y.; Zhang, J.; Shao, Y.; Jiang, X.; Pan, X.; et al. Activated carbon N-acetylcysteine microcapsule protects against nonalcoholic fatty liver disease in young rats via activating telomerase and inhibiting apoptosis. PLoS ONE 2018, 13, e0189856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.; Deng, X.; Zhang, N.; Liu, B.; Xin, S.; Li, G.; Xu, K. Baicalin attenuates non-alcoholic steatohepatitis by suppressing key regulators of lipid metabolism, inflammation and fibrosis in mice. Life Sci. 2018, 192, 46–54. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.-M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ozaki, S.; Gershwin, M.E.; Nakanuma, Y. Enhanced apoptosis relates to bile duct loss in primary biliary cirrhosis. Hepatology 1997, 26, 1399–1405. [Google Scholar] [CrossRef]
- Miyoshi, H.; Rust, C.; Roberts, P.J.; Burgart, L.J.; Gores, G.J. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 1999, 117, 669–677. [Google Scholar] [CrossRef]
- Schoemaker, M.H.; Conde de la Rosa, L.; Buist-Homan, M.; Vrenken, T.E.; Havinga, R.; Poelstra, K.; Haisma, H.J.; Jansen, P.L.M.; Moshage, H. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology 2004, 39, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Cubero, F.J.; Peng, J.; Liao, L.; Su, H.; Zhao, G.; Zoubek, M.E.; Macías-Rodríguez, R.; Ruiz-Margain, A.; Reißing, J.; Zimmermann, H.W.; et al. Inactivation of caspase 8 in liver parenchymal cells confers protection against murine obstructive cholestasis. J. Hepatol. 2018, 69, 1326–1334. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef]
- Canbay, A.; Higuchi, H.; Bronk, S.F.; Taniai, M.; Sebo, T.J.; Gores, G.J. Fas enhances fibrogenesis in the bile duct ligated mouse: A link between apoptosis and fibrosis. Gastroenterology 2002, 123, 1323–1330. [Google Scholar] [CrossRef]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; McMullen, M.R.; Pisano, S.G.; Liu, X.; Nagy, L.E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013, 57, 1773–1783. [Google Scholar] [CrossRef]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.-Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lv, X.; Hu, B.; Zhao, L.; Li, S.; Li, Z.; Qing, X.; Liu, H.; Xu, J.; Shao, Z. Critical contribution of RIPK1 mediated mitochondrial dysfunction and oxidative stress to compression-induced rat nucleus pulposus cells necroptosis and apoptosis. Apoptosis 2018, 23, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Heslop, K.A.; Rovini, A.; Hunt, E.G.; Fang, D.; Morris, M.E.; Christie, C.F.; Gooz, M.B.; DeHart, D.N.; Dang, Y.; Lemasters, J.J.; et al. JNK activation and translocation to mitochondria mediates mitochondrial dysfunction and cell death induced by VDAC opening and sorafenib in hepatocarcinoma cells. Biochem. Pharmacol. 2020, 171, 113728. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M. Oxidants, Antioxidants and Thiol Redox Switches in the Control of Regulated Cell Death Pathways. Antioxidants 2020, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Seehawer, M.; Heinzmann, F.; D’Artista, L.; Harbig, J.; Roux, P.-F.; Hoenicke, L.; Dang, H.; Klotz, S.; Robinson, L.; Doré, G.; et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018, 562, 69–75. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Chen, K.; Yuan, R.; Peng, L.; Maitra, U.; Diao, N.; Chen, C.; Zhang, Y.; Hu, Y.; Qi, C.-F.; et al. The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat. Commun. 2016, 7, 13436. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Dixit, V.M. Rescue from a fiery death: A therapeutic endeavor. Science 2019, 366, 688–689. [Google Scholar] [CrossRef]
