
The COP9 Signalosome Variant CSNCSN7A Stabilizes the Deubiquitylating Enzyme CYLD Impeding Hepatic Steatosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Silencing of CSN Subunits Reduces PPAR-γ Expression in Liver Cells
2.2. The Ub-Specific Protease CYLD Preferentially Binds to CSNCSN7A Variant
2.3. Reciprocal Expression of CYLD and CSN7A as Well as CSN7B
2.4. Overexpression of Flag-CSN7A in Liver Cells Increases CYLD Expression
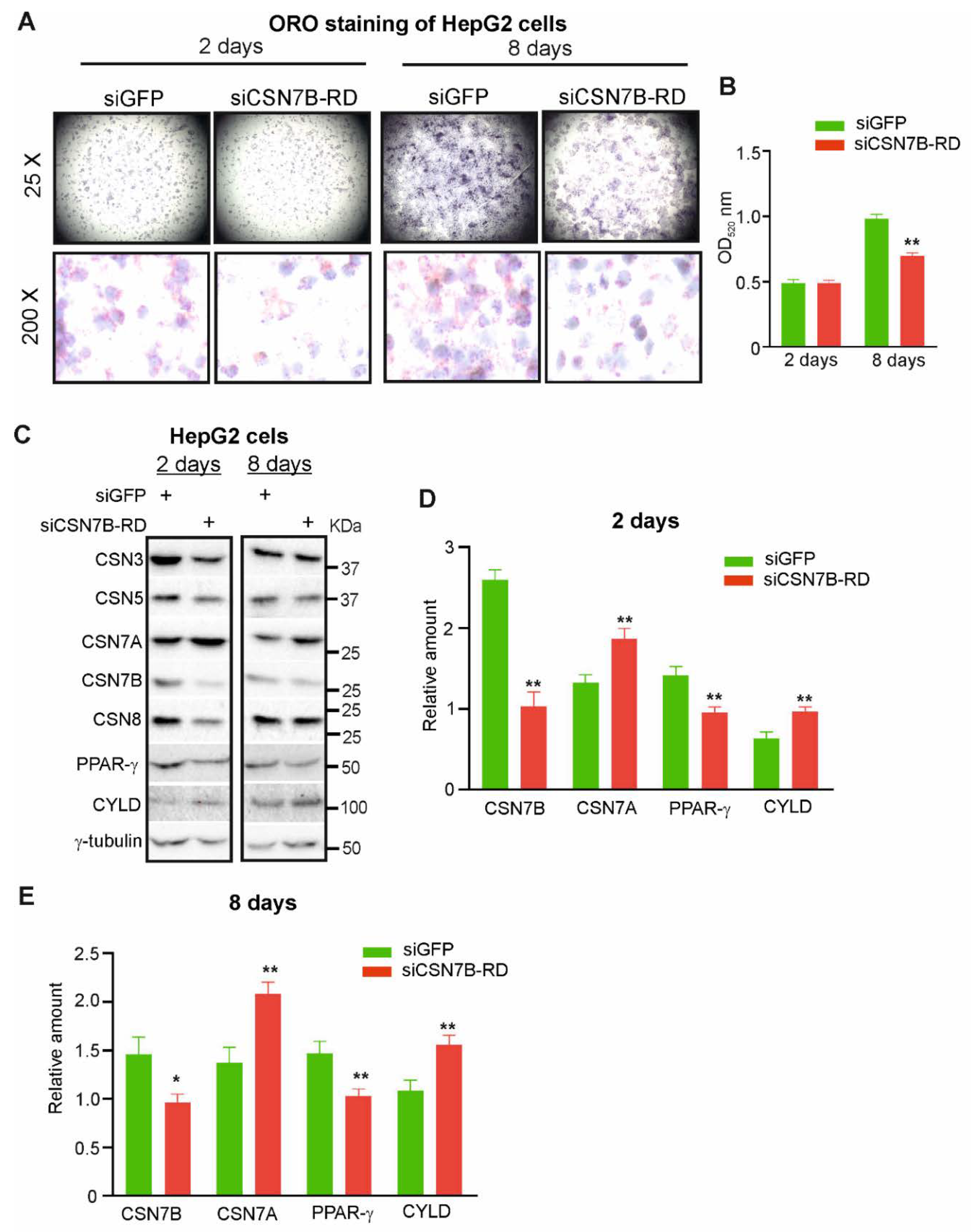
2.5. Downregulation of CSN7B Induces CYLD and Hinders Steatosis in HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Human Liver Histology
4.2. Preparation of Human Hepatocytes and Cell Culture
4.3. PA-Induced Steatosis in Cultured Hepatocytes
4.4. Knockouts of CSN7A or CSN7B and Downregulation of CSN Subunits and CYLD Using siRNA
4.5. Western Blot Analysis
4.6. Flag-Pulldowns
4.7. ORO Staining and Quantification of ORO
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the american association for the study of liver diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. Nafld, nash and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Osna, N.A.; Kharbanda, K.K. Treatment options for alcoholic and non-alcoholic fatty liver disease: A review. World J. Gastroenterol. 2017, 23, 6549–6570. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Guo, G.L. Role of fxr in liver inflammation during nonalcoholic steatohepatitis. Curr. Pharmacol. Rep. 2017, 3, 92–100. [Google Scholar] [CrossRef]
- Maratos-Flier, E. Fatty liver and fgf21 physiology. Exp. Cell Res. 2017, 360, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (nafld)-pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Ruiz, I.; Medina, M.A.; Martinez-Poveda, B. From food to genes: Transcriptional regulation of metabolism by lipids and carbohydrates. Nutrients 2021, 13, 1513. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Ni, W.; Lin, S.; Bian, S.; Zheng, W.; Qu, L.; Fan, Y.; Lu, C.; Xiao, M.; Zhou, P. Usp7 mediates pathological hepatic de novo lipogenesis through promoting stabilization and transcription of znf638. Cell Death Dis. 2020, 11, 843. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992, 61, 761–807. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. Ring domain e3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Dubiel, W.; Dubiel, D.; Wolf, D.A.; Naumann, M. Cullin 3-based ubiquitin ligases as master regulators of mammalian cell differentiation. Trends Biochem. Sci. 2018, 43, 95–107. [Google Scholar] [CrossRef]
