Abstract
In the present study we investigated the participation of hepatic peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α) in the metabolic programming of newborn rats exposed in utero to dexamethasone (DEX). On the 21st day of life, fasted offspring born to DEX-treated mothers displayed increased conversion of pyruvate into glucose with simultaneous upregulation of PEPCK (phosphoenolpyruvate carboxykinase) and G6Pase (glucose-6-phosphatase). Increased oxidative phosphorylation, higher ATP/ADP ratio and mitochondrial biogenesis and lower pyruvate levels were also found in the progeny of DEX-treated mothers. On the other hand, the 21-day-old progeny of DEX-treated mothers had increased hepatic triglycerides (TAG) and lower CPT-1 activity when subjected to short-term fasting. At the mechanistic level, rats exposed in utero to DEX exhibited increased hepatic PGC-1α protein content with lower miR-29a-c expression. Increased PGC-1α content was concurrent with increased association to HNF-4α and NRF1 and reduced PPARα expression. The data presented herein reveal that changes in the transcription machinery in neonatal liver of rats born to DEX-treated mothers leads to an inflexible metabolic response to fasting. Such programming is hallmarked by increased oxidative phosphorylation of pyruvate with impaired FFA oxidation and hepatic TAG accumulation.
1. Introduction
Several studies with animal models have shown that prenatal glucocorticoid (GC) excess resultant from either maternal caloric restriction or decreased placental activity of the 11beta-hydroxysteroid dehydrogenase 2 (11β-HSD2), is responsible for the low birth weight and the programming of metabolic and cardiovascular changes in the offspring. In humans, mutations in the gene of 11β-HDS2 are also associated with restriction in intrauterine growth [1].
In accordance with the above-mentioned studies, in utero overexposure to exogenous glucocorticoids is known to cause low birth weight and subsequent metabolic programming during adulthood. As such, rats exposed to dexamethasone (DEX) during the third week of pregnancy become glucose intolerant during adult life [2]. Metabolic programming is not seen in rats born to mothers exposed to DEX either during the first or the second weeks of pregnancy [2].
Glucose intolerance in rats born to DEX-treated mothers is not attributed to impaired insulin-induced glucose disposal by skeletal muscle but, instead, to a persistent increase in hepatic gluconeogenesis [3,4]. The expression of phosphoenolpyruvate carboxykinase (PEPCK), a limiting step for hepatic gluconeogenesis, is upregulated as early as the 5th day of life in rats exposed to DEX in utero [2]. Increased gluconeogenesis in the adult progeny of mothers treated with DEX during pregnancy is also attributed to impaired hepatic insulin signaling with ineffective inhibition of PEPCK expression [5,6]. Despite these consistent findings, the molecular mechanism leading to upregulation of gluconeogenic pathways in the liver of rats born to DEX-treated mothers remains to be determined.
A master regulator of gluconeogenesis enzymes and mitochondrial biogenesis in liver is the peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α). The ability of PGC-1α to regulate multiple biochemical pathways relies on its capacity to bind to several transcription factors such as PPAR (Peroxisome Proliferator-Activated Receptor) α and γ, ERRα (Estrogen Receptor-Related α), FoxO1 (Forkhead Box O1), HNF-4α (Hepatocyte Nuclear Factor 4α) and NRF1 (Nuclear Respiratory Factor 1) (reviewed in [7]).
PGC-1α plays a pivotal role by controlling the transcription of Pck1, the gene that encodes PEPCK [8]. PGC-1α was also reported to mediate HNF-4α transcriptional activity over G6pc, the gene that encodes glucose-6-phosphatase (G6Pase), another limiting step for gluconeogenesis [9]. Previous studies have also revealed that the association between PGC-1α and PPARs in different tissues induces changes in lipid metabolism through mitochondrial fatty acid oxidation control (reviewed in [10]).
The content of PGC-1α is fine-tuned by multiple transcriptional and post-transcriptional epigenetic mechanisms such as cytosine methylation at the promoter region and 3′UTR targeting by miRNAs [11,12,13]. Thus, the current study was carried out to investigate whether hepatic changes in PGC-1α expression and related targets have considerable impacts on glucose and lipid metabolism in prepubertal offspring exposed to DEX in utero.
2. Materials and Methods
2.1. Animals
All experimental procedures followed the guidelines of the Brazilian College for Animal Experimentation (COBEA) and were approved by the Ethics Committee on Animal Use at the Institute of Biomedical Sciences, University of Sao Paulo, Brazil (Certificate No. 5367250619). Adult male and female Wistar rats were obtained from the Animal Breeding Center at the University of Sao Paulo, Brazil, and kept under a 12-h light–dark cycle at 22 ± 2 °C with standard chow and water ad libitum.
2.2. Experimental Design
At 12 weeks of age, females were housed in individual cages with one male for 3 days. The presence of spermatozoa in a vaginal lavage indicated day 0 of gestation. Half of pregnant rats were treated with dexamethasone (DEX) (0.2 mg/kg body mass; Achê Pharmaceutical Laboratories, Guarulhos, SP, Brazil) diluted in drinking water from the 14th to the 19th day of pregnancy as previously described [4,14]. Untreated pregnant rats were used as controls (CTL). The number of pups was registered and adjusted to six per lactating mother within the first 24 h after delivery. Offspring were used on the 1st (L1), the 8th (L8) and the 21st (L21) days of life. As indicated, male and female pups were used for the experiments. Pups on the 21st day of life were subjected to a 6 h-fasting prior to the experimental procedures.
2.3. Analysis of Blood Parameters
Trunk blood samples were collected at L1, L8 and L21, and the serum was separated by centrifugation. Serum glucose, total cholesterol and triacylglycerol (TAG) levels were measured using commercially available kits (Labtest Diagnóstica SA, Lagoa Santa, Brazil).
2.4. Analysis of Tissue Parameters
Liver samples were quickly removed at L1, L8 and L21, washed with ice-cold phosphate-buffered saline (PBS), frozen with liquid nitrogen, and ground in a mortar. Powdered samples were maintained at −80 °C until used.
Lipids were extracted from liver samples as previously described [15]. Briefly, powdered samples were homogenized using a rotor-stator in a 4 mL solution of CHCl3 and methanol (2:1, v/v). The homogenate was subjected to extraction for at least 16 h at 4 °C, with gentle homogenization. After this, a 0.6% NaCl solution was added to the samples that were further centrifuged at 2000× g for 20 min. The organic layer was collected and dried in an Eppendorf Vacuum Concentrator Plus (Eppendorf, Hamburg, Germany). The lipids were solubilized in 200 µL of isopropanol and quantified using commercially available kits.
The activities of phosphoenolpyruvate carboxykinase (PEPCK; EC 4.1.1.32) and glucose-6-phosphatase (G6Pase; EC 3.1.3.9) were measured using spectrophotometric assays at 340 nm, following standard methods described elsewhere [16,17]. Citrate synthase (CS; EC 4.1.3.7) was assayed at 412 nm, as previously described [18].
2.5. Functional Tests
2.5.1. Intraperitoneal Glucose Tolerance Test (ip-GTT)
After fasting, 21-day-old rats received an intraperitoneal glucose injection (1 g/kg of a 20% solution of D-glucose). Blood samples were collected from the tail at 0, 5, 10, 15, 30, 60 and 120 min after the injection for the measurement of blood glucose levels. The area under the curve (AUC) of glycemia vs. time was calculated above each individual baseline (basal glycemia) to estimate glucose tolerance.
2.5.2. Intraperitoneal Insulin Tolerance Test (ip-ITT)
After fasting, 21-day-old rats received an intraperitoneal insulin injection (0.75 IU/kg). Blood samples were collected from the tail at 0, 5, 10, 15, 30 and 60 min after the injection for the measurement of serum glucose levels. The glucose disappearance rate (KITT) was calculated using the equation: 0.693/half-life. Glucose half-life was calculated from the slope of the least-squares analysis of the blood glucose concentrations during the linear phase of decay.