- Orning, P.; Lien, E.; Fitzgerald, K.A. Gasdermins and their role in immunity and inflammation. J. Exp. Med. 2019, 216, 2453–2465. [Google Scholar] [CrossRef]
- Al Mamun, A.; Akter, A.; Hossain, S.; Sarker, T.; Safa, S.A.; Mustafa, Q.G.; Muhammad, S.A.; Munir, F. Role of NLRP3 inflammasome in liver disease. J. Dig. Dis. 2020, 21, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Navarro, L.; Angosto-Bazarra, D.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. NLRP3 Inflammasome and Pyroptosis in Liver Pathophysiology: The Emerging Relevance of Nrf2 Inducers. Antioxidants 2022, 11, 870. [Google Scholar] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Ishida, K.; Kaji, K.; Sato, S.; Ogawa, H.; Takagi, H.; Takaya, H.; Kawaratani, H.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Sulforaphane ameliorates ethanol plus carbon tetrachloride-induced liver fibrosis in mice through the Nrf2-mediated antioxidant response and acetaldehyde metabolization with inhibition of the LPS/TLR4 signaling pathway. J. Nutr. Biochem. 2021, 89, 108573. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, Q.; Liu, H.; Song, Z.; Chen, W. Phytochemical compounds targeting on Nrf2 for chemoprevention in colorectal cancer. Eur. J. Pharmacol. 2020, 887, 173588. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Brol, M.J.; Magdaleno, F.; Schierwagen, R.; Uschner, F.E.; Klein, S.; Ortiz, C.; Tyc, O.; Bachtler, N.; Stunden, J.; et al. The Specific NLRP3 Antagonist IFM-514 Decreases Fibrosis and Inflammation in Experimental Murine Non-Alcoholic Steatohepatitis. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Colak, Y.; Hasan, B.; Erkalma, B.; Tandon, K.; Zervos, X.; Menzo, E.L.; Erim, T. Pathogenetic mechanisms of nonalcoholic fatty liver disease and inhibition of the inflammasome as a new therapeutic target. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101710. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Khanova, E.; Wu, R.; Wang, W.; Yan, R.; Chen, Y.; French, S.W.; Llorente, C.; Pan, S.Q.; Yang, Q.; Li, Y.; et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology 2018, 67, 1737–1753. [Google Scholar] [CrossRef] [PubMed]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef]
- Torres, S.; Segalés, P.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells 2022, 11, 1475. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.-Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Hendrikx, T.; Bieghs, V.; Walenbergh, S.M.A.; van Gorp, P.J.; Verheyen, F.; Jeurissen, M.L.J.; Steinbusch, M.M.F.; Vaes, N.; Binder, C.J.; Koek, G.H.; et al. Macrophage Specific Caspase-1/11 Deficiency Protects against Cholesterol Crystallization and Hepatic Inflammation in Hyperlipidemic Mice. PLoS ONE 2013, 8, e78792. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Haigh, W.G.; Thorning, D.; Savard, C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J. Lipid Res. 2013, 54, 1326–1334. [Google Scholar] [CrossRef]
- Koh, E.H.; Yoon, J.E.; Ko, M.S.; Leem, J.; Yun, J.-Y.; Hong, C.H.; Cho, Y.K.; Lee, S.E.; Jang, J.E.; Baek, J.Y.; et al. Sphingomyelin synthase 1 mediates hepatocyte pyroptosis to trigger non-alcoholic steatohepatitis. Gut 2021, 70, 1954–1964. [Google Scholar] [CrossRef]
