Abstract
Hepatic steatosis is a consequence of distorted lipid storage and plays a vital role in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). This study aimed to explore the role of the COP9 signalosome (CSN) in the development of hepatic steatosis and its interplay with the deubiquitylating enzyme (DUB) cylindromatosis (CYLD). CSN occurs as CSNCSN7A and CSNCSN7B variants regulating the ubiquitin proteasome system. It is a deneddylating complex and associates with other DUBs. CYLD cleaves Lys63-ubiquitin chains, regulating a signal cascade that mitigates hepatic steatosis. CSN subunits CSN1 and CSN7B, as well as CYLD, were downregulated with specific siRNA in HepG2 cells and human primary hepatocytes. The same cells were transfected with Flag-CSN7A or Flag-CSN7B for pulldowns. Hepatic steatosis in cell culture was induced by palmitic acid (PA). Downregulation of CSN subunits led to reduced PPAR-γ expression. Flag-pulldowns in both LiSa-2 and HepG2 cells and human primary hepatocytes revealed binding of CYLD preferentially to CSNCSN7A. This was influenced by PA treatment. Silencing of CSNCSN7B blocked lipid droplet formation caused a compensatory increase of CSNCSN7A stabilizing CYLD. Our results demonstrate that CSNCSN7A-mediated CYLD stabilization impedes hepatic steatosis. Therefore, stabilizing CSNCSN7A-CYLD interaction might be a strategy to retard hepatic steatosis.
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) has a worldwide prevalence of about 25% [1,2]. Many individuals with NAFLD progress from simple hepatic steatosis to nonalcoholic steatohepatitis (NASH) [3], which is characterized by severe lipid accumulation, inflammation, and fibrosis. NASH, accompanied by chronic inflammation, is regarded as a main risk factor for both cirrhosis and hepatocellular carcinoma (HCC) [4]. Due to the incomplete understanding of its pathogenesis, there is still no approved pharmacotherapy for NASH [1,5].
Hepatic steatosis results from an imbalance between lipid acquisition and lipid removal, which is regulated by four primary mechanisms: free fatty acid uptake, de novo lipogenesis, fatty acid oxidation, and export from hepatocytes as very-low-density lipoproteins. Disturbance in one or more of these pathways may result in the retention of fat in the liver, leading to hepatic steatosis and, finally, to NASH [6]. Primarily, fatty acids are stored as triacylglycerides (TAGs) in lipid droplets (LDs), which are distorted under disease conditions [7,8,9]. Hepatic steatosis can be caused by diet, gut microbiota, genetic factors, and de novo lipogenesis via the up-regulation of lipogenic transcription factors such as sterol regulatory binding protein-1c (SREBP1c), carbohydrate-responsive element-binding proteins (ChREBP), and peroxisome proliferator-activated receptor gamma (PPAR-γ) [8,10].
Although many open questions remain, it is known that the ubiquitin (Ub) proteasome system (UPS) plays a key role in the pathogenesis of NASH [11]. The ubiquitylation and deubiquitylation of proteins in hepatocytes has been shown to be involved in lipid metabolism [12]. In the UPS, a cascade of enzymes activates and transfers Ub to substrates as a signal for degradation. Poly-ubiquitylated proteins are subsequently degraded by the 26S proteasome [13]. Substrate specificity of the system is conferred by Ub ligases (E3s). A major representative subfamily of E3s are cullin-RING-ubiquitin ligases (CRLs) [14]. CRLs are multiprotein complexes organized around scaffolding cullins (Cullin 1-9, CUL1-9), containing a RING protein (e.g., RBX1 or RBX2) and associated with hundreds of exchangeable substrate-specific receptor (SR) proteins [14]. Based on their cognate cullin subunits, CRLs are divided into sub-families (CRL1 to CRL9) [15]. CRLs are activated by neddylation of their cullins [16,17]. The COP9 signalosome (CSN) is a master regulator of CRLs removing NEDD8 from cullins. Deneddylation inactivates CRLs and allows an exchange of SRs and, thus, adaptation to fluctuations in substrate availability [18,19,20,21,22,23,24,25]. The mammalian CSN consists of eight core subunits, CSN1 to CSN8, characterized either by the Proteasome lid-CSN-Initiation factor 3 (PCI) domain (CSN1-CSN4, CSN7, and CSN8) or by the MPR1/PAD1 N-terminal (MPN) domain (CSN5 and CSN6) [26]. All CSN subunits are required for the hydrolytic removal of NEDD8 [27,28], catalyzed by the JAMM/MPN+ domain localized to CSN5 [29]. Evolutionary diversification led to the occurrence of distinct CSN variants formed by the integration of different subunit paralogs/isoforms. Mammalian CSN variants containing CSN7A or CSN7B paralogs, CSNCSN7A and CSNCSN7B, co-exist in all mammalian cells possessing distinct functions in adipogenesis [30]. Recently, we discovered a variant-specific function of CSNCSN7B in DNA repair [31]. Because of its deneddylating activity, the CSN belongs to deubiquitylating enzyme (DUB) complexes [32] and is a member of the JAMM subfamily of DUBs. In addition, it is associated with a number of DUBs including USP15 and USP48 forming a multi-DUB complex [32].