2.5.3. Intraperitoneal Pyruvate Tolerance Test (ip-PTT)
After fasting, 21-day-old rats received an intraperitoneal injection of a sodium pyruvate solution (250 mg/mL) at a dosage of 1 g/kg. Glucose levels were determined in blood collected from the tail at 0, 15, 30, 60, 90 and 120 min after the injection. The AUC of glycemia vs. time was calculated using each individual baseline (basal glycemia) to estimate glucose production after a pyruvate load.
2.6. RNA Extraction and qPCR
Total RNA was extracted using TRIzol reagent. Aliquots of 2 µg were subjected to reverse transcription with random primers using High-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) for the analysis of mRNA expression. For miRNA expression analysis, 2 µg RNA samples were initially poly(A)-tailed and reverse transcribed with an adaptor linked to oligo-d(T) [19]. Values of mRNA and miRNA expression were normalized using internal control genes Rpl37a and Rnu43, respectively. Fold changes were calculated using the 2−ΔΔCT method. Primer sequences used for mRNA analysis were as follow (sense and antisense, respectively). Cd36 (AF072411) 5′-TCTTCCAGCCAACGCCTTTGC-3′, 5′-TGCACTTGCCAATGTCCAGCAC-3′; Cpt1 (NM_031559) 5′-ATTCCAGGAGAGTGCCAGGAGG-3′, 5′-CCCATGTCCTTGTAATGTGCGAG-3′; Dgat2 (NM_001012345) 5′-AAGCCCATCACCACCGTTG-3′, 5′-TTCCTTCCAGGAGCTGGCAC-3′; Dnmt3a (NM_001003958) 5′-GTCATGTGGTTCGGAGATGGC-3′, 5′-TGTATTCCAGACGGCACCCAG-3′; Fatp (NM_053580) 5′-GACAGATTGGCGAGTTCTACGGC-3′, 5′-GTACACATGCGTGAGGATACGGC-3′; G6pc (NM_013098) 5′-ACCTTCTTCCTGTTTGGTTTCGC-3′, 5′-CGGTACATGCTGGAGTTGAGGG-3′; Nrf1 (NM_001100708) 5′-GCCGTTGGAGCACTTACTGGAG-3′, 5′-CCGCCATAATGAATCCCTTTCC-3′; Pck1 (NM_198780) 5′-TGGTCTGGACTTCTCTGCCAAG-3′, 5′-AATGATGACCGTCTTGCTTTCG-3′; Ppargc1a (NM_031347) 5′-CCCATACACAACCGCAGTCG-3′, 5′-CTTCCTTTCCTCGTGTCCTCG-3′; Rpl37a (X14069) 5′-CAAGAAGGTCGGGATCGTCG-3′, 5′-ACCAGGCAAGTCTCAGGAGGTG-3′.
Primer sequences used for miRNA analysis were: Adapter linked to oligo d(T) 5′-GGCCACGCGTCGACTAGTAC(T)12-3′; Universal antisense 5′-GGCCACGCGTCGACTAGTAC-3′; rno-miR-29a (MIMAT0000802) 5′-TAGCACCATCTGAAATCGGTTA-3′; rno-miR-29b (MIMAT0000801) 5′-TAGCACCATTTGAAATCAGTGTT-3′; rno-miR-29c (MIMAT0000803 5′-TAGCACCATTTGAAATCGGTTA-3′; RNU43 snoRNA 5′-CTTATTGACGGGCGGACAGAAAC-3′.
2.7. Protein Extraction and Immunoblotting
Approximately 100 mg of powdered liver samples were processed for immunoblotting as described elsewhere [20]. Briefly, samples were homogenized in ice-cold solubilization buffer, and insoluble materials were removed by centrifugation at 15,000× g for 40 min at 4 °C. Supernatants were either immunoprecipitated with rabbit anti-HNF-4α or rabbit anti-NRF-1 antibodies plus protein A-Sepharose® 6 MB (GE Healthcare, Buckinghamshire, UK) [20], or processed for typical immunoblotting.
The primary antibodies used were as follows: anti-DNMT3A (Cat. #3598, RRID:AB_2277449) from Cell Signaling Technology (Danvers, MA, USA), anti-G6Pase-α Cat# sc-398155, RRID:AB_2813839), anti-PEPCK (Cat# sc-32879, RRID:AB_2160168), anti-HNF-4α (Cat# sc-8987, RRID:AB_2116913), anti-NRF-1 (Cat# sc-33771, RRID:AB_2153203), anti-PPARα (Cat# sc-1985, RRID:AB_2165740) and anti-PGC-1α (Cat# sc-517380, RRID:AB_2755043) from Santa Cruz Biotechnologies (Santa Cruz, TX, USA), anti-total OXPHOS (Cat# ab110413, RRID:AB_2629281) from Abcam (Cambridge, UK). Secondary antibodies conjugated to horseradish peroxidase (Bio-Rad Laboratories, Hercules, CA, USA) were employed for chemiluminescent detection of the protein bands, and a ChemiDocTM XRS+ imaging system (Bio-Rad Laboratories, Hercules, CA, USA) was employed for visualization. Target protein intensities were quantified by optical densitometry (OD) analysis using ImageJ software (version 1.53e). Before incubation with the primary antibodies, nitrocellulose membranes were stained with Ponceau S, scanned at high resolution and subjected to OD quantification of all labeled proteins in each lane. Subsequently, these values were applied to normalize the OD data of the target proteins detected in the respective membranes. This method has been validated as an appropriate loading control [21].
2.8. PGC-1α Gene Methylation
Methylated DNA was pulled down using the Methyl Hunter kit (MBL International, Japan; Cat. #5275) strictly following the manufacturer’s instructions. Briefly, 10 μg of DNA were sheared to 150–600 bp fragments with the use of ultra-sound. An aliquot was saved to use as input. For mCpG DNA pull-down, the sheared DNA was incubated with MDB1 protein overnight. Subsequently, anti-His-tag antibody-coated-magnetic beads were added to the samples and the mCpG DNA was separated using a magnetic rack. mCpG DNA was eluted and precipitated with ice-cold isopropanol, diluted in 20 μL of nuclease-free water and used for qPCR.
Primers used for amplification of the rat PGC-1α promoter region (GenBank Accession# AY382577) were designed to flank the putative methylation sites. As PGC-1α promoter region does not have CpG islands, we used CpG dinucleotides O/E (Observed/Expected) ratio higher than 0.6 to predict potential methylation sites [22]. Primer sequences were sense: 5′-GGTTTAGAGTTGGTGGCATTC-3′, and antisense: 5′-CTTACTGAGAGTGAACTGAAGGC-3′. The amplification was conducted as described above. Input DNA samples were used as internal control.
2.9. Relative mtDNA Content
The ratio of mitochondrial vs. nuclear DNA was determined by qPCR. Briefly, liver samples (approximately 100 mg) were used for phenol-chloroform-isoamylalcohol extraction of total DNA using standard procedures [23]. For PCR sample preparation, 20 ng of DNA was mixed with primers (10 μM), fluorescent dye SYBR Green (Platinum® SYBR® Green qPCR Supermix UDG, Invitrogen, Carlsbad, CA, USA), and nuclease-free water to reach a 12.5 μL final volume. The reaction was initiated at 94 °C for 10 min, followed by 40 cycles through 94 °C × 10 s, 60 °C × 30 s, and 94 °C × 10 s. All reactions were run in duplicate. Mitochondrial DNA content relative to nuclear DNA was calculated as described [24].
The following primers were designed to target: nDNA ACTB (V01217) 5′-TAATGAGGCTGGTGATAAATGGC-3′ and 5′-GGGCAGGTGAGACTGTAAGGAT-3′; and mtDNA COX I (JX105355) 5′-CAATTCCTACAGGCGTAAA-3′and 5′-CAATGTCAAGGGATGAGTTAG-3′.
