
Mitochondrial Dynamics in Drug-Induced Liver Injury




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Morphology
3. Mitochondrial Biogenesis
4. Mitochondrial Dynamics
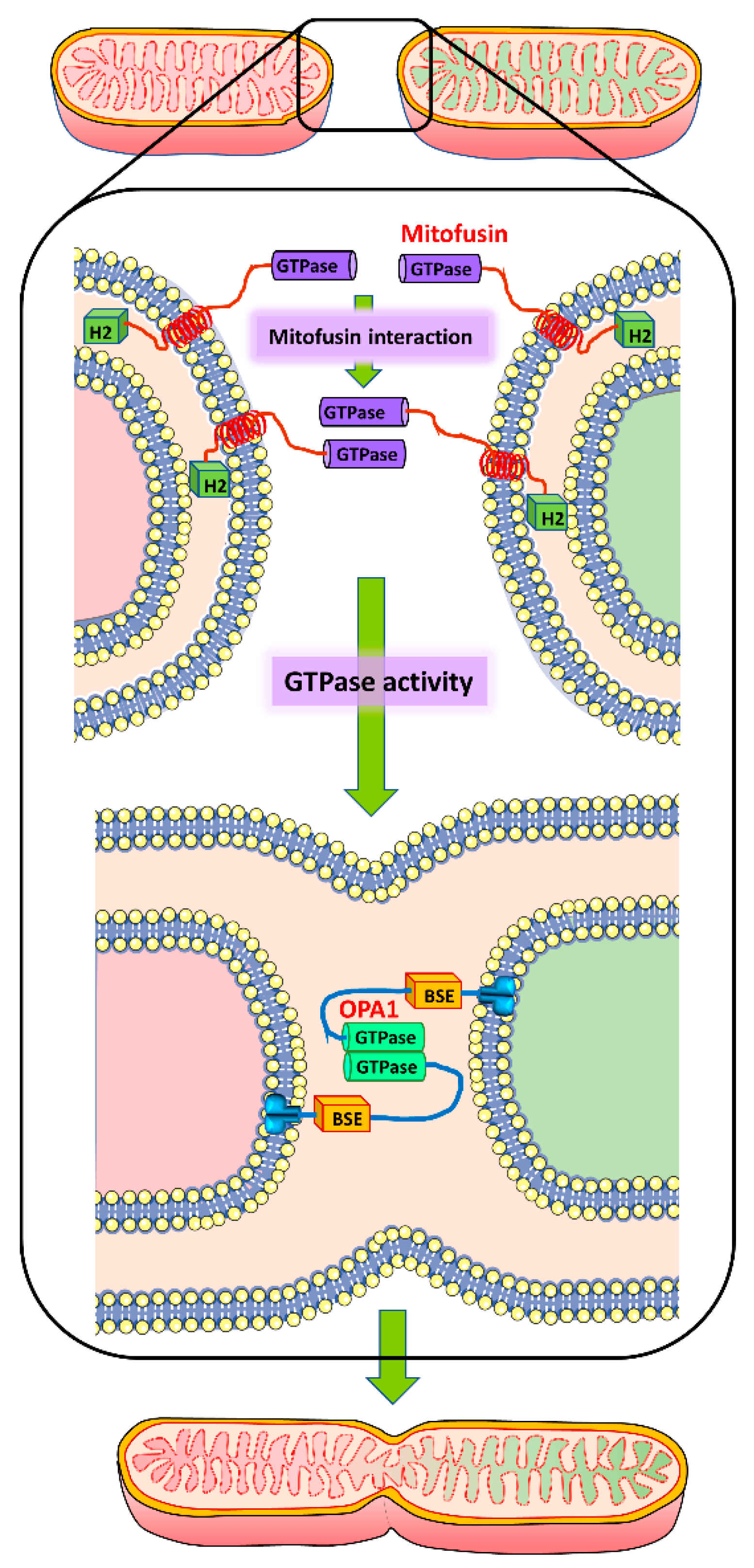
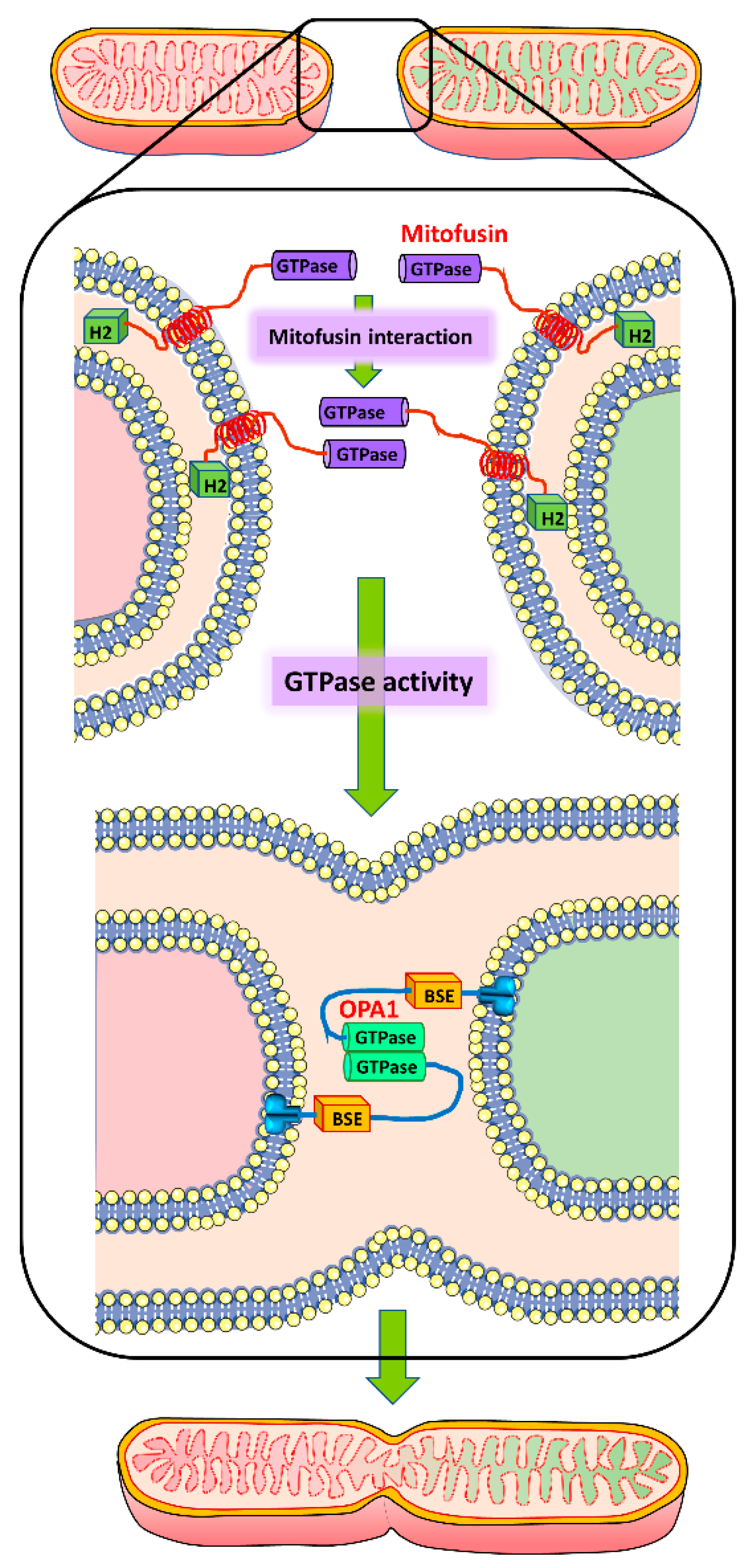
5. Mitochondrial Fusion Machinery
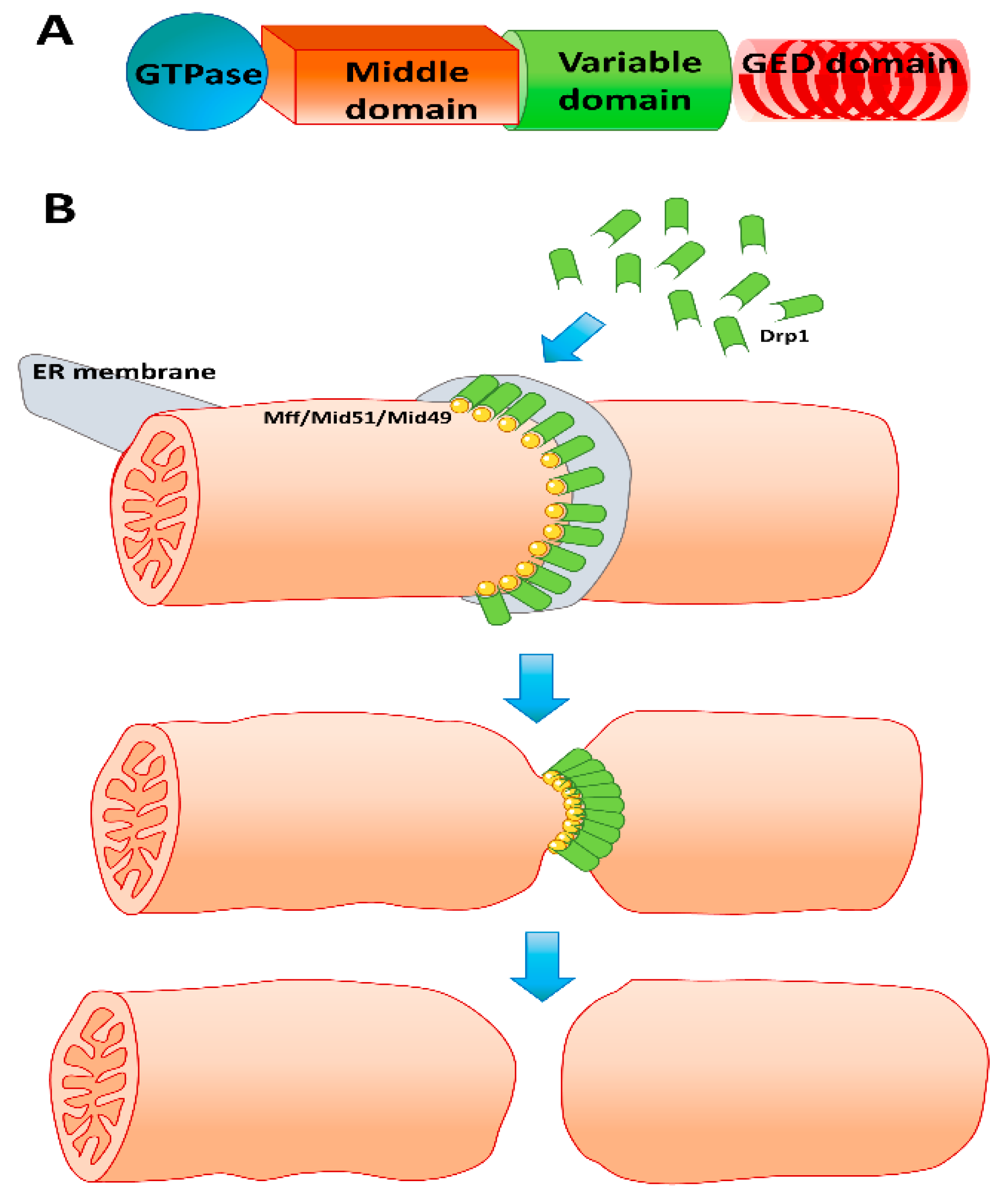
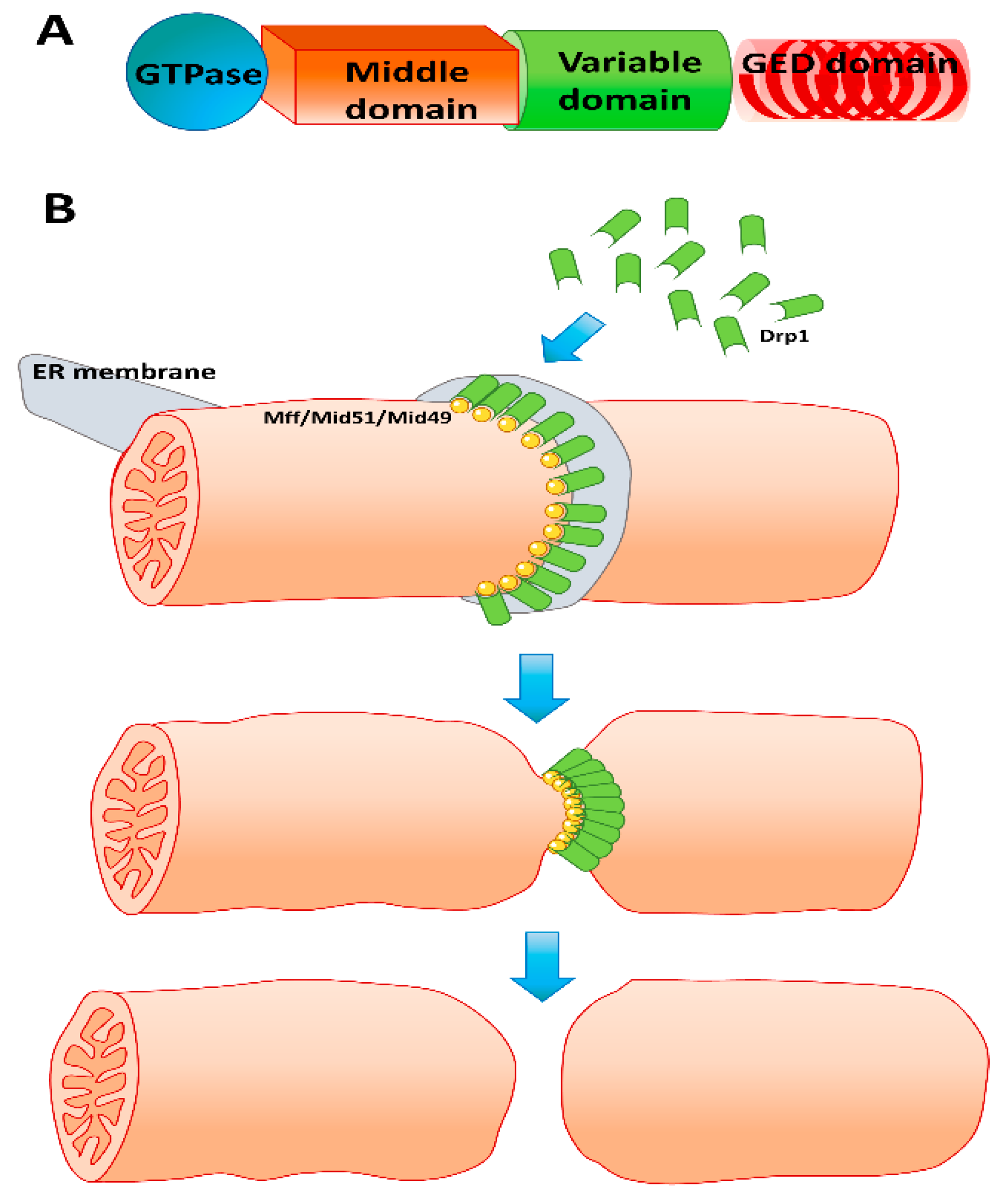
6. Mitochondrial Fission
7. Measurement of Mitochondrial Dynamics
8. Mitochondrial Dynamics in Hepatocytes
9. Mitochondrial Dynamics and Drug- or Toxin-Induced Liver Injury
10. Mitochondrial Biogenesis in Liver Injury
11. Mitochondrial Remodeling
12. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bernal, W.; Wendon, J. Acute liver failure. N. Engl. J. Med. 2013, 369, 2525–2534. [Google Scholar] [CrossRef]
- Abid, A.; Subhani, F.; Kayani, F.; Awan, S.; Abid, S. Drug induced liver injury is associated with high Mortality—A study from a tertiary care hospital in Pakistan. PLoS ONE 2020, 15, e0231398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.J.; Diaz, D.; O’Brien, P.J. Applications of cytotoxicity assays and pre-lethal mechanistic assays for assessment of human hepatotoxicity potential. Chem. Biol. Interact. 2004, 150, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Uetrecht, J. Mechanism of idiosyncratic drug induced liver injury (DILI): Unresolved basic issues. Ann. Transl. Med. 2021, 9, 730. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights into Mechanisms and Future Perspectives. Gene Expr. 2018, 18, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.I.; Imai, K.; Mima, K.; Nakagawa, S.; Hashimoto, D.; Chikamoto, A.; Baba, H. Idiosyncratic drug-induced liver injury: A short review. Hepatol. Commun. 2017, 1, 494–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boelsterli, U.A.; Lim, P.L. Mitochondrial Abnormalities—A link to idiosyncratic drug hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 220, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Jaeschke, H. Acetaminophen hepatotoxicity: A mitochondrial perspective. Adv. Pharmacol. 2019, 85, 195–219. [Google Scholar] [CrossRef]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Invest. 2012, 122, 1574–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, M.R.; Jaeschke, H. Biomarkers of mitotoxicity after acute liver injury: Further insights into the interpretation of glutamate dehydrogenase. J. Clin. Transl. Res. 2021, 7, 61–65. [Google Scholar]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef]