- Duda, D.M.; Borg, L.A.; Scott, D.C.; Hunt, H.W.; Hammel, M.; Schulman, B.A. Structural insights into nedd8 activation of cullin-ring ligases: Conformational control of conjugation. Cell 2008, 134, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Deshaies, R.J. Multimodal activation of the ubiquitin ligase scf by nedd8 conjugation. Mol. Cell 2008, 32, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, W.; Nhan, T.; Toth, J.I.; Petroski, M.D.; Wolf, D.A. Cand1 controls in vivo dynamics of the cullin 1-ring ubiquitin ligase repertoire. Nat. Commun. 2013, 4, 1642. [Google Scholar] [CrossRef]
- Zemla, A.; Thomas, Y.; Kedziora, S.; Knebel, A.; Wood, N.T.; Rabut, G.; Kurz, T. Csn- and cand1-dependent remodelling of the budding yeast scf complex. Nat. Commun. 2013, 4, 1641. [Google Scholar] [CrossRef]
- Pierce, N.W.; Lee, J.E.; Liu, X.; Sweredoski, M.J.; Graham, R.L.; Larimore, E.A.; Rome, M.; Zheng, N.; Clurman, B.E.; Hess, S.; et al. Cand1 promotes assembly of new scf complexes through dynamic exchange of f box proteins. Cell 2013, 153, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Dubiel, D.; Gierisch, M.E.; Huang, X.; Dubiel, W.; Naumann, M. Cand1-dependent control of cullin 1-ring ub ligases is essential for adipogenesis. Biochim. Biophys. Acta 2013, 1833, 1078–1084. [Google Scholar] [CrossRef]
- Reitsma, J.M.; Liu, X.; Reichermeier, K.M.; Moradian, A.; Sweredoski, M.J.; Hess, S.; Deshaies, R.J. Composition and regulation of the cellular repertoire of scf ubiquitin ligases. Cell 2017, 171, 1326–1339 e1314. [Google Scholar] [CrossRef]
- Liu, X.; Reitsma, J.M.; Mamrosh, J.L.; Zhang, Y.; Straube, R.; Deshaies, R.J. Cand1-mediated adaptive exchange mechanism enables variation in f-box protein expression. Mol. Cell 2018, 69, 773–786 e776. [Google Scholar] [CrossRef]
- Dubiel, D.; Ordemann, J.; Pratschke, J.; Dubiel, W.; Naumann, M. Cand1 exchange factor promotes keap1 integration into cullin 3-ring ubiquitin ligase during adipogenesis. Int. J. Biochem. Cell Biol. 2015, 66, 95–100. [Google Scholar] [CrossRef]
- Straube, R.; Shah, M.; Flockerzi, D.; Wolf, D.A. Trade-off and flexibility in the dynamic regulation of the cullin-ring ubiquitin ligase repertoire. PLoS Comput. Biol. 2017, 13, e1005869. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Bucher, P. The pci domain: A common theme in three multiprotein complexes. Trends Biochem. Sci. 1998, 23, 204–205. [Google Scholar] [CrossRef]
- Lingaraju, G.M.; Bunker, R.D.; Cavadini, S.; Hess, D.; Hassiepen, U.; Renatus, M.; Fischer, E.S.; Thoma, N.H. Crystal structure of the human cop9 signalosome. Nature 2014, 512, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Cavadini, S.; Fischer, E.S.; Bunker, R.D.; Potenza, A.; Lingaraju, G.M.; Goldie, K.N.; Mohamed, W.I.; Faty, M.; Petzold, G.; Beckwith, R.E.; et al. Cullin-ring ubiquitin e3 ligase regulation by the cop9 signalosome. Nature 2016, 531, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Cope, G.A.; Suh, G.S.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of predicted metalloprotease motif of jab1/csn5 in cleavage of nedd8 from cul1. Science 2002, 298, 608–611. [Google Scholar] [CrossRef]