2.10. ATP, ADP and Pyruvate Quantification
ATP, ADP, and pyruvate quantitation were analyzed in fresh liver. Samples were homogenized with perchloric acid (HClO4) (0.6 N) using sterile pistil (Axygen Scientific, Union City, CA, USA) on the same day of collection. The homogenate was centrifuged at 12,000 rpm, 4 °C, for 15 min. Aliquots of the supernatant were neutralized with 1 M potassium bicarbonate (KHCO3) for the ATP quantification. The buffer consisted of 75 mM Tris hydrochloride (Tris-HCl), 7.5 mM magnesium chloride (MgCl2), 0.8 mM EDTA, 1.5 mM potassium chloride (KCl), 4 mM β-mercaptoethanol, 0.05% TritonX100, 0.4 mM NADP+, 1 mM glucose and 10 units of hexokinase + glucose-6-phosphate dehydrogenase [25]. UV absorbance (340 nm) was read in a Bio-Tek uQuant Universal Microplate Spectrophotometer (BioTek Instruments, Inc., Winooski, VT, USA).
ADP was assayed as described [26]. The assay buffer (pH 8.2) consisted of 50 mM Tris-HCl, 2 mM MgSO4, 5 mM KCl, 0.2 mM NADH, 2 U lactate dehydrogenase, 4 U pyruvate kinase and 0.05% Triton X-100. The assay was initiated by addition of 2 mM phosphoenolpyruvate (PEP) and absorbance was measured at 340 nm.
Pyruvate was assayed according to Neville and Gelder [27]. Samples were deproteinized in perchloric acid (0.6 N) and the supernatant was neutralized using sodium bicarbonate solution (1 M). The assay buffer (pH 7.5) containing 50 mM Tris-HCl, 2 mM MgSO4, 5 mM KCl, 0.2 mM NADH and 2 units lactate dehydrogenase. The total assay volume was 165 µL (containing 10 µL of the homogenate) and absorbance was measured at 340 nm. We used a standard curve of pyruvate ranging from 0.1–0.6 mM.
2.11. Transmission Electron Microscopy
Liver fragments were cut into small pieces (approximately 2 mm3) and immersed in fixation buffer (glutaraldehyde 2% in 0.1 M sodium cacodylate buffer, pH 7.4) for at least 2 h at 4 °C. Samples were post-fixed with 1% osmium tetroxide (OsO4) for 2 h, washed three times with water, incubated overnight in a buffer with 1% uranyl acetate and 10% sucrose, dehydrated in grades of alcohol (2 × 15 min 70%; 2 × 15 min 95%; 4 × 15 min 100%) and embedded in epoxy resin.
Ultrathin sections were cut in a Reichert Ultracut-S microtome and post-stained with 4% aqueous uranyl-acetate and lead-citrate. Liver sections were analyzed in a FEI Tecnai G2 Spirit 200 kV transmission electron microscope (Field Electron and Ion Company-FEI, Hillsboro, OR, USA). Six images per animal were acquired at 3500× magnification and the lipid droplets density was measured using the Image J2 software (version 2.3.0) [28].
2.12. Statistical Analyses
All results are presented as the mean ± standard error of the mean (SEM). When parameters were assessed in multiples postnatal days, comparisons were made with two-way ANOVA considering (i) time after delivery (ii) maternal use of DEX. Tukey’s multiple comparison test was used to indicate differences among different postnatal days and Sidak’s multiple comparison test was used to indicate differences between CTL and DEX at specific time points. When the comparison was made between two groups, Mann–Whitney U or Student’s t-test were applied for nonparametric or parametric data, respectively. The normality of the data was checked using the Kolmogorov–Smirnov and Shaprio–Wilk tests. Statistical analyses were conducted using GraphPad Prism software version 8.4.3 (GraphPad Software Inc., San Diego, CA, USA). p-values < 0.05 indicate a significant difference.
3. Results
3.1. Offspring Born to DEX Mothers Have Reduced Body Mass at Birth
The maternal hydric intake during the last week of pregnancy was not altered by treatment with DEX (Supplementary Figure S1A). Treatment with DEX had no effect on litter size as well (Supplementary Figure S1B).
Body mass of the male offspring born to DEX mothers was lower than that of CTL at L1 (40% lower; p < 0.05). Such result validates the well-known effect of gestational glucocorticoid excess in offspring [2,4,29]. Body mass progressively increased over time in both groups, according to measurements at L8 and L21 (respectively 7.0- and 7.2-fold higher than L1; p < 0.0001). However, when compared to CTL, offspring of DEX mothers presented reduced body mass at L8 and at L21 (respectively 56% and 38% lower; p < 0.0001) (Figure 1A). Similar data were found in the female offspring born to DEX mothers at L1, L8 and L21 (respectively 48%, 63% and 41% lower than age-matched CTL; p < 0.05) (Supplementary Figure S2A).
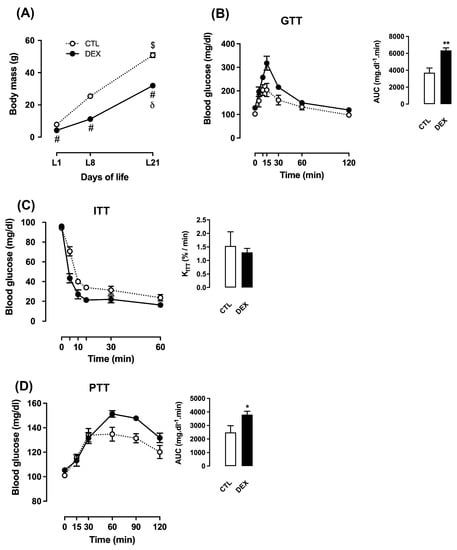
Figure 1.
Metabolic characteristics of rats born to DEX-treated mothers. Body mass of rats born to DEX-treated mothers and untreated mothers (CTL) was assessed at the first (L1), the eighth (L8) and the twenty-first (L21) days of life (A). At L21, rats were subjected to intraperitoneal glucose tolerance test (GTT) (B), insulin tolerance test (ITT) (C) and pyruvate tolerance test (PTT) (D). Data are presented as the mean ± SEM. * p < 0.05 and ** p < 0.01 vs. CTL at the same postnatal day. # p < 0.05 vs. CTL at the same time point. $ p < 0.05 vs. CTL at L1, and L8. δ p < 0.05 vs. DEX at L1 and L8. In (A), CTL and DEX (n = 10 to 15); in (B–D), CTL (n = 6), DEX (n = 8).
3.2. Offspring Born to DEX Mothers Develop Glucose Intolerance and Increased Whole-Body Conversion of Pyruvate into Glucose at Weaning
To determine the metabolic impacts caused by in utero exposure to DEX during early postnatal life, we have performed in vivo functional tests. Increased AUC values obtained from GTT demonstrated that the male offspring born to DEX mothers develop glucose intolerance at L21 (71% higher than CTL; p = 0.002) (Figure 1B). Increased AUC values from GTT were also found in female offspring born to DEX mothers (130% higher than CTL; p = 0.04) (Supplementary Figure S2B).
Glucose intolerance in the offspring of DEX mothers could not be attributed to changes in whole-body insulin sensitivity as no differences were found in the constant rate for glucose disappearance (KITT) in males (Figure 1C) and in females (Supplementary Figure S2C). On the other hand, whole-body conversion of pyruvate into glucose was significantly increased in the male offspring of DEX mothers, as revealed by higher AUC values obtained from the PTT (53% higher than CTL; p = 0.02) (Figure 1D).
3.3. Offspring of DEX Mothers Display Upregulated Gluconeogenesis-Limiting Enzymes in Liver at Weaning
With the attempt to determine if higher levels of hepatic gluconeogenesis was playing a role in increased whole-body conversion of pyruvate into glucose seen in the male offspring born to DEX mothers, we next assessed the expression and activity of limiting enzymes in this process. Our data revealed that either Pck1 mRNA expression, PEPCK protein content and PEPCK activity were upregulated in liver of the offspring born to DEX mothers at L21 (respectively 106%, 21% and 16% higher than CTL; p = 0.0006, p = 0.008 and p = 0.02) (respectively Figure 2A,C,E).