- Houten, S.M.; Auwerx, J. PGC-1alpha: Turbocharging mitochondria. Cell 2004, 119, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Abdelwahid, E. Mitochondrial dynamics regulate myocardial contractility and vice versa. Int. J. Cardiol. 2017, 247, 35. [Google Scholar] [CrossRef] [PubMed]
- Egner, A.; Jakobs, S.; Hell, S.W. Fast 100-nm resolution three-dimensional microscope reveals structural plasticity of mitochondria in live yeast. Proc. Natl. Acad Sci. USA 2002, 99, 3370–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 2001, 26, 23–29. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Pla-Martin, D.; Wiesner, R.J. Reshaping membranes to build mitochondrial DNA. PLoS Genet. 2019, 15, e1008140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva Ramos, E.; Motori, E.; Bruser, C.; Kuhl, I.; Yeroslaviz, A.; Ruzzenente, B.; Kauppila, J.H.K.; Busch, J.D.; Hultenby, K.; Habermann, B.H.; et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019, 15, e1008085. [Google Scholar] [CrossRef] [Green Version]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.M.; Tareste, D. Recent insights into the structure and function of Mitofusins in mitochondrial fusion. F1000 Res. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Mattie, S.; Krols, M.; McBride, H.M. The enigma of an interconnected mitochondrial reticulum: New insights into mitochondrial fusion. Curr. Opin. Cell Biol. 2019, 59, 159–166. [Google Scholar] [CrossRef]
- Choi, S.Y.; Huang, P.; Jenkins, G.M.; Chan, D.C.; Schiller, J.; Frohman, M.A. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 2006, 8, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Faelber, K.; Dietrich, L.; Noel, J.K.; Wollweber, F.; Pfitzner, A.K.; Muhleip, A.; Sanchez, R.; Kudryashev, M.; Chiaruttini, N.; Lilie, H.; et al. Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1. Nature 2019, 571, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, T.B.; Sanchez-Guerrero, A.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliuso, A.; Cossart, P.; Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef] [Green Version]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Frohlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, R.; Ji, W.K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 2018, 217, 251–268. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Itoh, K.; Yamada, T.; Cerveny, K.L.; Suzuki, T.L.; Macdonald, P.; Frohman, M.A.; Ramachandran, R.; Iijima, M.; Sesaki, H. Coincident Phosphatidic Acid Interaction Restrains Drp1 in Mitochondrial Division. Mol. Cell 2016, 63, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Ding, W.X. Mechanisms, pathophysiological roles and methods for analyzing mitophagy—Recent insights. Biol. Chem. 2018, 399, 147–178. [Google Scholar] [CrossRef] [Green Version]
- Umbaugh, D.S.; Nguyen, N.T.; Jaeschke, H.; Ramachandran, A. Mitochondrial Membrane Potential Drives Early Change in Mitochondrial Morphology After Acetaminophen Exposure. Toxicol. Sci. 2021, 180, 186–195. [Google Scholar] [CrossRef]
- Simula, L.; Campello, S. Monitoring the Mitochondrial Dynamics in Mammalian Cells. Methods Mol. Biol. 2018, 1782, 267–285. [Google Scholar] [CrossRef]
- Hodneland Nilsson, L.I.; Nitschke Pettersen, I.K.; Nikolaisen, J.; Micklem, D.; Avsnes Dale, H.; Vatne Rosland, G.; Lorens, J.; Tronstad, K.J. A new live-cell reporter strategy to simultaneously monitor mitochondrial biogenesis and morphology. Sci. Rep. 2015, 5, 17217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzapoiazova, T.; Li, H.; Nathan, A.; Srivstava, S.; Nasser, M.W.; Lennon, F.; Armstrong, B.; Mambetsariev, I.; Chu, P.G.; Achuthan, S.; et al. Monitoring and Determining Mitochondrial Network Parameters in Live Lung Cancer Cells. J. Clin. Med. 2019, 8, 1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohani, A.; Moore, J.H.; Kashatus, J.A.; Sesaki, H.; Kashatus, D.F.; Swami, N.S. Label-Free Quantification of Intracellular Mitochondrial Dynamics Using Dielectrophoresis. Anal. Chem. 2017, 89, 5757–5764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yang, Z.; Wu, Z.; He, Y.; Shan, C.; Chai, P.; Ma, C.; Tian, M.; Teng, J.; Jin, D.; et al. Mitochondrial dynamics quantitatively revealed by STED nanoscopy with an enhanced squaraine variant probe. Nat. Commun. 2020, 11, 3699. [Google Scholar] [CrossRef]
- Plucinska, G.; Paquet, D.; Hruscha, A.; Godinho, L.; Haass, C.; Schmid, B.; Misgeld, T. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J. Neurosci. 2012, 32, 16203–16212. [Google Scholar] [CrossRef]