Interestingly, the DUB cylindromatosis (CYLD) is involved in the regulation of different cellular processes including inflammation, fibrosis, and cancer and reverses the progression of NASH [33]. CYLD specifically affects the disassembly of Lys63-linked ubiquitin polymers [34]. It degrades Lys63-chains linked to components of different signaling pathways including NF-κB, Notch, and JNK [35]. It directly interacts with the kinase TAK1 and removes its Lys63-linked poly-ubiquitin chains, which blocks downstream activation of the JNK–p38 pathway, thereby retarding NASH [33]. CYLD contributes to hepatic homeostasis and restoration upon liver injury [36]. It is downregulated during NASH or NAFLD. An increase of CYLD mitigates NASH [33]. Interestingly, the stability of CYLD is controlled by CRL1β-TrCP ubiquitylation upon phosphorylation via IκB kinase [37]. On the other hand, during NASH the E3 ligase TRIM47 targets CYLD for degradation [33]. However, the exact interactome and stability regulation of CYLD during hepatic steatosis was unclear.
We provided evidence that the two CSN variants, CSNCSN7A and CSNCSN7B, have both general and specific functions during adipogenic differentiation [30,38]. Consequently, we were interested to see whether CSN plays a role in hepatic steatosis in human liver cells. For this purpose, an in-vitro model was used in which treatment of HepG2 cells with palmitic acid (PA) caused steatosis in cultured cells characterized by the accumulation of fatty acids in LDs and the overexpression of PPAR-γ [39,40,41]. Just as in adipocytes [30], the downregulation of CSN subunits in HepG2 cells and human primary hepatocytes led to the reduced expression of PPAR-γ. Moreover, we discovered that CYLD is associated with the CSN in LiSa-2 preadipocytes as well as hepatocytes. Closer examination revealed that CYLD binds to both CSNCSN7A and CSNCSN7B variants, albeit with preference to CSNCSN7A. PA treatment influences CYLD binding to CSNCSN7A and exacerbated hepatic steatosis. An increase of the CSNCSN7A variant elevated CYLD stability and reduced LD formation raising resistance to hepatic steatosis in HepG2 cells.
2. Results
2.1. Silencing of CSN Subunits Reduces PPAR-γ Expression in Liver Cells
Since CSN is a major player in adipogenesis [30,42], we were interested to see whether the regulator of CRLs is also involved in the pathogenesis of hepatic steatosis. To evaluate the role of CSN in hepatic steatosis, we compared healthy livers and fatty livers from patients by hematoxylin/eosin staining, Oil-Red-O (ORO) staining, and immunohistochemistry staining of both a paraffin-embedded liver and a frozen liver using the anti-CSN3 antibody (Figure 1A). As expected, ORO staining revealed an accumulation of fat in the fatty liver. Since CSN3 is an integrated subunit of the CSN holo-complex [32], staining with anti-CSN3 antibody visualized total CSN in the cells. Immunohistochemical analysis with both paraffin-embedded and frozen tissue revealed an increase of total CSN in fatty liver (Figure 1A, right two panels).
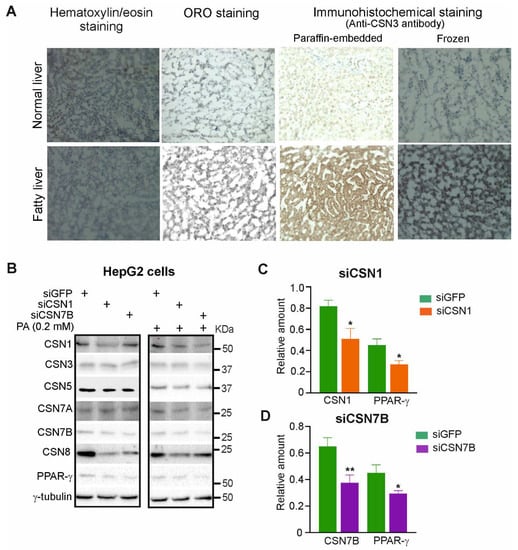
Figure 1.
Silencing of CSN1 or CSN7B reduces the expression of PPAR-γ, a regulator of hepatic steatosis. (A) Immunohistochemical staining using anti-CSN3 antibody in human normal and fatty liver tissue. Hematoxylin/eosin staining visualized nuclei, whereas ORO staining shows the number of LDs. Staining was visualized by light microscopy at 200-fold magnification. (B) Levels of the indicated proteins were assessed by Western blot analysis of HepG2 cells without (left panel) or with treatment using 0.2 mM PA (right panel) For 24 h. Control siRNA (siGFP) or siRNA directed against CSN1 (siCSN1) or CSN7B (siCSN7B) was transiently transfected into HepG2 cells 24 h before treatment with PA. (C,D) Blots as shown in (B) (left panel) were quantified by densitometry to measure the abundance of CSN1, CSN7B and PPAR-γ after transfection with siCSN1 (C) or siCSN7B (D). Data were normalized to γ-tubulin and are expressed as means of relative amounts plus/minus standard deviations (SD, n = 3). Unpaired Student’s t-test was used for statistical analysis. * p < 0.05, ** p < 0.01.