- Hong, C.H.; Ko, M.S.; Kim, J.H.; Cho, H.; Lee, C.-H.; Yoon, J.E.; Yun, J.-Y.; Baek, I.-J.; Jang, J.E.; Lee, S.E.; et al. Sphingosine 1-Phosphate Receptor 4 Promotes Nonalcoholic Steatohepatitis by Activating NLRP3 Inflammasome. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 925–947. [Google Scholar] [CrossRef]
- Samir, P.; Malireddi, R.K.S.; Kanneganti, T.-D. The PANoptosome: A Deadly Protein Complex Driving Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 238. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Alonso, C.; Fernández-Ramos, D.; Varela-Rey, M.; Martínez-Arranz, I.; Navasa, N.; Van Liempd, S.M.; Lavín Trueba, J.L.; Mayo, R.; Ilisso, C.P.; de Juan, V.G.; et al. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461.e7. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via SREBP1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef]
- Murase, T.; Misawa, K.; Minegishi, Y.; Aoki, M.; Ominami, H.; Suzuki, Y.; Shibuya, Y.; Hase, T. Coffee polyphenols suppress diet-induced body fat accumulation by downregulating SREBP-1c and related molecules in C57BL/6J mice. Am. J. Physiol. Metab. 2010, 300, E122–E133. [Google Scholar] [CrossRef]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kobayashi, Y.; Kawata, K.; Kawamura, K.; Sumiyoshi, S.; Noritake, H.; Watanabe, S.; Chida, T.; Souda, K.; Sakaguchi, T.; et al. Does Hepatic Oxidative Stress Enhance Activation of Nuclear Factor-E2-Related Factor in Patients with Nonalcoholic Steatohepatitis? Antioxid. Redox Signal. 2013, 20, 538–543. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Ziros, P.G.; Zaravinos, A.; Iskrenova, R.P.; Psyrogiannis, A.I.; Kyriazopoulou, V.E.; Sykiotis, G.P.; Habeos, I.G. Hepatic gene expression profiling in Nrf2 knockout mice after long-term high-fat diet-induced obesity. Oxid. Med. Cell. Longev. 2013, 2013, 340731. [Google Scholar] [CrossRef]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.J.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef]
- Wang, P.-X.; Zhang, R.; Huang, L.; Zhu, L.-H.; Jiang, D.-S.; Chen, H.-Z.; Zhang, Y.; Tian, S.; Zhang, X.-F.; Zhang, X.-D. Interferon regulatory factor 9 is a key mediator of hepatic ischemia/reperfusion injury. J. Hepatol. 2015, 62, 111–120. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, X.-H.; Zhang, X.-J.; Wang, P.-X.; Zhang, R.; Zhang, P.; Zhao, G.-N.; Gao, L.; Zhang, X.-F.; Tian, S. Targeting TRAF3 signaling protects against hepatic ischemia/reperfusions injury. J. Hepatol. 2016, 64, 146–159. [Google Scholar] [CrossRef]
- Tong, J.; Han, C.-J.; Zhang, J.-Z.; He, W.-Z.; Zhao, G.-J.; Cheng, X.; Zhang, L.; Deng, K.-Q.; Liu, Y.; Fan, H.-F.; et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology 2019, 69, 2471–2488. [Google Scholar] [CrossRef]
- Zhu, L.-H.; Wang, A.; Luo, P.; Wang, X.; Jiang, D.-S.; Deng, W.; Zhang, X.; Wang, T.; Liu, Y.; Gao, L. Mindin/Spondin 2 inhibits hepatic steatosis, insulin resistance, and obesity via interaction with peroxisome proliferator-activated receptor α in mice. J. Hepatol. 2014, 60, 1046–1054. [Google Scholar] [CrossRef]
- Wang, X.-A.; Deng, S.; Jiang, D.; Zhang, R.; Zhang, S.; Zhong, J.; Yang, L.; Wang, T.; Hong, S.; Guo, S. CARD3 deficiency exacerbates diet-induced obesity, hepatosteatosis, and insulin resistance in male mice. Endocrinology 2013, 154, 685–697. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Kawahara, H.; Fukura, M.; Tsuchishima, M.; Takase, S. Mutation of Mitochondrial DNA in Livers From Patients With Alcoholic Hepatitis and Nonalcoholic Steatohepatitis. Alcohol. Clin. Exp. Res. 2007, 31, S54–S60. [Google Scholar] [CrossRef]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueño, A.L.; Fernández Gianotti, T.; Castaño, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator–activated receptor γ coactivator 1α promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- LEGHI, G.; Domenici, F.; Vannucchi, H. Influence of oxidative stress and obesity in patients with nonalcoholic steatohepatitis. Arq. Gastroenterol. 2015, 52, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, A.; Duseja, A.; Das, A.; Dhiman, R.K.; Chawla, Y.K.; Kohli, K.K.; Bhansali, A. Patients with Nonalcoholic Fatty Liver Disease (NAFLD) have Higher Oxidative Stress in Comparison to Chronic Viral Hepatitis. J. Clin. Exp. Hepatol. 2013, 3, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative Stress and Enzymatic Antioxidant Status in Patients with Nonalcoholic Steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar] [PubMed]
- Svegliati-Baroni, G.; Pierantonelli, I.; Torquato, P.; Marinelli, R.; Ferreri, C.; Chatgilialoglu, C.; Bartolini, D.; Galli, F. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2019, 144, 293–309. [Google Scholar] [CrossRef]
- Liu, S.; Shi, W.; Li, G.; Jin, B.; Chen, Y.; Hu, H.; Liu, L.; Xie, F.; Chen, K.; Yin, D. Plasma reactive carbonyl species levels and risk of non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2011, 26, 1010–1015. [Google Scholar] [CrossRef]
- Comporti, M. Effect of in vivo and in vitro ethanol administration on liver lipid peroxidation. Lab. Investig. 1967, 16, 616–624. [Google Scholar]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef]
- Ambade, A.; Mandrekar, P. Oxidative Stress and Inflammation: Essential Partners in Alcoholic Liver Disease. Int. J. Hepatol. 2012, 2012, 853175. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Defective TNF-α-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J. Clin. Investig. 2003, 111, 197–208. [Google Scholar] [CrossRef]
- Saheki, T.; Kobayashi, K.; Iijima, M.; Moriyama, M.; Yazaki, M.; Takei, Y.; Ikeda, S.-I. Metabolic derangements in deficiency of citrin, a liver-type mitochondrial aspartate-glutamate carrier. Hepatol. Res. 2005, 33, 181–184. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Iron and CYP2E1-dependent oxidative stress and toxicity. Alcohol 2003, 30, 115–120. [Google Scholar] [CrossRef]
- Venkatraman, A.; Shiva, S.; Davis, A.J.; Bailey, S.M.; Brookes, P.S.; Darley-Usmar, V.M. Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology 2003, 38, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Federico, A. Oxidative stress in viral and alcoholic hepatitis. Free Radic. Biol. Med. 2003, 34, 1–10. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef]
- von Montfort, C.; Matias, N.; Fernandez, A.; Fucho, R.; de la Rosa, L.C.; Martinez-Chantar, M.L.; Mato, J.M.; Machida, K.; Tsukamoto, H.; Murphy, M.P.; et al. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J. Hepatol. 2012, 57, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.N.; Wulff, J.; Comerford, M.; Vuppalanchi, R.; Chalasani, N. Serum metabolic signatures of primary biliary cirrhosis and primary sclerosing cholangitis. Liver Int. 2015, 35, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Varatharajalu, R.; Garige, M.; Leckey, L.C.; Arellanes-Robledo, J.; Reyes-Gordillo, K.; Shah, R.; Lakshman, M.R. Adverse signaling of scavenger receptor class B1 and PGC1s in alcoholic hepatosteatosis and steatohepatitis and protection by betaine in rat. Am. J. Pathol. 2014, 184, 2035–2044. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial cholesterol accumulation in alcoholic liver disease: Role of ASMase and endoplasmic reticulum stress. Redox Biol. 2014, 3, 100–108. [Google Scholar] [CrossRef]
