1. Introduction
Congenital hepatic fibrosis (CHF), Caroli syndrome (CS), and polycystic liver disease (PLD) are liver diseases caused by ductal plate malformation, a developmental abnormality of the portobiliary system [
1]. The exact incidence and prevalence are not known, but they are considered rare diseases.
CHF is associated with autosomal recessive polycystic liver disease and genetic mutations in the polycystic kidney and hepatic disease 1 (PKHD1) gene, leading to dysfunctional fibrocystin found in cholangiocyte primary cilia. These alterations are involved in disease pathogenesis and progression. CS is a rare congenital liver disease characterized by non-obstructive cystic dilatations of the intra-hepatic and rarely extra-hepatic bile ducts. PLD is a genetic disorder characterized by the appearance of numerous cysts spread throughout the liver and that in most cases is described as autosomal dominant polycystic liver disease (ADPCLD).
As other inherited disorders, these diseases come with multiorgan involvement (e.g., kidneys and central nervous system). The most common clinical manifestations are portal hypertension with a variable degree of hypersplenism and variceal bleeding, and recurrent cholangitis, frequently involving multidrug-resistant (MDR) microorganisms leading to difficulties in finding the right antimicrobial treatment. Acute cholangitis should be suspected in patients with fever, abdominal pain, and jaundice (known as Charcot’s triad). Diagnosis can be confirmed by transabdominal ultrasound and computed tomography scan, along with invasive procedure such as endoscopic retrograde cholangiopancreatography (ERCP).
However, the clinical manifestations of these congenital disorders are nonspecific, and the diagnosis is extremely difficult. Symptoms and signs onset is variable and ranges from early childhood through adulthood. Radiology is the mainstay of diagnosis [
2] but liver biopsy is found of help mostly in CHF [
3]. Histological findings show porto-portal peribiliary fibrosis rather than porto-central bridging, more typical of other cirrhosis etiologies [
4,
5].
A clear consensus on how to manage these conditions is missing, symptomatic medical treatment is useful until the episodes of cholangitis are so frequent that other measures must be implemented. The best option in case of diffuse and uncontrollable involvement is orthotopic liver transplant (OLT) [