Next, we were interested to see whether the downregulation of CSN subunits had an impact on PPAR-γ, a major player in hepatic steatosis [8]. To silence CSN subunits siRNAs against siGFP (control), siCSN1 or siCSN7B were transfected in HepG2 cells with a final concentration of 50 nM. After 24 h, cells were incubated with or without PA for another 24 h. As demonstrated before, treatment of HepG2 cells with PA caused an increase of lipid accumulation and decrease in lipolysis as well as an elevation of PPAR-γ expression [39,40]. As shown in Figure 1B, the downregulation of CSN1 (Figure 1C) as well as CSN7B (Figure 1D) by about 40% led to reduction of PPAR-γ expression confirming data obtained in LiSa-2 cells [30,42]. Transfection with siCSN1 and siCSN7B and subsequent treatment with PA caused similar downregulation of CSN subunits and PPAR-γ as without PA (Figure 1B).
To optimize the downregulation of CSN7B, a new siRNA was designed called siCSN7B-RD (Figure 2A). Silencing of CSN7B using siCSN7B-RD in HepG2 cells was much more efficien, t reducing its expression by ~70% as revealed by densitometry and quantification of three independent experiments (Figure 2C). Silencing of CSN7B was accompanied by a significant downregulation of PPAR-γ. Most interestingly, under this condition the expression of CSN7A increased (Figure 2B,C). Similar results were obtained after treatment of HepG2 cells with PA (Figure 2B). In human primary hepatocytes, the downregulation of CSN7B by siCSN7B-RD was less efficient. Nevertheless, a significant decrease of PPAR-γ and an increase of CSN7A was measured (Figure 2D,E). After treatment with PA, similar effects were observed (Figure 2D).
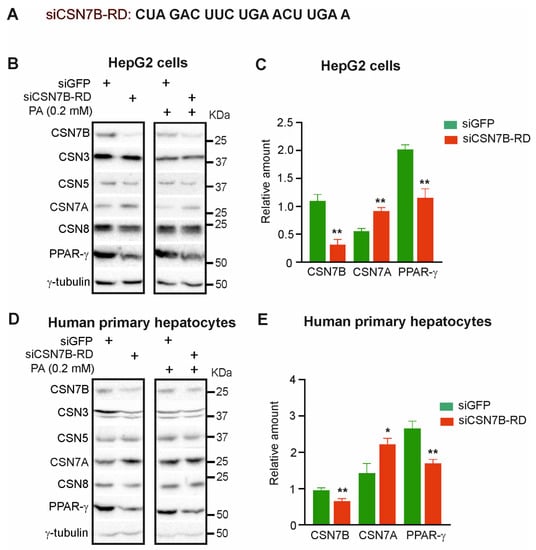
Figure 2.
Silencing by siCSN7B-RD increases CSN7A expression and reduces PPAR-γ in HepG2 cells and in human primary hepatocytes. (A) The sequence of siCSN7B-RD. (B) Western blot of HepG2 cells transfected with siGFP or siCSN7B-RD for 24 h followed by treatment without (left panel) or with 0.2 mM of PA (right panel) for 24 h. Antibodies were used as indicated. (C) Blots as shown in (B) (left panel) were quantified by densitometry to measure the abundance of CSN7B, CSN7A and PPAR-γ after transfection with siGFP (control) or siCSN7B-RD. Data were normalized to γ-tubulin and expressed as means of relative amounts plus/minus standard deviations (SD, n = 3). Unpaired Student’s t-test was used for statistical analysis. ** p < 0.01. (D) Western blot of human primary hepatocytes transfected with siGFP or siCSN7B-RD for 24 h, followed by treatment without (left panel) or with 0.2 mM of PA (right panel) for 24 h. Antibodies were used as indicated. (E) Blots as shown in (D) (left panel) were quantified by densitometry to measure the abundance of CSN7B, CSN7A and PPAR-γ after transfection with siGFP (control) or siCSN7B-RD. Data were normalized to γ-tubulin and expressed as means of relative amounts plus/minus standard deviations (SD, n = 3). Unpaired Student’s t-test was used for statistical analysis. * p < 0.05, ** p < 0.01.
In summary, the downregulation of CSN7B caused a reduction of PPAR-γ, the inducer of hepatic steatosis. Simultaneously, CSN7B silencing led to a compensatory increase of CSN7A independent of PA treatment.