- Roskams, T.; Yang, S.Q.; Koteish, A.; Durnez, A.; DeVos, R.; Huang, X.; Achten, R.; Verslype, C.; Diehl, A.M. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am. J. Pathol. 2003, 163, 1301–1311. [Google Scholar] [CrossRef]
- Milkiewicz, P. Liver transplantation in primary biliary cirrhosis. Clin. Liver Dis. 2008, 12, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Gershwin, M.E.; Gish, R.G.; Invernizzi, P.; Jones, D.E.J.; Lindor, K.; Ma, X.; Mackay, I.R.; Parés, A.; Tanaka, A.; et al. Changing nomenclature for PBC: From ‘cirrhosis’ to ‘cholangitis. ’ Hepatology 2015, 62, 1620–1622. [Google Scholar] [CrossRef] [PubMed]
- Cash, W.J.; McCance, D.R.; Young, I.S.; McEneny, J.; Cadden, I.S.; McDougall, N.I.; Callender, M.E. Primary biliary cirrhosis is associated with oxidative stress and endothelial dysfunction but not increased cardiovascular risk. Hepatol. Res. 2010, 40, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Gershwin, M.E. The Immunobiology and Pathophysiology of Primary Biliary Cirrhosis. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 303–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Ma, X.; Tsuneyama, K.; Huang, S.; Takahashi, T.; Chalasani, N.P.; Bowlus, C.L.; Yang, G.-X.; Leung, P.S.C.; Ansari, A.A.; et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: Implications for therapy. Hepatology 2014, 59, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, P.; Terracciano, L.; D’Angelo, S.; Ferbo, U.; Bracigliano, A.; Tarantino, L.; Perrella, A.; Perrella, O.; Chiara, G.; Panico, L.; et al. Oxidative stress and steatosis are cofactors of liver injury in primary biliary cirrhosis. J. Gastroenterol. 2010, 45, 1053–1062. [Google Scholar] [CrossRef]
- Grattagliano, I.; Calamita, G.; Cocco, T.; Wang, D.Q.-H.; Portincasa, P. Pathogenic role of oxidative and nitrosative stress in primary biliary cirrhosis. World J. Gastroenterol. 2014, 20, 5746–5759. [Google Scholar] [CrossRef]
- Salunga, T.L.; Cui, Z.-G.; Shimoda, S.; Zheng, H.-C.; Nomoto, K.; Kondo, T.; Takano, Y.; Selmi, C.; Alpini, G.; Gershwin, M.E.; et al. Oxidative stress-induced apoptosis of bile duct cells in primary biliary cirrhosis. J. Autoimmun. 2007, 29, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Kobayashi, Y.; Souda, K.; Kawamura, K.; Sumiyoshi, S.; Takahashi, Y.; Noritake, H.; Watanabe, S.; Suehiro, T.; Nakamura, H. Enhanced hepatic Nrf2 activation after ursodeoxycholic acid treatment in patients with primary biliary cirrhosis. Antioxid. Redox Signal. 2010, 13, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Palmieri, V.O.; Portincasa, P.; Minerva, F.; Palasciano, G. Long-term ursodeoxycholate improves circulating redox changes in primary biliary cirrhotic patients. Clin. Biochem. 2011, 44, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Wasik, U.; Milkiewicz, M.; Kempinska-Podhorodecka, A.; Milkiewicz, P. Protection against oxidative stress mediated by the Nrf2/Keap1 axis is impaired in Primary Biliary Cholangitis. Sci. Rep. 2017, 7, 44769. [Google Scholar] [CrossRef] [PubMed]