6].
Other options may be hepatectomy or transjugular intrahepatic portosystemic shunt, but the latter should be considered a bridging option before transplantation.
Here, we report a case of a patient with recurrent episodes of cholangitis, with occasional systemic involvement leading to bloodstream infection, who ultimately received a diagnosis of CHF. We also provide a review of the literature regarding the infectious events occurring in patients with CHF, CS, and PLD, presenting an overview of the microorganism involved and the antibiotic employed.
2. Methods
2.1. Histology
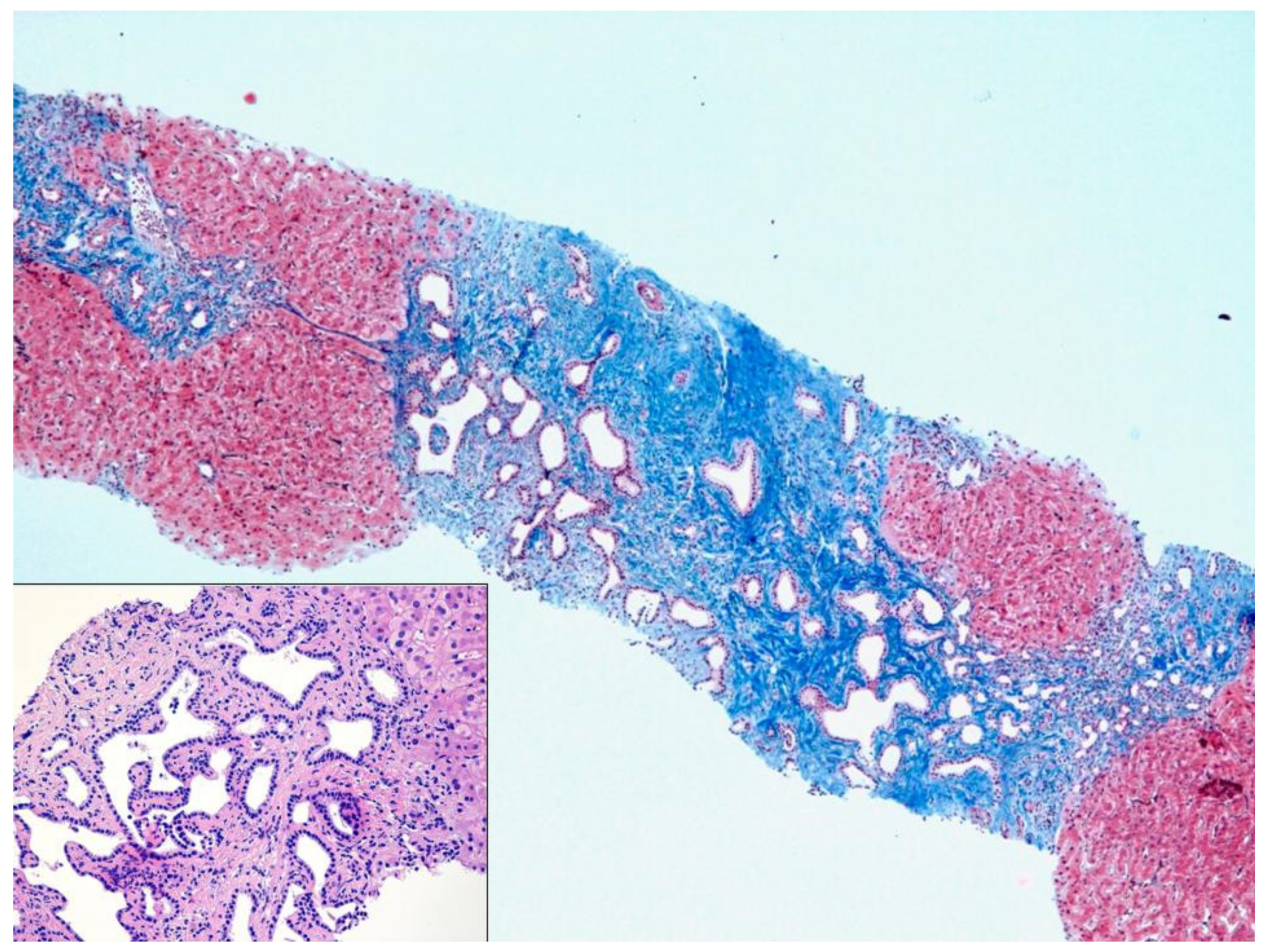
The liver biopsy was routinely formalin-fixed, paraffin-embedded and Hematoxylin-Eosin stained. Additional stainings for Masson’s trichrome, PAS-diastase, and cytokeratin 7 was also prepared.
Liver parenchyma demonstrated diffuse architectural alteration due to portal tracts fibrous expansion, with diffuse portal-to-portal septa and focal nodular delimitation. The fibrous septa showed diffuse biliary duct proliferation, sometimes dilated and with tortuous profile, without any cytological atypia, associated with venous portal hypoplasia. Inflammatory infiltration was inconspicuous; liver trabeculae and centrilobular veins were unremarkable. The morphology was typical for CHF.
2.2. Literature Search
The following strings were employed to identify relevant studies indexed on PubMed: ((congenital hepatic fibrosis [Title/Abstract]) AND (recurrent cholangitis [Title/Abstract])) AND (english[Language]); ((Caroli syndrome[Title/Abstract]) AND (recurrent cholangitis[Title/Abstract])) AND (english[Language]); ((polycystic liver disease [Title/Abstract]) AND (recurrent cholangitis[Title/Abstract])) AND (english[Language]). The literature search was performed on the 1 June 2021.
3. Case Report
We report the case of a 50-year-old woman with a history of recurring cholangitis and abnormality of the portobiliary system. Since 2019 the patient was hospitalized on several occasions for cholangitis and bloodstream infections with isolation on blood and bile fluid of Enterobacterales (Escherichia coli, Klebsiella oxytoca, extended beta-lactamase producing Klebsiella pneumoniae, multidrug-resistant Pseudomonas aeruginosa), Enterococcus faecium, Acinetobacter pittii, and Candida orthopsilosis.