2.2. The Ub-Specific Protease CYLD Preferentially Binds to CSNCSN7A Variant
For comparison, we studied the expression of CSN subunits in preadipogenic LiSa-2 cells. Recently, we established Lisa-2 cell lines stably expressing either empty Flag-vector, Flag-CSN7A, or Flag-CSN7B [30]. The preadipogenic cell lines were stained with ORO (Figure 3A) or lysed for western blots (Figure 3C). As visualized by ORO staining in Figure 3A and as demonstrated before [30], there was an increase of lipids accumulated in LDs in LiSa-2 cells stably expressing Flag-CSN7B (Figure 3B). The Western blot showed a typical protein pattern. While stably transfected Flag-CSN7A was weakly expressed, stably transfected Flag-CSN7B was produced more abundantly. Both Flag-CSN7A and Flag-CSN7B overexpression induced an increase of other CSN subunits compared to control (Figure 3C).
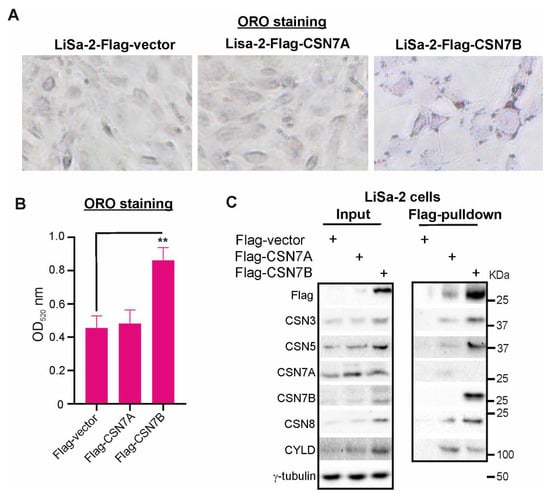
Figure 3.
The deubiquitylating enzyme CYLD is associated with the CSN and preferentially binds to CSNCSN7A variant. (A) Increased formation of LDs in LiSa-2 preadipocytes stably expressing Flag-CSN7B compared to control (Flag-vector) and Flag-CSN7A expressing cells. The three cell lines were stained with ORO and LD formation was visualized by light microscopy at 200-fold magnification. (B) Quantification of ORO stain as shown in (A). Cells were lysed and supernatants were measured spectrophotometrically at 520 nm. Results are expressed as means plus/minus standard deviations (SD, n = 3). Unpaired Student’s t-test was used for statistical analysis. ** p < 0.01. (C) Left panel: Western blot of LiSa-2-Flag, LiSa-2-Flag-CSN7A, and LiSa-2-Flag-CSN7B cells. Antibodies were used as indicated. Right panel: Flag-pulldowns of LiSa-2-Flag, LiSa-2-Flag-CSN7A, and LiSa-2-Flag-CSN7B cells.
Flag-pulldowns were performed to evaluate the interactomes of CSNFlag-CSN7A and CSNFlag-CSN7B in LiSa-2 cells (Figure 3C, right panel). Since the CSN is a multi-DUB complex [32], we were curious to see whether the CSN variants interact with CYLD, a DUB involved in hepatic homeostasis [36]. Strikingly, as shown in Figure 3C (left panel), CYLD was induced in Flag-CSN7A-LiSa-2 as well as in Flag-CSN7B-LiSa-2 cells. Flag-pulldowns revealed binding of CYLD to both CSN variants (Figure 3C, right panel). However, more CYLD protein was associated with precipitated CSNFlag-CSN7A as compared to CSNFlag-CSN7B, which was precipitated in larger amounts. Based on this data, we concluded that CYLD preferentially binds to the CSNFlag-CSN7A variant.
2.3. Reciprocal Expression of CYLD and CSN7A as Well as CSN7B
Interestingly, the downregulation of CSN7B using siCSN7B-RD in HepG2 cells led to an increase of CYLD (Figure 4A, input). Since the reduction of CSN7B caused a compensatory increase of CSN7A, we assumed that binding to CSNCSN7A stabilized CYLD protein. As expected, the subsequent immunoprecipitation of CSN7B revealed low amounts of CYLD associated with the CSNCSN7B complex, indicating that the stimulating effect of CSN7B silencing on CYLD expression is most likely due to the compensatory increase of CSN7A.
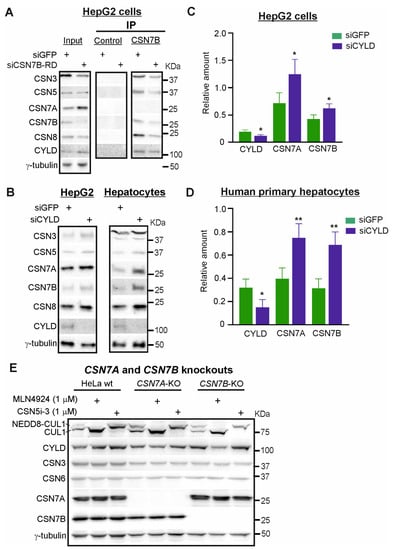
Figure 4.