- Kilanczyk, E.; Banales, J.M.; Wunsch, E.; Barbier, O.; Avila, M.A.; Mato, J.M.; Milkiewicz, M.; Milkiewicz, P. S-adenosyl-L-methionine (SAMe) halts the autoimmune response in patients with primary biliary cholangitis (PBC) via antioxidant and S-glutathionylation processes in cholangiocytes. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165895. [Google Scholar] [CrossRef]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- Zhuang, X.; Forde, D.; Tsukuda, S.; D’Arienzo, V.; Mailly, L.; Harris, J.M.; Wing, P.A.C.; Borrmann, H.; Schilling, M.; Magri, A.; et al. Circadian control of hepatitis B virus replication. Nat. Commun. 2021, 12, 1658. [Google Scholar] [CrossRef]
- Bäuerle, J.; Laguno, M.; Mauss, S.; Mallolas, J.; Murillas, J.; Miquel, R.; Schmutz, G.; Setzer, B.; Gatell, J.M.; Walker, U.A. Mitochondrial DNA depletion in liver tissue of patients infected with hepatitis C virus: Contributing effect of HIV infection? HIV Med. 2005, 6, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Venturini, D.; Simão, A.N.C.; Barbosa, D.S.; Lavado, E.L.; Narciso, V.E.S.; Dichi, I.; Dichi, J.B. Increased Oxidative Stress, Decreased Total Antioxidant Capacity, and Iron Overload in Untreated Patients with Chronic Hepatitis C. Dig. Dis. Sci. 2010, 55, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Sumida, Y.; Yoh, T.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Kashima, K.; Nakamura, H.; Yodoi, J. Thioredoxin Levels in the Sera of Untreated Viral Hepatitis Patients and Those Treated with Glycyrrhizin or Ursodeoxycholic Acid. Antioxid. Redox Signal. 2000, 2, 687–694. [Google Scholar] [CrossRef]
- Qi, L.; Zou, Z.-Q.; Wang, L.-Y.; Gao, S.; Fan, Y.-C.; Long, B.; Guo, Y.-M.; Xu, A.-L.; Han, J.; Li, T.; et al. Methylation of the Glutathione-S-Transferase M3 Gene Promoter is Associated with Oxidative Stress in Acute-on-Chronic Hepatitis B Liver Failure. Tohoku J. Exp. Med. 2012, 228, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Honda, A.; Miyazaki, T.; Kohjima, M.; Nakamuta, M.; Matsuzaki, Y. Increased serum oxysterol concentrations in patients with chronic hepatitis C virus infection. Biochem. Biophys. Res. Commun. 2014, 446, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Ivanova, O.N.; Bartosch, B.; Valuev-Elliston, V.T.; Mukhtarov, F.; Kochetkov, S.N.; Ivanov, A. V Hepatitis C Virus NS5A Protein Triggers Oxidative Stress by Inducing NADPH Oxidases 1 and 4 and Cytochrome P450 2E1. Oxid. Med. Cell. Longev. 2016, 2016, 8341937. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Okawa, O.; Shirahashi, R.; Tokutomi, N.; Tamano, M. Changes in serum zinc levels in hepatitis C patients before and after treatment with direct-acting antiviral agents. Hepatol. Res. 2019, 49, 1353–1356. [Google Scholar] [CrossRef]
- Alavian, S.; Showraki, A. Hepatitis B and its Relationship With Oxidative Stress. Hepat. Mon. 2016, 16, e37973. [Google Scholar] [CrossRef]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. New concepts on the clinical course and stratification of compensated and decompensated cirrhosis. Hepatol. Int. 2018, 12, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Valle, V.; Chávez-Tapia, N.C.; Uribe, M.; Méndez-Sánchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Sancho-bru, P.; Ginès, P.; Lora, J.M.; Al-garawi, A.; Solé, M.; Colmenero, J.; Nicolás, J.M.; Jiménez, W.; Weich, N.; et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 2003, 125, 117–125. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Mariño, Z.; Perelló, C.; Iñarrairaegui, M.; Ribeiro, A.; Lens, S.; Díaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, R.; Llovet, J.M.; Han, G.; Tak, W.Y.; Yang, J.; Guglielmi, A.; Paik, S.W.; Reig, M.; Kim, D.Y.; Chau, G.