During these hospital stays the patient was administered several antibiotic cycles, always with a good and fast clinical response. The patient repeated on several occasion magnetic resonance imaging of the biliary tree, without the detection of any abnormalities. Disease-free period became shorter and shorter between each hospitalization, severely hampering the patient’s well-being. In occasion of her last hospitalization, due to a cholangitis recurrence with no microbiological isolation, the patient was treated with empirical antibiotic therapy (piperacillin/tazobactam) and it was decided to perform a liver biopsy. This showed a histological pattern compatible with CHF (
Figure 1). Furthermore,
Enterococcus faecium was isolated from culture performed on the specimen. The patient continued antibiotic therapy with linezolid, for a total of 10 days, and piperacillin/tazobactam, for a total of 14 days, in our outpatient clinic.
She was finally placed on waiting list for OLT while receiving a chronic suppressive antibiotic therapy with amoxicillin/clavulanate, with no evidence of recurrent cholangitis at 4 weeks since start of prophylaxis.
4. Review of Literature
Many studies led from the early 1980s up to the present day confirmed the recurrency of cholangitis in subjects affected by CHF and/or Caroli’s disease [
7].
Table 1 summarizes the microorganisms isolated and the management proposed for the cases identified.
Sugiura et al. [
8] described the case of a 4-year-old patient suffering from choledochal cyst and pancreaticobiliary malfunction (PBM). She was treated by resection of the choledochal cyst and Roux-en-Y reconstruction. After surgery she suffered from recurrent cholangitis and was not responsive to several antibiotics. Liver biopsy was essential in diagnosing CHF as it showed expanded portal areas with fibrous tissue and increased bile ducts. She underwent living-donor liver transplant and three species of multidrug-resistant
Pseudomonas aeruginosa were cultured in the explanted liver.
Previously, Howlett et al. [
9] reported the case of a 5-month-old girl with story of unknown fever and hepatosplenomegaly. Liver biopsy showed CHF with acute suppurative cholangitis. As in the previous case, an Enterobacterales was the culprit microorganism, with
Klebsiella aerogenes isolated from two blood cultures. The patient was treated with two weeks of IV gentamycin followed by oral cephalexin, but fever recurred.
In their case series, Rawat et al. [
10] in 2013 described the clinical manifestations and outcomes in 40 children affected by CHF and Caroli’s disease between 1990 and 2009. Recurrent episodes of cholangitis were found in three patients, including one with recurrent cholangitis after kidney transplant at 2 years of age and under antibiotic prophylaxis.
Similar findings were reported by Shorbagi et al. [
11] who, throughout the years 1974–2009, diagnosed CHF in a total of 26 patients. A history of recurrent cholangitis was found in six patients (23%). In eight patients CHF was associated with Caroli’s disease (the combination is known as Caroli’s syndrome). Eventually, all patients who suffered from cholangitis presented Caroli’s disease, where cholangitis is the main manifestation.
The same results were presented by Nakanuma et al. [
12], who performed liver biopsy in six patients (three affected by PLD and three affected by CHF) and found in 5 out of 6 multiple cystic dilatations of the intrahepatic biliary tree, referred to as Caroli’s disease. The three patients presenting both CHF and Caroli’s disease showed recurrent episodes of cholangitis. Additionally, Summerfield and Nagafuchi, in their review of 51 patients affected by fibropolycystic disease, found the combination of both CHF and Caroli’s disease to be the most striking: biopsies from five subjects showed an overlap between the two main histopathological features. All of these subjects had recurrent episodes of cholangitis during life [
13].
5. Discussion
As highlighted by our case and the several reports shown above, in liver diseases characterized by ductal plate malformation recurrent cholangitis is an extremely common manifestation.
The reason of this higher risk of developing infections can be found firstly in the impaired structure of the biliary ducts, that favors cholelithiasis, a known risk factor for bacterial colonization and subsequent infection. Moreover, congenital liver diseases affecting the ductal plate can affect the delicate microbiological balance characterizing the liver–gut axes, causing gut dysbiosis and promoting bacterial overgrowth with relative abundance of pathogen taxa. Lastly, this imbalance can damage the intestinal barrier, favoring bacterial translocation and exposing the patient to a higher risk of bloodstream involvement.
These infections are usually caused by Enterobacterales, which frequently harbors genes conferring resistance to several antibiotic classes, thus complicating the clinical course.