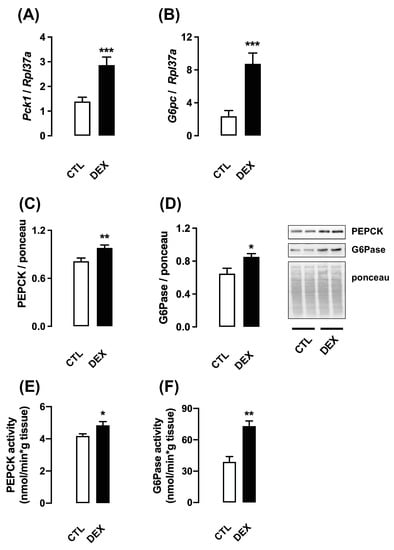
Figure 2.
Expression and activity of enzymes related to gluconeogenesis in rats born to DEX-treated mothers. Liver fragments were removed from rats born to DEX-treated mothers and untreated mothers (CTL) at the twenty-first day of life. One set of samples was processed for RNA extraction and qPCR detection of Pck1 (A) and G6pc (B). Another set of samples was processed for Western blot detection of PEPCK (C) and G6Pase (D). Band intensities were normalized by the signal from Ponceau S staining. PEPCK (E) and G6Pase (F) activities were also assessed. Data are presented as the mean ± SEM. * p < 0.05, ** p < 0.01, and *** p < 0.001 vs. CTL. CTL (n = 8), DEX (n = 8).
Similarly, the expression of G6pc mRNA, G6Pase protein content and G6Pase activity were also increased in liver of the offspring born to DEX mothers at L21 (respectively 270%, 32% and 89% higher than CTL; p = 0.0008, p = 0.019 and p = 0.001) (respectively Figure 2B,D,F).
3.4. Offspring Born to DEX Mothers Display Increased PGC-1α Content and Interaction with HNF-4α in Liver at Weaning
To understand the mechanisms underlying the increased expression PEPCK and G6Pase, we investigated PGC-1α expression and its association with the nuclear factor HNF-4α. Hepatic protein content of PGC-1α was significantly increased in the male offspring born to DEX mothers at L21 (43% higher than CTL; p = 0.006). No changes were detected in PGC-1α protein content at L1 and at L8 (Figure 3A).
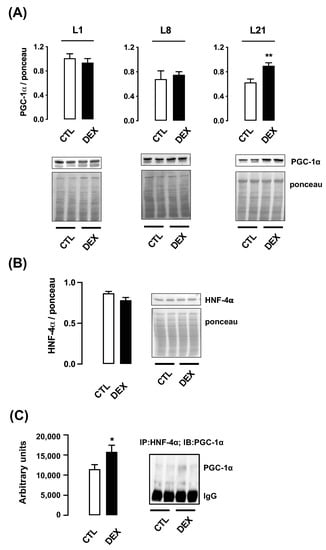
Figure 3.
Hepatic PGC-1α content and association with HNF-4α in rats born to DEX-treated mothers. Liver fragments were removed from rats born to DEX-treated mothers and untreated mothers (CTL) at the first (L1), the eighth (L8) and the twenty-first (L21) days of life and processed for Western blot detection of PGC-1α (A). Samples from L21 rats were also processed for Western blot detection of HNF-4α (B) and HNF-4α/PGC-1α association (C). Band intensities were normalized by the signal from Ponceau S staining. Data are presented as the mean ± SEM. * p < 0.05 and ** p < 0.01 vs. CTL. In (A), CTL and DEX (n = 5 to 6); in (B,C), CTL and DEX (n = 7 to 9).
3.5. Offspring of DEX Mothers Display Reduced Ppargc1a mRNA Expression and Increased DNA Methylation in Ppargc1a Promoter
In contrast to its protein content, hepatic mRNA expression of Ppargc1a was markedly increased in the male offspring of DEX mothers at L1 (2.6-fold higher than that of CTL; p < 0.0001) and presented a progressive reduction at L8 and at L21 (respectively 6.7-fold lower than L1 and 6.1-fold lower than L8; p < 0.0001 and p = 0.0019) (Figure 4A).
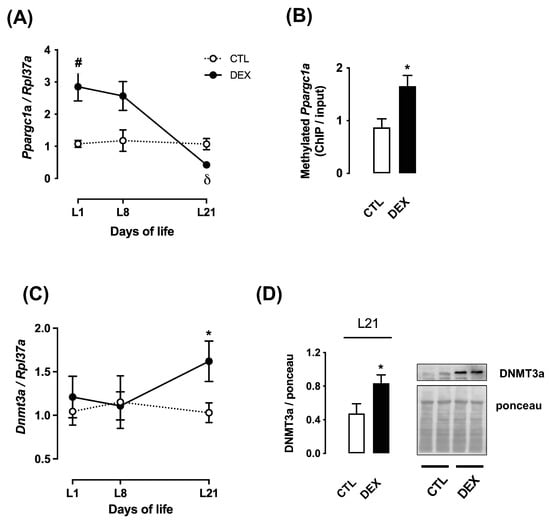
Figure 4.
Ppargc1a expression and parameters related to its methylation in rats born to DEX-treated mothers. Liver fragments were removed from rats born to DEX-treated mothers and untreated mothers (CTL) at the first (L1), the eighth (L8) and the twenty-first (L21) days of life and processed for RNA extraction and qPCR detection of Ppargc1a (A) and Dnmt3a (C) expressions. Samples from L21 rats were also processed for chromatin immunoprecipitation and quantification of the Ppargc1a promoter methylation (B) and Western blot detection of DNMT3a (D). Band intensities were normalized by the signal from Ponceau S staining. Data are presented as the mean ± SEM. # p < 0.05 vs. CTL at the same time point δ p < 0.05 vs. DEX at L1 and L8. * p < 0.05 vs. CTL. In (A), CTL and DEX (n = 6); in (B–D), CTL and DEX (n = 5).
We hypothesized that this effect would be due to DNA methylation in the Ppargc1a promoter. Accordingly, we have found that cytosine methylation at the promoter region of the Ppargc1a gene was significantly enhanced in liver of offspring of DEX mothers at L21 (90% higher than CTL; p = 0.01) (Figure 4B).
To collect further evidence to support that hepatic Ppargc1a mRNA expression is controlled by DNA methylation, we next evaluated Dnmt3a mRNA expression and DNMT3a content. No changes in the hepatic Dnmt3a mRNA expression in both groups were detected at L1 or L8. However, mRNA expression of Dnmt3a was significantly increased in the offspring of DEX mothers at L21 (57% higher than CTL; p < 0.05) (Figure 4C). The offspring of DEX mothers also exhibited increased protein content of DNMT3a at L21 (76% higher than CTL; p = 0.04) (Figure 4D).
3.6. Offspring of DEX Mothers Display Changes in Hepatic Expression of miR-29 Family Members
Since the rise in hepatic PGC-1α protein content in the offspring of DEX mothers at L21 could not be explained by corresponding changes in its mRNA, we investigated miRNAs known to modulate PGC-1α during early postnatal life. Based on a previous microarray analysis (data not shown), we have focused on the miR-29 family members.
At L1, hepatic expression of miR-29a, miR-29b and miR-29c were markedly increased in the male offspring of DEX mothers when compared to CTL (respectively 2.2-fold higher; 4.8-fold higher and 2-fold higher; p = 0.004; p < 0.0001; p = 0.001). However, a progressive decline in the expression of miR-29 family members was observed over time, reaching the lowest values at L21 (respectively 3.5-fold lower for miR-29a; 8.0-fold lower for miR-29b and 3.9-fold lower for miR-29c; p < 0.0001). Furthermore, offspring of DEX mothers at L21 presented reduced expression of miR-29c when compared to CTL (2.5-fold lower; p = 0.01) (Figure 5A–C).