Silencing of CYLD stabilizes CSN7A as well as CSN7B in HepG2 cells and in human primary hepatocytes. (A) Immunoprecipitation of CSN7B. Western blot of HepG2 cells transfected with siGFP or siCSN7B-RD for 48 h (input, left panel) and IP of CSN7B (middle and right panel). Antibodies were used as indicated. (B) Western blot of HepG2 cells (left panel) or human primary hepatocytes (right panel) transiently transfected with siGFP or siCYLD for 48 h. Antibodies were used as in (A). (C,D) Western blots as shown in (B) left panel, (C) or in (B) right panel, (D) were quantified by densitometry to measure the abundance of CYLD, CSN7A and CSN7B after transfection with siGFP (control) or siCYLD. Data were normalized to γ-tubulin and expressed as means of relative amounts plus/minus standard deviations (SD, n = 4). Unpaired Student’s t-test was used for statistical analysis. * p < 0.05, ** p < 0.01. (E) Western blots of HeLa wildtype (wt), HeLa-CSN7A-KO and HeLa-CSN7B-KO cells treated without or with MLN4924 (1 µM) or CSN5i-3 (1 µM) for 16 h. Antibodies were used as indicated.
To study the reciprocal effect of CYLD on CSN subunit expression, specific siRNA against CYLD (siCYLD) was transfected in HepG2 cells or human primary hepatocytes with a final concentration of 50 nM. After 48 h, cells were lysed for Western blots. Downregulation of CYLD by about 40–50% (Figure 4C,D) led to an increase of CSN subunits expression including CSN7A and CSN7B in HepG2 cells as well as in human primary hepatocytes. Increased expression of CSN7A and CSN7B induced by CYLD downregulation is more pronounced in human primary hepatocytes and amounts to 1.8-fold for CSN7A and 2.2-fold for CSN7B (Figure 4D).
To see whether the stability of CYLD depends on one of the CSN variants, we used CSN7A and CSN7B knockout cells which we established by CRISPR/Cas9 strategy in HeLa cells [31]. In addition, we used CSN5i-3, a specific inhibitor of CSN-mediated deneddylation [43] and the neddylation inhibitor MLN4924 [44], to ask whether degradation of CYLD depends on CSN-CRL axis. Neither the knockout of CSN7A nor that of CSN7B caused complete loss of CYLD, indicating that CSN variants have largely redundant functions in controlling CYLD stability. However, as shown in Figure 4E, CYLD expression in HeLa-CSN7B-KO cells was higher than in HeLa wt or HeLa-CSN7A-KO cells, perhaps induced by overexpression of CSN7A. In all HeLa derived cells MLN4924 (1 μM), completely blocked neddylation of cullin 1 (CUL1) whereas CSN5i-3 (1 μM) inhibited CSN-mediated deneddylation of CUL1. In both cases, prevention of the CSN-CRL pathway led to stabilization of CYLD in the parental HeLa cells, indicating degradation of CYLD at least in part occurred via the CSN-CRL-proteasome pathway.
2.4. Overexpression of Flag-CSN7A in Liver Cells Increases CYLD Expression
Flag-CSN7A or Flag-CSN7B were transiently transfected into HepG2 cells. After 24 h, transfected cells were treated with PA using indicated concentrations or control medium. After another 24 h, cells were harvested for Western blot and Flag-pulldowns. In lysates (input), the overexpression of Flag-CSN7A as well as Flag-CSN7B led to an increase of CSN subunit expression (Figure 5A, input), as it has been shown before in LiSa-2 cells stably overexpressing Flag-CSN7A or Flag-CSN7B (Figure 3C). CYLD expression was stimulated predominately by Flag-CSN7A independently of PA treatment. Flag pulldowns revealed that Flag-CSN7A and Flag-CSN7B were integrated into endogenous CSN complexes after 48 h of transfection, because all tested CSN subunits were co-precipitated. As shown before, more CYLD was associated with CSNFlag-CSN7A as compared to CSNFlag-CSN7B. Interestingly, PA treatment led to a reduction of CYLD, which is, however, accompanied by a similar decline of Flag-CSN7A and CSN3 (Figure 5A,B). CSNFlag-CSN7B-CYLD interaction was not affected by PA treatment (Figure 5A,C). Similar results were obtained with human primary hepatocytes (Figure 5D–F). The overexpression of Flag-CSN7A stimulated CYLD expression. Flag-pulldowns confirmed that CYLD preferentially binds to CSNFlag-CSN7A (Figure 5D, Flag-pulldowns). PA treatment caused decrease of CSNFlag-CSN7A accompanied by a decline of CYLD (Figure 5E), whereas CYLD binding to CSNFlag-CSN7B was not affected by PA (Figure 5F).