- Huang, X.; Ordemann, J.; Pratschke, J.; Dubiel, W. Overexpression of cop9 signalosome subunits, csn7a and csn7b, exerts different effects on adipogenic differentiation. FEBS Open Bio 2016, 6, 1102–1112. [Google Scholar] [CrossRef]
- Wang, J.; Dubiel, D.; Wu, Y.; Cheng, Y.; Wolf, D.A.; Dubiel, W. Csn7b defines a variant cop9 signalosome complex with distinct function in DNA damage response. Cell Rep. 2021, 34, 108662. [Google Scholar] [CrossRef]
- Dubiel, W.; Chaithongyot, S.; Dubiel, D.; Naumann, M. The cop9 signalosome: A multi-dub complex. Biomolecules 2020, 10, 1082. [Google Scholar] [CrossRef]
- Ji, Y.X.; Huang, Z.; Yang, X.; Wang, X.; Zhao, L.P.; Wang, P.X.; Zhang, X.J.; Alves-Bezerra, M.; Cai, L.; Zhang, P.; et al. The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat. Med. 2018, 24, 213–223. [Google Scholar] [CrossRef]
- Komander, D.; Lord, C.J.; Scheel, H.; Swift, S.; Hofmann, K.; Ashworth, A.; Barford, D. The structure of the cyld usp domain explains its specificity for lys63-linked polyubiquitin and reveals a b box module. Mol. Cell 2008, 29, 451–464. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. Cyld is a deubiquitinating enzyme that negatively regulates nf-kappab activation by tnfr family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Hellerbrand, C.; Massoumi, R. Cylindromatosis--a protective molecule against liver diseases. Med. Res. Rev. 2016, 36, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fukushima, H.; North, B.J.; Nagaoka, Y.; Nagashima, K.; Deng, F.; Okabe, K.; Inuzuka, H.; Wei, W. Scfbeta-trcp regulates osteoclastogenesis via promoting cyld ubiquitination. Oncotarget 2014, 5, 4211–4221. [Google Scholar] [CrossRef] [PubMed]
- Dubiel, D.; Bintig, W.; Kahne, T.; Dubiel, W.; Naumann, M. Cul3 neddylation is crucial for gradual lipid droplet formation during adipogenesis. Biochim. Biophys. Acta 2017, 1864, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.Q.; Li, X.Y.; Wang, L.; Feng, Z.L.; Li, X.F.; Wen, Y.F.; Han, J.X. Palmitate induces fat accumulation by activating c/ebpbeta-mediated g0s2 expression in hepg2 cells. World J. Gastroenterol. 2017, 23, 7705–7715. [Google Scholar] [CrossRef]
- Labrie, M.; Lalonde, S.; Najyb, O.; Thiery, M.; Daneault, C.; Des Rosiers, C.; Rassart, E.; Mounier, C. Apolipoprotein d transgenic mice develop hepatic steatosis through activation of ppargamma and fatty acid uptake. PLoS ONE 2015, 10, e0130230. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to nash pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef]
- Huang, X.; Ordemann, J.; Muller, J.M.; Dubiel, W. The cop9 signalosome, cullin 3 and keap1 supercomplex regulates chop stability and adipogenesis. Biol. Open 2012, 1, 705–710. [Google Scholar] [CrossRef]