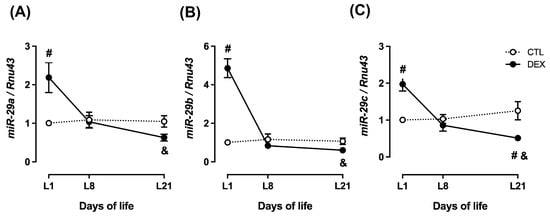
Figure 5.
Hepatic miR-29 expression in rats born to DEX-treated mothers. Liver fragments were removed from rats born to DEX-treated mothers and untreated mothers (CTL) at the first (L1), the eighth (L8) and the twenty-first (L21) days of life and processed for RNA extraction and qPCR detection of miR-29a (A), miR-29b (B) and miR-29c (C) expressions. Data are presented as the mean ± SEM. # p < 0.05 vs. CTL at the same time point. & p < 0.05 vs. DEX at L1. CTL (n = 6), DEX (n = 6).
3.7. Offspring of DEX Mothers Display Increased Hepatic Mitochondrial Biogenesis at Weaning
To evaluate whether changes in PGC-1α content were associated with biological outcomes in the male offspring of DEX mothers, we assessed key markers of mitochondrial biogenesis and mitochondrial respiratory function. Hepatic relative mitochondrial DNA (mtDNA) content was unaffected at either L1 or L8 in both groups. On the other hand, a significant increase was observed in the offspring of DEX mothers at L21 (2.8-fold higher than CTL; p < 0.0001). When an intragroup analysis was performed, offspring of DEX mothers at L21 displayed increased relative mtDNA content compared to those at L1 and L8 (respectively 4.0- and 9.7-fold higher; p < 0.0001) (Figure 6A).
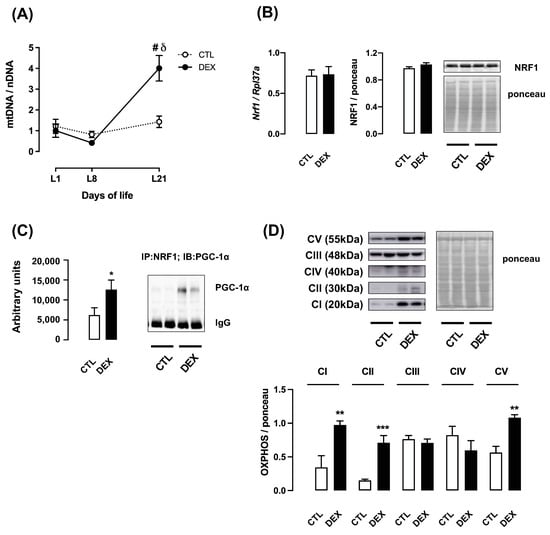
Figure 6.
Relative mtDNA content and markers of mitochondrial function in liver from rats born to DEX-treated mothers. Liver fragments were removed from rats born to DEX-treated mothers and untreated mothers (CTL) at the first (L1), the eighth (L8) and the twenty-first (L21) days of life. A set of samples was processed for total DNA extraction and mitochondrial DNA quantification (A). Samples from L21 rats were also processed for qPCR detection of Nrf1 (B) and Western blot detection of NRF1 (B). Protein extracts of samples from L21 rats were also used to assess the PGC-1α/NRF1 association (C) and the content of oxidative phosphorylation (OXPHOS) complexes (D). Band intensities were normalized by the signal from Ponceau S staining. Data are presented as the mean ± SEM. # p < 0.05 vs. CTL at the same time point; δ p < 0.05 vs. DEX at L1 and L8; * p < 0.05, ** p < 0.01 and *** p < 0.001 vs. CTL. In (A,D), CTL and DEX (n = 5 to 6); in (B,C), CTL and DEX (n = 8 to 10).
Next, we analyzed the nuclear respiratory factor 1 (NRF1), a transcription factor involved in mitochondrial DNA transcription and replication. Although no changes were observed in Nrf1 mRNA expression and NRF1 protein content at L21 (Figure 6B), the offspring of DEX mothers showed increased hepatic association of NRF1 with PGC-1α (102% higher than CTL; p = 0.04) (Figure 6C).
To assess mitochondrial respiration at L21, we evaluated the protein content of oxidative phosphorylation (OXPHOS) complexes. Our data revealed a significant increase in protein content of OXPHOS complexes I, II and V in the offspring of DEX mothers (respectively 182%, 375% and 92% higher than CTL; p = 0.0068; p = 0.0008 and p = 0.001). No difference was observed in OXPHOS complexes III and IV (Figure 6D).
Quantitative analysis of ATP in fresh tissue showed an increase in hepatic ATP content of the offspring of DEX mothers (40% higher than CTL; p = 0.016) (Figure 7A). In parallel, ADP and pyruvate content decreased (respectively 64% and 65% lower than CTL; p = 0.002) in the liver of offspring of DEX mothers (Figure 7B,C). ATP/ADP ratio was 2.9-fold higher in liver samples of DEX group.
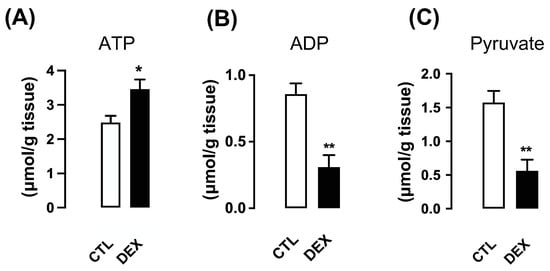
Figure 7.
Hepatic ATP, ADP and pyruvate contents in rats born to DEX-treated mothers. Liver fragments were removed from L21 rats born to DEX-treated mothers and untreated mothers (CTL), and ATP (A), ADP (B) and pyruvate (C) contents were assessed. Data are presented as the mean ± SEM. * p < 0.05 and ** p < 0.01 vs. CTL (n = 5 to 10).
3.8. Offspring of DEX Mothers Have Reduced Hepatic PPARα Protein Content Accompanied by Increased Lipid Density at Weaning
To explore additional biochemical consequences of increased PGC-1α content in the male offspring of DEX mothers, we analyzed potential changes in hepatic lipid metabolism. A major regulator of mitochondrial fatty acid β-oxidation, peroxisome proliferator-activated receptor alpha (PPARα), was previously reported to be modulated by PCG-1α [30]. Our data demonstrated that PPARα content was significantly reduced in the offspring of DEX mothers at L21 (37% lower than CTL; p < 0.0001) (Figure 8A).
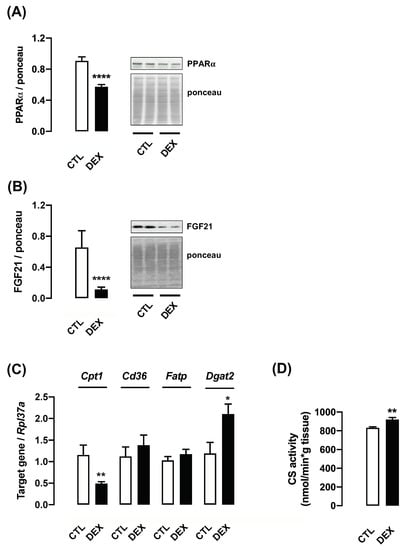
Figure 8.
Parameters associated with lipid metabolism in liver from rats born to DEX-treated mothers. Liver fragments were removed from L21 rats born to DEX-treated mothers and untreated mothers (CTL) and processed for Western blot detection of PPARα (A) and FGF21 (B). Another set of samples was processed for qPCR detection of Cpt1, Cd36, Slc27a1/Fatp and Dgat2 (C) and measurement of citrate synthase activity (D). Band intensities were normalized by the signal from Ponceau S staining. Data are presented as the mean ± SEM. * p < 0.05, ** p < 0.01 and **** p < 0.0001 vs. CTL. In (A,C), CTL and DEX (n = 11 to 13); in (B,D), CTL and DEX (n = 7 to 10).
PPARα is known to regulate the expression of the hepatokine fibroblast growth factor 21 (FGF21), which was reported to be crucial for the improvement of whole-body energy metabolism [31]. Accordingly, our results showed reduced expression of FGF21 in the offspring of DEX mothers at L21 (83% lower than CTL; p < 0.0001) (Figure 8B).