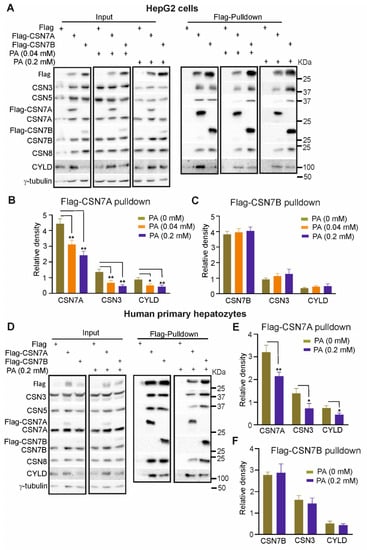
Figure 5.
Overexpression of CSN7A, but not of CSN7B, stabilizes CYLD. (A) Western blots of 1.2 × 107 HepG2 cells transiently transfected with 60 μg Flag-vector (control), Flag-CSN7A or Flag-CSN7B. 24 h after transfection, cells were treated with 0, 0.04 or 0.2 mM PA for another 24 h. Cells were harvested for Western blots (left three panels) or Flag pulldowns (right three panels). Antibodies were used as indicated. (B,C) Blots of Flag-pulldowns as shown in (A) (right three panels) were quantified by densitometry to measure the abundance of CSN7A, CSN3, and CYLD in Flag-CSN7A pulldowns (B) or of CSN7B, CSN3 and CYLD in Flag-CSN7B pulldowns (C) after treatment with 0, 0.04 and 0.2 mM PA. Data are expressed as means of relative density plus/minus standard deviations (SD, n = 3). Unpaired Student’s t-test was used for statistical analysis. * p < 0.05, ** p < 0.01. (D) Western blots of human primary hepatocytes transiently transfected with Flag-vector (control), Flag-CSN7A or Flag-CSN7B as in (A). Immediately after transfection, cells were treated with 0 or 0.2 mM PA for 24 h. Cells were harvested for Western blots (left two panels) or Flag pulldowns (right two panels). Antibodies were used as in (A). (E,F) Blots of Flag-pulldowns as shown in (D) (right two panels) were quantified by densitometry to measure the abundance of CSN7A, CSN3 and CYLD in Flag-CSN7A pulldowns (E) or CSN7B, CSN3 and CYLD in Flag-CSN7B pulldowns (F) after treatment with 0 and 0.2 mM PA. Data are expressed as means of relative density plus/minus standard deviations (SD, n = 3). Unpaired Student’s t-test was used for statistical analysis. * p < 0.05, ** p < 0.01.
2.5. Downregulation of CSN7B Induces CYLD and Hinders Steatosis in HepG2 Cells
Mild steatosis in HepG2 cells can be induced by endogenous lipids after five to eight days. Steatosis in HepG2 cells was monitored for two and eight days by ORO staining (Figure 6A,B). In the upper panel, the entire well was directly photographed from a six-well plate (25-fold amplification), whereas the lower panel is a 200-fold amplification. On day two, the number of lipid loaded LDs did not differ between control and siCSN7B-RD sample. However, on day eight steatosis was developed as indicated by lipid loaded LDs, and the downregulation of CSN7B significantly reduced the number of LDs formed indicating retardation of steatosis. This result was confirmed by quantification of ORO staining (Figure 6B).
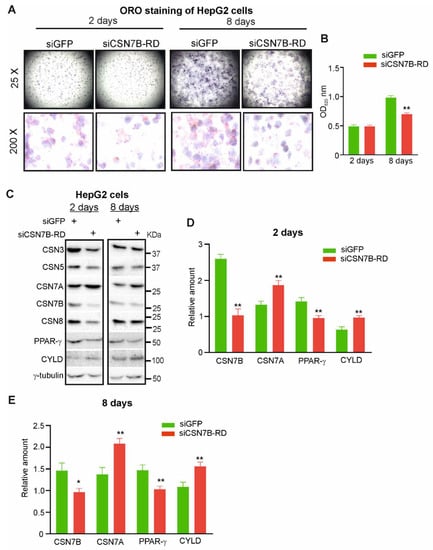
Figure 6.
Silencing of CSN7B retards hepatic steatosis in HepG2 cells by elevated CSNCSN7A stabilizing CYLD. (A) siGFP (control) or siCSN7B-RD were transiently transfected into HepG2 cells. Cells were harvested on day 2 and day 8 of hepatic steatosis and stained with ORO to visualize LD formation by light microscopy with a magnification of 25-fold (upper panels) or 200-fold (lower panels). (B) Quantification of ORO stain as shown in (A). Cells were lysed and supernatants were measured spectrophotometrically at 520 nm. Results are expressed as means plus/minus standard deviations (SD, n = 3). Unpaired Student’s t-test was used for statistical analysis. ** p < 0.01. (C) Western blot of HepG2 cells transfected with siGFP (control) or siCSN7B-RD and harvested after 2 days (left panel) or 8 days (right panel) of hepatic steatosis. Antibodies were used as indicated. (D,E) Blots of HepG2 cells transfected with siGFP (control) or siCSN7B-RD after two days of steatosis (D) or 8 days of steatosis (E) were quantified by densitometry to measure the abundance of CSN7A, CSN7B, PPAR-γ and CYLD. Data were normalized to γ-tubulin and expressed as means of relative amounts plus/minus standard deviations (SD, n = 3). Unpaired Student’s t-test was used for statistical analysis. * p < 0.05, ** p < 0.01.