Due to the congenital and progressive nature of these diseases, OLT represents the only curative treatment. Recurrent cholangitis with an unexplained cause should induce the clinician in suspecting the presence of a congenital alteration of the biliary tree and prompt the decision to perform a liver biopsy. Chronic suppressive antibiotic therapy could be considered while the patient is in the waiting list for OLT to reduce the risk of major infectious events but should be protracted for the shorter possible time.
Moreover, further studies are needed to assess the impact of gut–liver homeostasis in congenital plate duct diseases and its possible role in pathogenesis and clinical progression, with particular attention to infectious complications, assessing the possible role of microbiome modulation in shaping the patient outcome.
Author Contributions
Conceptualization, E.P., G.V. and A.L.; methodology, A.L. and M.M.; writing—original draft preparation, E.P., G.V., M.M. and A.L.; writing—review and editing, D.M., R.L., B.A., D.D., M.I. and A.L.F.; supervision, A.L., A.B. and A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gunay-Aygun, M. Liver and kidney disease in ciliopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Ramond, M.J.; Huguet, C.; Danan, G.; Rueff, B.; Benhamou, J.P. Partial Hepatectomy in the Treatment of Caroli’s Disease-Report of a Case and Review of the Literature. Dig. Dis. Sci. 1984, 29, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, A.; Migliorati, R.; Vajro, P.; Caldore, M.; Vecchione, R. Congenital Hepatic Fibrosis with Gastrointestinal Bleeding in Early Infancy. Clin. Pediatr. 1982, 21, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Luoto, T.T.; Pakarinen, M.P.; Jahnukainen, T.; Jalanko, H. Liver Disease in Autosomal Recessive Polycystic Kidney Disease: Clinical Characteristics and Management in Relation to Renal Failure. J. Pediatr. Gastroenterol. Nutr. 2014, 59, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Lasagni, A.; Cadamuro, M.; Morana, G.; Fabris, L.; Strazzabosco, M. Fibrocystic Liver Disease: Novel Concepts and Translational Perspectives. Transl. Gastroenterol. Hepatol. 2021, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Tallón Aguilar, L.; Sánchez Moreno, L.; Barrera Pulido, L.; Pareja Ciuró, F.; Suárez Artacho, G.; Álamo Matinez, J.M.; Bernal Bellido, C.; Garía González, I.; Serrano Díaz-Canedo, J.; Gómez Bravo, M.A.; et al. Liver Transplantation Consequential to Caroli’s Syndrome: A Case Report. Transplant. Proc. 2008, 40, 3121–3122. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S.; Yi, N.J.; Suh, K.S.; Seo, J.K. Pediatric Liver Transplantation for Fibropolycystic Liver Disease. Pediatr. Transplant. 2012, 16, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Endo, T.; Ito, K.; Goto, K.; Sato, Y.; Kondo, S.; Suzuki, T.; Hashimoto, T. Recurrent Cholangitis with Congenital Hepatic Fibrosis and Pancreaticobiliary Maljunction after Roux-En-Y Reconstruction. Eur. J. Pediatr. Surg. Rep. 2013, 1, 043–045. [Google Scholar] [CrossRef][Green Version]
- Howlett, S.A.; Shulman, S.T.; Ayoub, E.M.; Alexander, R.A.; Donnelly, W.H.; Cerda, J.J. Cholangitis Complicating Congenital Hepatic Fibrosis. Am. J. Dig. Dis. 1975, 20, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Rawat, D.; Kelly, D.A.; Milford, D.V.; Sharif, K.; Lloyd, C.; McKiernan, P.J. Phenotypic Variation and Long-Term Outcome in Children with Congenital Hepatic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Shorbagi, A.; Bayraktar, Y. Experience of a Single Center with Congenital Hepatic Fibrosis: A Review of the Literature. World J. Gastroenterol. 2010, 16, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Terada, T.; Ohta, G.; Kurachi, M.; Matsubara, F. Caroli’s Disease in Congenital Hepatic Fibrosis and Infantile Polycystic Disease. Liver 1982, 2, 346–354. [Google Scholar] [CrossRef]
- Summerfield, J.A.; Nagafuchi, Y.; Sherlock, S.; Cadafalch, J.; Scheuer, P.J. Hepatobiliary Fibropolycystic Diseases. A Clinical and Histological Review of 51 Patients. J. Hepatol. 1986, 2, 141–156. [Google Scholar] [CrossRef]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 



{kind=link}