We also evaluated several putative PPARα target genes related to fatty acid oxidation (FAO) [32]. The mRNA expression of carnitine palmitoyltransferase 1 (Cpt1), a rate-limiting enzyme of FAO, was markedly reduced in liver of the offspring born to DEX mothers at L21 (2.4-fold lower than CTL; p = 0.007). Additionally, this group also displayed increased diacylglycerol acyltransferase 2 (Dgat2) mRNA expression, which represents an important step for triglyceride synthesis (1.8-fold higher than CTL; p = 0.02). Changes were not detected for Cd36, a scavenger receptor for fatty acid uptake and Slc27a1/Fat, a fatty acid transporter (Figure 8C). Citrate synthase activity was significantly increased in the liver of offspring of DEX mothers at L21 (11% higher than CTL; p = 0.003) (Figure 8D).
Male offspring of DEX mothers exhibited increased serum and hepatic TAG levels (respectively 2.4-fold higher than CTL; p = 0.0017 and 2.8-fold higher than CTL; p = 0.0001) (Figure 9A,C), and unaltered serum total cholesterol concentration (Figure 9B).
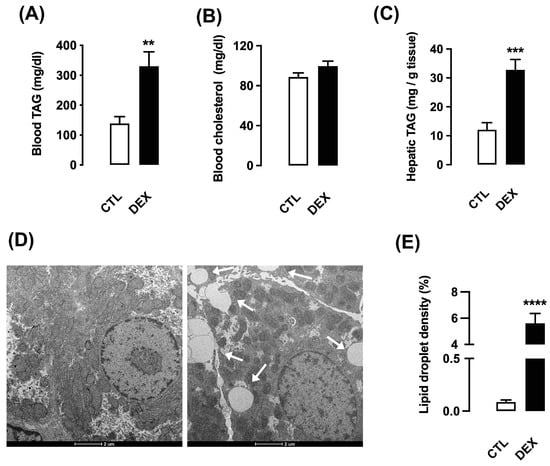
Figure 9.
Hepatic triacylglycerol content in rats born to DEX-treated mothers. Serum and liver fragments were removed from L21 rats born to DEX-treated mothers and untreated mothers (CTL) and used for measurement of circulating triacylglycerol (TAG) (A) and cholesterol (B). Hepatic levels of TAG were also assessed (C). Another set of samples of liver was processed for transmission electron microscopy and lipid droplet density analysis (D,E). The images were acquired under a final magnification at 3500×. Horizontal bar represents 2 µm. Data are presented as the mean ± SEM. ** p < 0.01, *** p < 0.001 and **** p < 0.0001 vs. CTL. In (A), CTL and DEX (n = 8 to 10); in (B), CTL and DEX (n = 6 to 9); in (C), CTL and DEX (n = 10 to 15); in (D,E), CTL and DEX (n = 4).
We also evaluated the presence of hepatic lipid droplets by electron microscopy analysis. The offspring of DEX mothers exhibited easily detectable intracellular fat accumulation when compared to CTL. The quantitative analysis revealed a marked increase in hepatic lipid density in offspring of DEX mothers at L21 (65-fold higher than CTL; p = 0.0003) (Figure 9E). Electron microscopy representative images are shown in Figure 9D.
4. Discussion
The present investigation reports novel mechanisms that explain how the exposure to DEX during pregnancy exerts hepatic metabolic programming during early postnatal life. Our findings show that rises in oxidative phosphorylation and gluconeogenesis and increased hepatic TG in the offspring of DEX mothers at L21 correlate with upregulated protein levels of PGC-1α. The present study also reveals that increased content of PGC-1α concurs with higher relative mitochondrial DNA, a key readout of mitochondrial biogenesis. Importantly, mitochondrial biogenesis has been reported to act as an adaptive response of the liver to drug-induced injury [33].
Glucose intolerance and increased hepatic PEPCK expression in the offspring of DEX mothers at the 21st day of life was previously reported in a former study [2]. Besides PEPCK, our experiments showed that antenatal exposure to DEX leads to a concomitant increase in hepatic G6Pase expression. Notably, this is the first study to further describe that the offspring of DEX mothers also have increased whole-body conversion of pyruvate into glucose at weaning. This description is of relevance because it shows that the already known upregulation of hepatic PEPCK programmed by in utero exposure to DEX is accompanied by increased conversion of pyruvate into glucose. Importantly, we previously described that insulin levels are not modulated in 21-day old offspring born to DEX-treated mothers [29]. Hence, it is plausible to assume that, rather than reduced insulin levels, transcriptional changes in the liver are the putative causes of increased gluconeogenesis presently described in the offspring born to DEX-treated mothers.
The transcriptional mechanism underlying a long-lasting increase of PEPCK in liver of DEX offspring is relatively unexplored. A direct action of DEX in the fetal liver is the triggering event for such mechanism, as increased PEPCK was still detected in rats born to DEX-treated mothers and breastfed by vehicle-treated mothers [34]. A more recent study described that the offspring of DEX mothers exhibit increased serum glucagon levels during lactation period until the 21st day of life [29]. This data may help to explain the already described upregulation of hepatic PEPCK and G6Pase, as these enzymes are classically known as being stimulated by glucagon.
Exposure to DEX during late pregnancy has been reported to increase hepatic expression of HNF-4α during the rat fetal life and to increase glucocorticoid receptor (GR) expression at the 5th day after birth [2,35]. In this context, our findings demonstrating increased PGC-1α association with HNF-4α in liver of the offspring of DEX mothers at L21 is of pivotal relevance, as this nuclear co-activator was described to boost both GR and HNF-4α transcriptional activity over the Pck1 gene [36]. Importantly, PGC-1α also plays a determinant role in modulating HNF-4α transcriptional activity over G6pc gene in liver cells [37].
We also found that epigenetic regulation of hepatic Ppargc1a promoter was associated with a decreased expression of this gene in the prepubertal offspring of DEX mothers. A reduction in Ppargc1a mRNA expression was already noted in liver of adult rats born to mothers either exposed to DEX or subjected to prenatal stress [38,39]. The present investigation reveals that this consistent suppression in hepatic Ppargc1a mRNA seen in rats exposed to antenatal DEX correlates with an increase in Ppargc1a promoter cytosine methylation. This mechanism has already been reported in skeletal muscle, where cytosine methylation of the Ppargc1a promoter within non-CpG islands was described to repress PGC-1α expression [11]. Methylation of the Ppargc1a promoter was also reported to be acutely inhibited by exercise, leading to a short term increased in PGC-1α expression in skeletal muscle [12]. However, these interesting data contrast with the presently found high levels of protein PGC-1α in the liver the offspring born to DEX mothers.
Hence, we hypothesized that an additional post-transcriptional mechanism would prevail in the liver the offspring of DEX mothers so that an increase of PGC-1α protein content occur at weaning, despite the reduced Ppargc1a transcription. This hypothesis revealed itself to be plausible, considering that we found a progressive decrease in miR-29a-c expression in liver of the offspring of DEX mothers, reaching minimal values on the 21st day of life.
Supporting this proposition, it has previously been demonstrated that PGC-1α is a validated target for miR-29a-c. Overexpression of miR-29a-c in liver cells was able to reduce PGC-1α protein content without affecting Ppargc1a mRNA levels. Moreover, overexpression of miR-29a-c reduced G6Pase expression in liver cells and pyruvate conversion into glucose by obese mice [13]. Besides its regulation by miR-29a-c, it must be stressed that PGC-1α content is degraded by the ubiquitin proteasome system [40]. Such mechanism is increased in the adipose tissue of obese individuals [41]. Therefore, current experiments do not rule out that changes in PGC-1α degradation might be taking place in the liver rats born to DEX-treated mothers.
Aside from its role in the control of gluconeogenesis, PGC-1α is also recognized as a crucial regulator of mitochondrial biogenesis [42]. In accordance with high levels of PGC-1α, our results evidenced the enhanced relative content of mitochondrial DNA and respiration markers in liver of the offspring born to DEX mothers.