In Figure 6C–E, using the above model, siGFP (control) or siCSN7B-RD were transfected in HepG2 cells and analyzed by Western blot after day two or day eight of steatosis. On day two, as expected, CSN7B was downregulated by about 60% causing an increase of CSN7A and of CYLD by about 30–40% and a concomitant decrease of PPAR-γ by more than 30% (Figure 6C,D). Although CSN7B downregulation on day eight was not as efficient anymore as on day two, there was still a significant increase of CSN7A as well as of CYLD and a considerable reduction of PPAR-γ (Figure 6E).
Collectively, our data demonstrate that steatosis can be retarded by CSN7B silencing causing a compensatory increase of CSN7A accompanied with CYLD stabilization and with the reduction of PPAR-γ.
3. Discussion
Data show that the CSN variants and their associated CYLD have a role in the pathogenesis of human fatty liver tissue. The CSN is a deneddylating enzyme complex and a major regulator of CRLs, conferring specifically to the UPS. As shown before in [31] and here, CSNCSN7A and CSNCSN7B variants substitute each other in terms of CSN-mediated deneddylating activity and binding to CRLs. Beyond that, the CSN variants are associated with additional proteins, which might account for specific functions in different locations, as shown in plants [45]. The CSN is a DUB complex belonging to the JAMM sub-family and it is associated with additional DUBs [32]. We have shown that the CSN binds to USP15 [46] and silencing of the CSN is accompanied by a reduction of USP15-mediated protection of CRL3-bound KEAP1 leading to inhibition of PPAR-γ expression and a blockade of adipogenesis [42]. In addition, USP48 is a predominantly nuclear CSN-associated DUB, stabilizing the nuclear pool of NF-κB transcription factor RelA upon TNF stimulation [47].
Here we demonstrate, for the first time, the association of the DUB CYLD with the DUB CSN. It has been demonstrated before that CYLD suppresses NASH in mice and monkeys [33]. Reduced CYLD expression in fatty liver tissue is associated with the severity of NAFLD. Under these conditions, it is believed that tripartite-motif-containing protein 47 (TRIM47) ubiquitylates CYLD following its degradation via 26S proteasome [33]. Supplementing CYLD represents a promising therapeutic strategy for NASH [33,48]. CYLD directly interacts with the kinase TAK1 and removes Lys63-ubiquitin chains. This impedes hyperactivation of JNK–p38 signaling mitigating NASH [33].
We observed a stable pool of CYLD binding to both CSNCSN7A as well as CSNCSN7B variants. On the other hand, there is a mobile CYLD pool, which binds exclusively to CSNCSN7A. Downregulation of CSN7B and knockout of CSN7B in HeLa cells is accompanied by a compensatory increase of CSN7A complex stabilizing CYLD. Likewise, the overexpression of CSN7A in liver cells protects CYLD. We conclude that CYLD preferentially interacts with the CSNCSN7A variant, which stabilizes the DUB. In our cell model of hepatic steatosis, downregulation of CSN7B in HepG2 cells after two and eight days revealed an increase of CSN7A that stabilized CYLD. Moreover, PPAR-γ was reduced. Interestingly, in other tissues increased PPAR-γ protects from triglyceride accumulation and insulin resistance [49]. In HepG2 cells, the stabilization of CYLD and decline of PPAR-γ led to a retardation of hepatic steatosis demonstrated by significant reduction of LD formation after eight days.
Our data shows that an increase of CSN7A, be it by downregulation of CSN7B or by overexpression of CSN7A, stabilizes CYLD and prevents development of hepatic steatosis. To our knowledge, there is currently no pharmacological treatment approved for NAFLD/NASH and weight loss. Following a diet and lifestyle intervention is the only therapy to reduce liver fat and insulin resistance [50,51]. Therefore, stabilizing CSNCSN7A and its interaction with CYLD might be a strategy to retard the pathogenesis of human fatty liver tissue.
4. Materials and Methods
4.1. Human Liver Histology
Human liver tissue was obtained from liver segments resected for medical reasons and examined by two pathologists from patients with fatty livers at the Department of Surgery at Charité—Universitätsmedizin Berlin, Campus Virchow Klinikum. All experiments were performed with the informed consent of the patients, in accordance with the declaration of Helsinki, and following the institutional and ethical guidelines (approval of the local Ethics Committee: EA1/289/16). Liver sections were stained with hematoxylin/eosin by standard methods. Sections from liver tissue were prepared, formalin-fixed, and stained with Oil-Red-O (ORO). Immunohistochemistry staining was performed with anti-CSN3 antibody (Abcam, ab12321) on liver sections, which were paraffin-embedded or frozen in optimal cutting temperature media.