-Y.; et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: The SPACE trial. J. Hepatol. 2016, 64, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Ande, S.R.; Nguyen, K.H.; Grégoire Nyomba, B.L.; Mishra, S. Prohibitin-induced, obesity-associated insulin resistance and accompanying low-grade inflammation causes NASH and HCC. Sci. Rep. 2016, 6, 23608. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Fatty acid-induced T cell loss greases liver carcinogenesis. Cell Metab. 2016, 23, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Suna, N.; Boyacioglu, A.S. Management of Hepatocellular Carcinoma: Prevention, Surveillance, Diagnosis, and Staging. Exp. Clin. 2017, 15, 31–35. [Google Scholar]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2006, 401, 1–11. [Google Scholar] [CrossRef]
- Ko, C.; Siddaiah, N.; Berger, J.; Gish, R.; Brandhagen, D.; Sterling, R.K.; Cotler, S.J.; Fontana, R.J.; McCashland, T.M.; Han, S.H.B.; et al. Prevalence of hepatic iron overload and association with hepatocellular cancer in end-stage liver disease: Results from the National Hemochromatosis Transplant Registry. Liver Int. 2007, 27, 1394–1401. [Google Scholar] [CrossRef]
- Marrogi, A.J.; Khan, M.A.; van Gijssel, H.E.; Welsh, J.A.; Rahim, H.; Demetris, A.J.; Kowdley, K.V.; Hussain, S.P.; Nair, J.; Bartsch, H.; et al. Oxidative Stress and p53 Mutations in the Carcinogenesis of Iron Overload-Associated Hepatocellular Carcinoma. JNCI J. Natl. Cancer Inst. 2001, 93, 1652–1655. [Google Scholar] [CrossRef]
- Tanaka, S.; Miyanishi, K.; Kobune, M.; Kawano, Y.; Hoki, T.; Kubo, T.; Hayashi, T.; Sato, T.; Sato, Y.; Takimoto, R.; et al. Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J. Gastroenterol. 2013, 48, 1249–1258. [Google Scholar] [CrossRef]
- Ribas, V.; de la Rosa, L.C.; Robles, D.; Núñez, S.; Segalés, P.; Insausti-Urkia, N.; Solsona-Vilarrasa, E.; Fernández-Checa, J.C.; García-Ruiz, C. Dietary and Genetic Cholesterol Loading Rather Than Steatosis Promotes Liver Tumorigenesis and NASH-Driven HCC. Cancers 2021, 13, 4091. [Google Scholar] [CrossRef]
- Sun, L.; Beggs, K.; Borude, P.; Edwards, G.; Bhushan, B.; Walesky, C.; Roy, N.; Manley, M.W., Jr.; Gunewardena, S.; O’Neil, M.; et al. Bile acids promote diethylnitrosamine-induced hepatocellular carcinoma via increased inflammatory signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G91–G104. [Google Scholar] [CrossRef]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basañez, G.; Antonsson, B.; Prieto, J.; García-Ruiz, C.; Colell, A.; et al. Mitochondrial Cholesterol Contributes to Chemotherapy Resistance in Hepatocellular Carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef]
- de la Rosa, L.C.; Garcia-Ruiz, C.; Vallejo, C.; Baulies, A.; Nuñez, S.; Monte, M.J.; Marin, J.J.G.; Baila-Rueda, L.; Cenarro, A.; Civeira, F.; et al. STARD1 promotes NASH-driven HCC by sustaining the generation of bile acids through the alternative mitochondrial pathway. J. Hepatol. 2021, 74, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Silva, T.; Andrade, B.P.; Borges, F. Alzheimer’s Disease and Antioxidant Therapy: How Long How Far? Curr. Med. Chem. 2013, 20, 2939–2952. [Google Scholar] [CrossRef]
- Silva, C.; Pinto, M.; Fernandes, C.; Benfeito, S.; Borges, F. Antioxidant Therapy and Neurodegenerative Disorders: Lessons From Clinical Trials; Wolkenhauer, O.B.T.-S.M., Ed.; Academic Press: Oxford, UK, 2021; pp. 97–110. ISBN 978-0-12-816078-7. [Google Scholar]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Jampana, S.C.; Weinman, S.A. Antioxidants as therapeutic agents for liver disease. Liver Int. 2011, 31, 1432–1448. [Google Scholar] [CrossRef] [PubMed]