In agreement with the higher mitochondrial DNA content, we observed an increase in both NRF1-PGC-1α association and the expression of key enzymes of mitochondrial respiratory chain. In skeletal muscle, it was reported that PGC-1α binds to and coactivates the transcription factor NRF1, leading to mitochondrial replication and transcription [43]. Mitochondrial ATP synthase (Complex V) and ATP content are also increased in the liver of DEX mother offspring’s, which probably cope with maximal coupled respiration. Indeed, the higher ATP/ADP ratio (essential to inhibit glycolysis) and the lower pyruvate content (indicative of mitochondrial pyruvate metabolism) suggest that hepatocytes of the offspring born to DEX mothers display a prominent oxidative metabolism at L21. This higher oxidative rate seems to be concurrent with reduced glycolysis (as evidenced by higher ATP/ADP ratio), which might contribute to the increased gluconeogenesis flux seen in the liver of offspring born to DEX mothers at L21.
In addition to the high rate of oxidative phosphorylation and gluconeogenesis, the present study suggests reduced fatty acid oxidation in liver of offspring of DEX mothers at L21 (increased hepatic TG and reduced CPT-1 activity). Such metabolic adaptation is likely to be linked to decreased levels of PPARα, a factor reported to promote transcriptional regulation of pivotal genes involved in hepatic lipid metabolism [10]. In accordance with the present data, adult rats born to DEX-treated mothers were reported to display increased hepatic triglycerides accumulation when exposed to long-term fasting, liquid fructose or high fat diet [4,14,38,44]. On the other hand, our data on increased citrate synthase activity favor the proposition that increased fatty acid syntheses may also contribute to hepatic steatosis in the progeny exposed to DEX in utero.
It is still unclear if there is a postnatal window during which specific interventions can be employed to revert/prevent the hepatic steatosis programmed by antenatal DEX. Noteworthy, postnatal treatment with leptin was described to prevent hepatic steatosis seen in the offspring born to mothers exposed to caloric restriction, a model known to increase endogenous glucocorticoid production [45].
The present data also reveal that PPARα may underlie a mechanism governing the content of the metabolic regulator FGF21 in the liver of the offspring of DEX mothers at the moment of weaning. FGF21 is a well-characterized target of PPARα and has evident antidiabetic and triglyceride-lowering effects in animal models of diabetes and obesity [46].
5. Conclusions
Data presented herein reveal inflexible energy metabolism in liver of short-term fasted prepubertal rats born to DEX mothers. At this age, the hepatic metabolism of rats exposed to DEX in utero is unable to increase FFA oxidation while enhanced mitochondrial pyruvate oxidative phosphorylation seems to meet energy demand. Changes in transcription machinery (i.e., increased PGC-1α content and association with HNF-4α and NRF1 and reduced PPARα) are concordant with this metabolic phenotype and likely to contribute TG accumulation and increased gluconeogenesis at the age of weaning.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/livers1040016/s1, Figure S1: Water intake of untreated (CTL) and treated (DEX) pregnant rats at the 3rd week of pregnancy (A) (n = 12). Litter size of CTL and DEX mothers (n = 12 to 18). Figure S2: Metabolic characteristics of female rats born to DEX-treated mothers. Body mass of rats born to DEX-treated mothers and untreated mothers (CTL) was assessed at the first (L1), the eighth (L8) and the twenty-first (L21) days of life (A). At L21, rats were subjected to intraperitoneal glucose tolerance test (GTT) (B) and insulin tolerance test (ITT) (C). Data are presented as the mean ± SEM. * p < 0.05 vs. CTL at the same postnatal day. # p < 0.05 vs. CTL at the same time point (n = 5).
Author Contributions
C.V.C. and S.B. developed the concept, designed experiments, and interpreted the data. C.V.C., C.J.T., T.B.P., A.R.C., G.M.M., A.G.A., L.C.P., F.S.S., M.M.O., L.d.S.A., G.A.P. and D.S.S.F.G. conducted experiments and collected data. C.V.C., C.J.T., S.B., L.R.S. and G.F.A. analyzed data. S.B. and G.F.A. prepared the manuscript. C.V.C., C.J.T., G.M.M., L.R.S. and D.S.S.F.G. made revisions to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2013/07607-8, 2014/02977-4, 2016/22605-0, 2016/23008-5, 2019/03196-0; 2020/06397-3, 2020/09717-9), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES—Finance Code 001).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Brazilian College for Animal Experimentation (COBEA) and were approved by the Ethics Committee on Animal Use at the Institute of Biomedical Sciences, University of Sao Paulo, Brazil (Certificate No. 5367250619).
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw data is available upon request.
Acknowledgments
The authors are grateful for the technical assistance from José R. de Mendonça, Renata S. Mendes and Fabio T. Sato.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Seckl, J.R.; Meaney, M.J. Glucocorticoid programming. Ann. N. Y. Acad. Sci. 2004, 1032, 63–84. [Google Scholar] [CrossRef]
- Nyirenda, M.J.; Lindsay, R.S.; Kenyon, C.J.; Burchell, A.; Seckl, J.R. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J. Clin. Invest. 1998, 101, 2174–2181. [Google Scholar] [CrossRef]
- Cleasby, M.E.; Kelly, P.A.; Walker, B.R.; Seckl, J.R. Programming of rat muscle and fat metabolism by in utero overexposure to glucocorticoids. Endocrinology 2003, 144, 999–1007. [Google Scholar] [CrossRef]
- Pantaleão, L.C.; Murata, G.; Teixeira, C.J.; Payolla, T.B.; Santos-Silva, J.C.; Duque-Guimaraes, D.E.; Sodré, F.S.; Lellis-Santos, C.; Vieira, J.C.; de Souza, D.N.; et al. Prolonged fasting elicits increased hepatic triglyceride accumulation in rats born to dexamethasone-treated mothers. Sci. Rep. 2017, 7, 10367. [Google Scholar] [CrossRef]
- Buhl, E.S.; Neschen, S.; Yonemitsu, S.; Rossbacher, J.; Zhang, D.; Morino, K.; Flyvbjerg, A.; Perret, P.; Samuel, V.; Kim, J.; et al. Increased hypothalamic-pituitary-adrenal axis activity and hepatic insulin resistance in low-birth-weight rats. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1451–E1458. [Google Scholar] [CrossRef]
- O’Brien, K.; Sekimoto, H.; Boney, C.; Malee, M. Effect of fetal dexamethasone exposure on the development of adult insulin sensitivity in a rat model. J. Matern. Fetal Neonatal Med. 2008, 21, 623–628. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef]
- Barthel, A.; Schmoll, D. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E685–E692. [Google Scholar] [CrossRef]
- Dankel, S.N.; Hoang, T.; Flågeng, M.H.; Sagen, J.V.; Mellgren, G. cAMP-mediated regulation of HNF-4alpha depends on the level of coactivator PGC-1alpha. Biochim. Biophys. Acta. 2010, 1803, 1013–1019. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Barrès, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Liu, C.; Qiao, A.; Cui, Y.; Zhang, H.; Cui, A.; Zhang, S.; Yang, Y.; Xiao, X.; Chen, Y.; et al. MicroRNA-29a-c decrease fasting blood glucose levels by negatively regulating hepatic gluconeogenesis. J. Hepatol. 2013, 58, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Payolla, T.B.; Teixeira, C.J.; Sato, F.T.; Murata, G.M.; Zonta, G.A.; Sodré, F.S.; Campos, C.V.; Mesquita, F.N.; Anhê, G.F.; Bordin, S. In Utero Dexamethasone Exposure Exacerbates Hepatic Steatosis in Rats That Consume Fructose During Adulthood. Nutrients 2019, 11, 2114. [Google Scholar] [CrossRef]