- Mandal, A.; Pinter, K.; Drerup, C.M. Analyzing Neuronal Mitochondria In Vivo Using Fluorescent Reporters in Zebrafish. Front Cell Dev. Biol. 2018, 6, 144. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Hajnoczky, N.; Antony, A.N.; Csordas, G.; Gaspers, L.D.; Clemens, D.L.; Hoek, J.B.; Hajnoczky, G. Mitochondrial morphology and dynamics in hepatocytes from normal and ethanol-fed rats. Pflugers Arch 2012, 464, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Pezet, M.G.; Gomez-Duran, A.; Klimm, F.; Aryaman, J.; Burr, S.; Wei, W.; Saitou, M.; Prudent, J.; Chinnery, P.F. Oxygen tension modulates the mitochondrial genetic bottleneck and influences the segregation of a heteroplasmic mtDNA variant in vitro. Commun. Biol. 2021, 4, 584. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Martin-Levilain, J.; Jimenez-Sanchez, C.; Karaca, M.; Foti, M.; Martinou, J.C.; Maechler, P. In vivo stabilization of OPA1 in hepatocytes potentiates mitochondrial respiration and gluconeogenesis in a prohibitin-dependent way. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [Green Version]
- Hsu, W.H.; Lee, B.H.; Pan, T.M. Leptin-induced mitochondrial fusion mediates hepatic lipid accumulation. Int. J. Obes. 2015, 39, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Khraiwesh, H.; Lopez-Dominguez, J.A.; Lopez-Lluch, G.; Navas, P.; de Cabo, R.; Ramsey, J.J.; Villalba, J.M.; Gonzalez-Reyes, J.A. Alterations of ultrastructural and fission/fusion markers in hepatocyte mitochondria from mice following calorie restriction with different dietary fats. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Perez, M.; Simoni-Nieves, A.; Rosales, P.; Nuno-Lambarri, N.; Rosas-Lemus, M.; Souza, V.; Miranda, R.U.; Bucio, L.; Uribe Carvajal, S.; Marquardt, J.U.; et al. Cholesterol burden in the liver induces mitochondrial dynamic changes and resistance to apoptosis. J. Cell Physiol. 2019, 234, 7213–7223. [Google Scholar] [CrossRef] [PubMed]
- Galloway, C.A.; Lee, H.; Brookes, P.S.; Yoon, Y. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am. J. Physiol Gastrointest. Liver Physiol. 2014, 307, G632–G641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Wang, L.; Lee, H.; O’Brien, D.K.; Bronk, S.F.; Gores, G.J.; Yoon, Y. Decreasing mitochondrial fission prevents cholestatic liver injury. J. Biol. Chem. 2014, 289, 34074–34088. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hong, J.Y.; Rockwell, C.E.; Copple, B.L.; Jaeschke, H.; Klaassen, C.D. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int. 2012, 32, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, Y.; Chang, X.; Zhang, X. Imbalance in mitochondrial dynamics induced by low PGC-1alpha expression contributes to hepatocyte EMT and liver fibrosis. Cell Death Dis. 2020, 11, 226. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lv, L.; Jiang, Z.; Yang, H.; Li, S.; Jiang, Y. Mitofusin 2 protects hepatocyte mitochondrial function from damage induced by GCDCA. PLoS ONE 2013, 8, e65455. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef] [PubMed]
- He, G.W.; Gunther, C.; Kremer, A.E.; Thonn, V.; Amann, K.; Poremba, C.; Neurath, M.F.; Wirtz, S.; Becker, C. PGAM5-mediated programmed necrosis of hepatocytes drives acute liver injury. Gut 2017, 66, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, D.; Zhang, T.; Tong, Q.; Ye, R.D.; Lin, L. SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging and mitochondrial integrity. Cell Death Dis. 2017, 8, e3158. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, A.; Duan, L.; Akakpo, J.Y.; Jaeschke, H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: Current understanding and future perspectives. J. Clin. Transl. Res. 2018, 4. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. A mitochondrial journey through acetaminophen hepatotoxicity. Food Chem. Toxicol. 2020, 140, 111282. [Google Scholar] [CrossRef]
- Gunawan, B.K.; Liu, Z.X.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006, 131, 165–178. [Google Scholar] [CrossRef]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, A.; McGill, M.R.; Xie, Y.; Ni, H.M.; Ding, W.X.; Jaeschke, H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013, 58, 2099–2108. [Google Scholar] [CrossRef] [Green Version]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.W.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhou, J.; Li, Y.; Sun, K.; Chen, J. Mitochondrial Damage and Drp1 Overexpression in Rifampicin- and Isoniazid-induced Liver Injury Cell Model. J. Clin. Transl. Hepatol. 2019, 7, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Sun, W.; Chen, J. IL-2 augments the sorafenib-induced apoptosis in liver cancer by promoting mitochondrial fission and activating the JNK/TAZ pathway. Cancer Cell Int. 2018, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Dirks-Naylor, A.J.; Kouzi, S.A.; Bero, J.D.; Phan, D.T.; Taylor, H.N.; Whitt, S.D.; Mabolo, R. Doxorubicin alters the mitochondrial dynamics machinery and mitophagy in the liver of treated animals. Fundam. Clin. Pharmacol. 