- Delanty, N.; Dichter, M.A. Antioxidant Therapy in Neurologic Disease. Arch. Neurol. 2000, 57, 1265–1270. [Google Scholar] [CrossRef]
- Sacco, R.; Eggenhoffner, R.; Giacomelli, L. Glutathione in the treatment of liver diseases: Insights from clinical practice. Minerva Gastroenterol. Dietol. 2016, 62, 316–324. [Google Scholar]
- Torres, S.; Matías, N.; Baulies, A.; Nuñez, S.; Alarcon-Vila, C.; Martinez, L.; Nuño, N.; Fernandez, A.; Caballeria, J.; Levade, T.; et al. Mitochondrial GSH replenishment as a potential therapeutic approach for Niemann Pick type C disease. Redox Biol. 2017, 11, 60–72. [Google Scholar] [CrossRef]
- Basu, P.; Shah, N.; Aloysius, M.; Junior, R. Effect of Vitamin E and Alpha Lipoic Acid in Nonalcoholic Fatty Liver Disease: A Randomized, Placebo-Controlled, Open-Label, Prospective Clinical Trial (VAIN Trial). Open J. Gastroenterol. 2014, 4, 199–207. [Google Scholar] [CrossRef]
- Parasassi, T.; Brunelli, R.; Costa, G.; De Spirito, M.; Krasnowska, E.; Lundeberg, T.; Pittaluga, E.; Ursini, F. Thiol Redox Transitions in Cell Signaling: A Lesson from N-Acetylcysteine. ScientificWorldJournal. 2010, 10, 905927. [Google Scholar] [CrossRef]
- Usman, M.; Bakhtawar, N. Vitamin E as an Adjuvant Treatment for Non-alcoholic Fatty Liver Disease in Adults: A Systematic Review of Randomized Controlled Trials. Cureus 2020, 12, e9018. [Google Scholar] [CrossRef]
- Benfeito, S.; Oliveira, C.; Soares, P.; Fernandes, C.; Silva, T.; Teixeira, J.; Borges, F. Antioxidant therapy: Still in search of the ‘magic bullet’. Mitochondrion 2013, 13, 427–435. [Google Scholar] [CrossRef]
- Mantle, D.; Hargreaves, I. Coenzyme Q10 supplementation in non-alcoholic fatty liver disease: An overview. Gastrointest. Nurs. 2020, 18, 22–27. [Google Scholar] [CrossRef]
- Alavian, K.N.; Dworetzky, S.I.; Bonanni, L.; Zhang, P.; Sacchetti, S.; Mariggio, M.A.; Onofrj, M.; Thomas, A.; Li, H.; Mangold, J.E.; et al. Effects of dexpramipexole on brain mitochondrial conductances and cellular bioenergetic efficiency. Brain Res. 2012, 1446, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Pérez, L.C.; Padilla-Martínez, I.I.; Cruz, A.; Mendieta-Wejebe, J.E.; Tamay-Cach, F.; Rosales-Hernández, M.C. Evaluation of a new benzothiazole derivative with antioxidant activity in the initial phase of acetaminophen toxicity. Arab. J. Chem. 2019, 12, 3871–3882. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conde de la Rosa, L.; Goicoechea, L.; Torres, S.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Role of Oxidative Stress in Liver Disorders. Livers 2022, 2, 283-314. https://doi.org/10.3390/livers2040023
Conde de la Rosa L, Goicoechea L, Torres S, Garcia-Ruiz C, Fernandez-Checa JC. Role of Oxidative Stress in Liver Disorders. Livers. 2022; 2(4):283-314. https://doi.org/10.3390/livers2040023
Chicago/Turabian StyleConde de la Rosa, Laura, Leire Goicoechea, Sandra Torres, Carmen Garcia-Ruiz, and José C. Fernandez-Checa. 2022. "Role of Oxidative Stress in Liver Disorders" Livers 2, no. 4: 283-314. https://doi.org/10.3390/livers2040023
APA StyleConde de la Rosa, L., Goicoechea, L., Torres, S., Garcia-Ruiz, C., & Fernandez-Checa, J. C. (2022). Role of Oxidative Stress in Liver Disorders. Livers, 2(4), 283-314. https://doi.org/10.3390/livers2040023