- Vicente, J.M.; Teixeira, C.J.; Santos-Silva, J.C.; de Souza, D.N.; Tobar, N.; Furtuoso, F.S.; Adabo, I.G.; Sodré, F.S.; Murata, G.; Bordin, S.; et al. The absence of lactation after pregnancy induces long-term lipid accumulation in maternal liver of mice. Life Sci. 2019, 217, 261–270. [Google Scholar] [CrossRef]
- Opie, L.H.; Newsholme, E.A. The activities of fructose 1,6-diphosphatase, phosphofructokinase and phosphoenolpyruvate carboxykinase in white muscle and red muscle. Biochem. J. 1967, 103, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Zammit, V.A.; Newsholme, E.A. The maximum activities of hexokinase, phosphorylase, phosphofructokinase, glycerol phosphate dehydrogenases, lactate dehydrogenase, octopine dehydrogenase, phosphoenolpyruvate carboxykinase, nucleoside diphosphatekinase, glutamate-oxaloacetate transaminase and arginine kinase in relation to carbohydrate utilization in muscles from marine invertebrates. Biochem. J. 1976, 160, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Alp, P.R.; Newsholme, E.A.; Zammit, V.A. Activities of citrate synthase and NAD+-linked and NADP+-linked isocitrate dehydrogenase in muscle from vertebrates and invertebrates. Biochem. J. 1976, 154, 689–700. [Google Scholar] [CrossRef]
- Gomes, P.R.; Graciano, M.F.; Pantaleão, L.C.; Rennó, A.L.; Rodrigues, S.C.; Velloso, L.A.; Latorraca, M.Q.; Carpinelli, A.R.; Anhê, G.F.; Bordin, S. Long-term disruption of maternal glucose homeostasis induced by prenatal glucocorticoid treatment correlates with miR-29 upregulation. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E109–E120. [Google Scholar] [CrossRef]
- Anhê, G.F.; Hirabara, S.M.; Turrer, T.C.; Caperuto, L.C.; Anhê, F.F.; Ribeiro, L.M.; Marçal, A.C.; Carvalho, C.R.; Curi, R.; Machado, U.F.; et al. Postpartum glycemic homeostasis in early lactating rats is accompanied by transient and specific increase of soleus insulin response through IRS2/AKT pathway. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R2225–R2233. [Google Scholar] [CrossRef][Green Version]
- Romero-Calvo, I.; Ocón, B.; Martínez-Moya p Suárez, M.D.; Zarzuelo, A.; Martínez-Augustin, O.; de Medina, F.S. Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal. Biochem. 2010, 401, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Strauss, W.M. Preparation of genomic DNA from mammalian tissue. In Current Protocols in Molecular Biology; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; John Wiley & Sons: New York, NY, USA, 1995; pp. 2–13. [Google Scholar] [CrossRef]
- Rooney, J.P.; Ryde, I.T.; Sanders, L.H.; Howlett, E.H.; Colton, M.D.; Germ, K.E.; Mayer, G.D.; Greenamyre, J.T.; Meyer, J.N. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 2015, 1241, 23–38. [Google Scholar] [CrossRef]
- Lamprecht, W.; Trautschold, I. Determination with Hexokinase and Glucose-6-phosphate Dehydrogenase. In Methods of Enzymatic Analyses, 2nd ed.; Bergmeyer, H.U., Ed.; Academic Press: New York, NY, USA, 1965; pp. 543–551. [Google Scholar]
- Adam, H. Adenosine-5′-diphosphate and adenosine-5′-monophosphate. In Methods of Enzymatic Analyses, 2nd ed.; Bergmeyer, H.U., Ed.; Academic Press: New York, NY, USA, 1965; pp. 573–577. [Google Scholar]
- Neville, J.F., Jr.; Gelder, R.L. Modified enzymatic methods for the determination of L-(+)-lactic and pyruvic acids in blood. Am. J. Clin. Pathol. 1971, 55, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Santos-Silva, J.C.; da Silva, P.M.R.; de Souza, D.N.; Teixeira, C.J.; Bordin, S.; Anhê, G.F. In utero exposure to dexamethasone programs the development of the pancreatic β- and α-cells during early postnatal life. Life Sci. 2020, 255, 117810. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 2000, 1868–1876. [Google Scholar] [CrossRef]
- Goto, T.; Hirata, M.; Aoki, Y.; Iwase, M.; Takahashi, H.; Kim, M.; Li, Y.; Jheng, H.F.; Nomura, W.; Takahashi, N.; et al. The hepatokine FGF21 is crucial for peroxisome proliferator-activated receptor-α agonist-induced amelioration of metabolic disorders in obese mice. J. Biol. Chem. 2017, 292, 9175–9190. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef]
- Ramachandran, A.; Umbaugh, D.S.; Jaeschke, H. Mitochondrial Dynamics in Drug-Induced Liver Injury. Livers 2021, 1, 102–115. [Google Scholar] [CrossRef]
- Nyirenda, M.J.; Welberg, L.A.; Seckl, J.R. Programming hyperglycaemia in the rat through prenatal exposure to glucocorticoids-fetal effect or maternal influence? J. Endocrinol. 2001, 170, 653–660. [Google Scholar] [CrossRef]
- Dean, S.; Tang, J.I.; Seckl, J.R.; Nyirenda, M.J. Developmental and tissue-specific regulation of hepatocyte nuclear factor 4-alpha (HNF4-alpha) isoforms in rodents. Gene Expr. 2010, 14, 337–344. [Google Scholar] [CrossRef]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef]
- Boustead, J.N.; Stadelmaier, B.T.; Eeds, A.M.; Wiebe, P.O.; Svitek, C.A.; Oeser, J.K.; O’Brien, R.M. Hepatocyte nuclear factor-4 alpha mediates the stimulatory effect of peroxisome proliferator-activated receptor gamma co-activator-1 alpha (PGC-1 alpha) on glucose-6-phosphatase catalytic subunit gene transcription in H4IIE cells. Biochem. J. 2003, 369, 17–22. [Google Scholar] [CrossRef]
- Drake, A.J.; Raubenheimer, P.J.; Kerrigan, D.; McInnes, K.J.; Seckl, J.R.; Walker, B.R. Prenatal dexamethasone programs expression of genes in liver and adipose tissue and increased hepatic lipid accumulation but not obesity on a high-fat diet. Endocrinology 2010, 151, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Brunton, P.J.; Sullivan, K.M.; Kerrigan, D.; Russell, J.A.; Seckl, J.R.; Drake, A.J. Sex-specific effects of prenatal stress on glucose homoeostasis and peripheral metabolism in rats. J. Endocrinol. 2013, 217, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Trausch-Azar, J.; Leone, T.C.; Kelly, D.P.; Schwartz, A.L. Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1{alpha} via the N-terminal pathway. J. Biol. Chem. 2010, 285, 40192–40200. [Google Scholar] [CrossRef] [PubMed]
- Bombassaro, B.; Ignacio-Souza, L.M.; Nunez, C.E.; Razolli, D.S.; Pedro, R.M.; Coope, A.; Araujo, E.P.; Chaim, E.A.; Velloso, L.A. A20 deubiquitinase controls PGC-1α expression in the adipose tissue. Lipids Health Dis. 2018, 17, 90. [Google Scholar] [CrossRef]
- Uldry, M.; Yang, W.; St-Pierre, J.; Lin, J.; Seale, P.; Spiegelman, B.M. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell. Metab. 2006, 3, 333–341. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Carbone, D.L.; Zuloaga, D.G.; Hiroi, R.; Foradori, C.D.; Legare, M.E.; Handa, R.J. Prenatal dexamethasone exposure potentiates diet-induced hepatosteatosis and decreases plasma IGF-I in a sex-specific fashion. Endocrinology 2012, 153, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Szostaczuk, N.; Priego, T.; Palou, M.; Palou, A.; Picó, C. Oral leptin supplementation throughout lactation in rats prevents later metabolic alterations caused by gestational calorie restriction. Int. J. Obes. Lond. 2017, 41, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Lundåsen, T.; Hunt, M.C.; Nilsson, L.M.; Sanyal, S.; Angelin, B.; Alexson, S.E.; Rudling, M. PPARalpha is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).