- Schlierf, A.; Altmann, E.; Quancard, J.; Jefferson, A.B.; Assenberg, R.; Renatus, M.; Jones, M.; Hassiepen, U.; Schaefer, M.; Kiffe, M.; et al. Targeted inhibition of the cop9 signalosome for treatment of cancer. Nat. Commun. 2016, 7, 13166. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Sintchak, M.D.; Gavin, J.M.; Liao, H.; Bruzzese, F.J.; Bump, N.J.; Soucy, T.A.; Milhollen, M.A.; Yang, X.; Burkhardt, A.L.; et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: The nedd8 e1 inhibitor mln4924 forms a nedd8-amp mimetic in situ. Mol. Cell 2010, 37, 102–111. [Google Scholar] [CrossRef]
- Jin, D.; Li, B.; Deng, X.W.; Wei, N. Plant cop9 signalosome subunit 5, csn5. Plant Sci. 2014, 224C, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hetfeld, B.K.; Helfrich, A.; Kapelari, B.; Scheel, H.; Hofmann, K.; Guterman, A.; Glickman, M.; Schade, R.; Kloetzel, P.M.; Dubiel, W. The zinc finger of the csn-associated deubiquitinating enzyme usp15 is essential to rescue the e3 ligase rbx1. Curr. Biol. 2005, 15, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Naumann, M. Csn-associated usp48 confers stability to nuclear nf-kappab/rela by trimming k48-linked ub-chains. Biochim. Biophys. Acta 2015, 1853, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Li, H. Innate immune regulatory networks in hepatic lipid metabolism. J. Mol. Med. 2019, 97, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Chen, M.M.; Cai, J.J.; Yu, Y.; She, Z.G.; Li, H. Current and emerging approaches for nonalcoholic steatohepatitis treatment. Gene Expr. 2019, 19, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Horner, R.; Gassner, J.; Kluge, M.; Tang, P.; Lippert, S.; Hillebrandt, K.H.; Moosburner, S.; Reutzel-Selke, A.; Pratschke, J.; Sauer, I.M.; et al. Impact of percoll purification on isolation of primary human hepatocytes. Sci. Rep. 2019, 9, 6542. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Dubiel, D.; Dubiel, W. The COP9 Signalosome Variant CSNCSN7A Stabilizes the Deubiquitylating Enzyme CYLD Impeding Hepatic Steatosis. Livers 2021, 1, 116-131. https://doi.org/10.3390/livers1030011
Huang X, Dubiel D, Dubiel W. The COP9 Signalosome Variant CSNCSN7A Stabilizes the Deubiquitylating Enzyme CYLD Impeding Hepatic Steatosis. Livers. 2021; 1(3):116-131. https://doi.org/10.3390/livers1030011
Chicago/Turabian StyleHuang, Xiaohua, Dawadschargal Dubiel, and Wolfgang Dubiel. 2021. "The COP9 Signalosome Variant CSNCSN7A Stabilizes the Deubiquitylating Enzyme CYLD Impeding Hepatic Steatosis" Livers 1, no. 3: 116-131. https://doi.org/10.3390/livers1030011
APA StyleHuang, X., Dubiel, D., & Dubiel, W. (2021). The COP9 Signalosome Variant CSNCSN7A Stabilizes the Deubiquitylating Enzyme CYLD Impeding Hepatic Steatosis. Livers, 1(3), 116-131. https://doi.org/10.3390/livers1030011