2014, 28, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Hasnat, M.; Yuan, Z.; Naveed, M.; Khan, A.; Raza, F.; Xu, D.; Ullah, A.; Sun, L.; Zhang, L.; Jiang, Z. Drp1-associated mitochondrial dysfunction and mitochondrial autophagy: A novel mechanism in triptolide-induced hepatotoxicity. Cell Biol. Toxicol. 2019, 35, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, H.; Ni, H.M.; Xiong, A.; Wang, Z.; Sesaki, H.; Ding, W.X.; Yang, L. Inhibition of Drp1 protects against senecionine-induced mitochondria-mediated apoptosis in primary hepatocytes and in mice. Redox Biol. 2017, 12, 264–273. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Lin, Y.; Han, P.Y.; Jiang, S.; Che, L.; He, C.Y.; Lin, Y.C.; Lin, Z.N. HBx combined with AFB1 triggers hepatic steatosis via COX-2-mediated necrosome formation and mitochondrial dynamics disorder. J. Cell Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, K.; Ramachandran, A.; McGill, M.R.; Mansouri, A.; Asselah, T.; Farhood, A.; Woolbright, B.L.; Ding, W.X.; Jaeschke, H. Induction of mitochondrial biogenesis protects against acetaminophen hepatotoxicity. Food Chem. Toxicol. 2017, 108, 339–350. [Google Scholar] [CrossRef]
- Degli Esposti, D.; Hamelin, J.; Bosselut, N.; Saffroy, R.; Sebagh, M.; Pommier, A.; Martel, C.; Lemoine, A. Mitochondrial roles and cytoprotection in chronic liver injury. Biochem. Res. Int. 2012, 2012, 387626. [Google Scholar] [CrossRef] [PubMed]
- Shlomai, A.; Paran, N.; Shaul, Y. PGC-1alpha controls hepatitis B virus through nutritional signals. Proc. Natl. Acad Sci. USA 2006, 103, 16003–16008. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Joe, Y.; Rah, S.Y.; Kim, S.K.; Park, S.U.; Park, J.; Kim, J.; Ryu, J.; Cho, G.J.; Surh, Y.J.; et al. Carbon monoxide-induced TFEB nuclear translocation enhances mitophagy/mitochondrial biogenesis in hepatocytes and ameliorates inflammatory liver injury. Cell Death Dis. 2018, 9, 1060. [Google Scholar] [CrossRef]
- Carchman, E.H.; Whelan, S.; Loughran, P.; Mollen, K.; Stratamirovic, S.; Shiva, S.; Rosengart, M.R.; Zuckerbraun, B.S. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. FASEB J. 2013, 27, 4703–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haden, D.W.; Suliman, H.B.; Carraway, M.S.; Welty-Wolf, K.E.; Ali, A.S.; Shitara, H.; Yonekawa, H.; Piantadosi, C.A. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 2007, 176, 768–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inata, Y.; Kikuchi, S.; Samraj, R.S.; Hake, P.W.; O’Connor, M.; Ledford, J.R.; O’Connor, J.; Lahni, P.; Wolfe, V.; Piraino, G.; et al. Autophagy and mitochondrial biogenesis impairment contribute to age-dependent liver injury in experimental sepsis: Dysregulation of AMP-activated protein kinase pathway. FASEB J. 2018, 32, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.W.; Choi, H.S.; Lee, S.M. Resolvin D1 attenuates liver ischaemia/reperfusion injury through modulating thioredoxin 2-mediated mitochondrial quality control. Br. J. Pharmacol. 2018, 175, 2441–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joe, Y.; Zheng, M.; Kim, H.J.; Uddin, M.J.; Kim, S.K.; Chen, Y.; Park, J.; Cho, G.J.; Ryter, S.W.; Chung, H.T. Cilostazol attenuates murine hepatic ischemia and reperfusion injury via heme oxygenase-dependent activation of mitochondrial biogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G21–G29. [Google Scholar] [CrossRef] [Green Version]
- Prakash, C.; Kumar, V. Chronic Arsenic Exposure-Induced Oxidative Stress is Mediated by Decreased Mitochondrial Biogenesis in Rat Liver. Biol. Trace Elem. Res. 2016, 173, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Pi, H.; Xu, S.; Zhang, L.; Li, Y.; Li, M.; Cao, Z.; Tian, L.; Xie, J.; Li, R.; et al. Melatonin Improves mitochondrial function by promoting MT1/SIRT1/PGC-1 alpha-dependent mitochondrial biogenesis in cadmium-induced hepatotoxicity in vitro. Toxicol Sci. 2014, 142, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Tiao, M.M.; Lin, T.K.; Liou, C.W.; Wang, P.W.; Chen, J.B.; Kuo, F.Y.; Huang, C.C.; Chou, Y.M.; Chuang, J.H. Early transcriptional deregulation of hepatic mitochondrial biogenesis and its consequent effects on murine cholestatic liver injury. Apoptosis 2009, 14, 890–899. [Google Scholar] [CrossRef]
- Tiao, M.M.; Lin, T.K.; Chen, J.B.; Liou, C.W.; Wang, P.W.; Huang, C.C.; Chou, Y.M.; Huang, Y.H.; Chuang, J.H. Dexamethasone decreases cholestatic liver injury via inhibition of intrinsic pathway with simultaneous enhancement of mitochondrial biogenesis. Steroids 2011, 76, 660–666. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Bernstein, D. Mitochondrial remodeling: Rearranging, recycling, and reprogramming. Cell Calcium 2016, 60, 88–101. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E.; Ragavan, M.; Palavicini, J.P.; Fourcaudot, M.; Bakewell, T.M.; Valdez, I.A.; Ayala, I.; Jin, E.S.; Madesh, M.; Han, X.; et al. Insulin resistance is mechanistically linked to hepatic mitochondrial remodeling in non-alcoholic fatty liver disease. Mol. Metab. 2021, 45, 101154. [Google Scholar] [CrossRef]
- Garcia, J.; Decker, C.W.; Sanchez, S.J.; Ouk, J.M.; Siu, K.M.; Han, D. Obesity and steatosis promotes mitochondrial remodeling that enhances respiratory capacity in the liver of ob/ob mice. FEBS Lett. 2018, 592, 916–927. [Google Scholar] [CrossRef]
- Wilson-Fritch, L.; Nicoloro, S.; Chouinard, M.; Lazar, M.A.; Chui, P.C.; Leszyk, J.; Straubhaar, J.; Czech, M.P.; Corvera, S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J. Clin. Invest. 2004, 114, 1281–1289. [Google Scholar] [CrossRef]
- Han, D.; Johnson, H.S.; Rao, M.P.; Martin, G.; Sancheti, H.; Silkwood, K.H.; Decker, C.W.; Nguyen, K.T.; Casian, J.G.; Cadenas, E.; et al. Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free Radic. Biol. Med. 2017, 102, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Wanet, A.; Remacle, N.; Najar, M.; Sokal, E.; Arnould, T.; Najimi, M.; Renard, P. Mitochondrial remodeling in hepatic differentiation and dedifferentiation. Int. J. Biochem. Cell Biol. 2014, 54, 174–185. [Google Scholar] [CrossRef]
- Santulli, G.; Monaco, G.; Parra, V.; Morciano, G. Editorial: Mitochondrial Remodeling and Dynamic Inter-Organellar Contacts in Cardiovascular Physiopathology. Front. Cell Dev. Biol. 2021, 9, 679725. [Google Scholar] [CrossRef]
- Han, D.; Dara, L.; Win, S.; Than, T.A.; Yuan, L.; Abbasi, S.Q.; Liu, Z.X.; Kaplowitz, N. Regulation of drug-induced liver injury by signal transduction pathways: Critical role of mitochondria. Trends Pharmacol. Sci. 2013, 34, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramachandran, A.; Umbaugh, D.S.; Jaeschke, H. Mitochondrial Dynamics in Drug-Induced Liver Injury. Livers 2021, 1, 102-115. https://doi.org/10.3390/livers1030010
Ramachandran A, Umbaugh DS, Jaeschke H. Mitochondrial Dynamics in Drug-Induced Liver Injury. Livers. 2021; 1(3):102-115. https://doi.org/10.3390/livers1030010
Chicago/Turabian StyleRamachandran, Anup, David S. Umbaugh, and Hartmut Jaeschke. 2021. "Mitochondrial Dynamics in Drug-Induced Liver Injury" Livers 1, no. 3: 102-115. https://doi.org/10.3390/livers1030010
APA StyleRamachandran, A., Umbaugh, D. S., & Jaeschke, H. (2021). Mitochondrial Dynamics in Drug-Induced Liver Injury. Livers, 1(3), 102-115. https://doi.org/10.3390/livers1030010