4.2. Preparation of Human Hepatocytes and Cell Culture
Hepatocytes were isolated using an established two-step-perfusion protocol with collagenase P [52]. Briefly, under aseptic conditions the liver tissue was perfused through the vena cava with liver perfusion medium for 15 min at 37 °C. Thereafter, digestion media containing 1 mg collagenase P/mL (Roche Diagnostics GmbH, Mannheim, Germany) at 37 °C were infused for 8–12 min through the vena cava. The resulting cell suspension was then centrifuged (5 min, 50× g, 4 °C) to eliminate cell debris. After resuspending in culture medium (phenol red free Williams E with supplements: 1 µM insulin (Lilly, Indianapolis), 1 µM fortecortin (Merck Serono GmbH, Darmstadt, Germany), 1 mM sodium pyruvate, 10 mM HEPES-Buffer and 10% fetal calf serum (Biochrom)) the remaining cell suspension was subjected to percoll density separation by centrifugation through a percoll layer (25% percoll, Biochrom GmbH, Berlin, Germany) in phosphate buffered saline (1 g/mL, 300 mOsm) for 20 min at 1500× g and 4 °C. Cell pellets were re-suspended in ice-cold PBS and centrifuged (5 min, 50× g, 4 °C) to eliminate percoll particles. After resuspension, cells were counted and seeded onto collagen-coated culture plates (Biochrom GmbH, Berlin, Germany). Human HepG2 cells were culture as human hepatocytes (see above). Cell lines were tested on a regular basis with PCR Mycoplasma Test Kit I/C (PromoCell) to exclude mycoplasma contamination.
4.3. PA-Induced Steatosis in Cultured Hepatocytes
To induce steatosis under cell culture conditions, we treated HepG2 cells or human primary hepatocytes with PA. PA-induced steatosis is a routine method in our laboratory. The model is highly reproducible and stable under our conditions. To prepare a 20 mM stock solution, PA (Sigma P-9767) was complexed to a 40% BSA solution. The final mixture was sterilized and stored at 4 °C. To induce steatosis, cells were exposed to 0.04 or 0.2 mM PA for 24 h.
4.4. Knockouts of CSN7A or CSN7B and Downregulation of CSN Subunits and CYLD Using siRNA
Knockouts of CSN7A and CSN7B in HeLa cells were performed using the CRISPR/Cas9 strategy according to a protocol from Santa Cruz [31]. HeLa wildtype, HeLa-CSN7A-KO and HeLa-CSN7B-KO cells were treated with the deneddylation inhibitor CSN5i-3 (1 µM) (Novartis) and the neddylation inhibitor MLN4924 (1 µM) (Biochem) for 16 h.
For downregulation, cells were transfected with purchased siRNA using Lipofectamine® 2000 reagent (Invitrogen) following the reverse transfection protocol by the manufacturer. Commercial siRNA against CSN7B was purchased from Cell Signaling (Denver, CO, USA). SiCSN7B-RD was selected from a set of designed siRNAs synthesized by Eurofins (Germany). SiCYLD was purchased from Santa Cruz (sc-37326). Cells were plated at 50% confluency and then transfected with siRNA (50 nM) and subsequently incubated for 48 h, if not stated otherwise.
4.5. Western Blot Analysis
Cell lysates were prepared by standard methods and subjected to immunoblot analysis with the antibodies against the following proteins: CSN1 (BIOMOL, PW8285), CSN3 (Abcam, ab12321), CSN5 (Gene Tex, GTX70207), CSN6 (Santa Cruz, sc-137153), CSN7A (Santa Cruz, sc-398882), CSN7B (Abcam, ab11895), CSN8 (AFFINITI, PW8290), PPAR-γ (Santa Cruz, sc-7196), CYLD (Cell Signaling, #8462), Flag (Merck, F1804), and loading control γ-tubulin (Santa Cruz, sc-17787).
4.6. Flag-Pulldowns
Flag pulldowns were performed using Flag beads (Sigma, A2220) with the anti-Flag antibody as described before [30]. Briefly, cells were transfected with pCMV-3Tag-1a control vector (Stratagene, La Jolla, CA, USA) encoding 3 N-terminal Flag-tags or 3 × Flag-CSN7A cDNA or 3 × Flag-CSN7B cDNA. After 48 h, cells were lysed and the lysates were used for pulldowns. Experiments were replicated at least three times. Stable transfections with Flag, Flag-CSN7A, or Flag-CSN7B in LiSa-2 cells were established in our lab [30].
4.7. ORO Staining and Quantification of ORO
ORO staining was performed using the Thermo Scientific HyClone complete AdvanceSTEM™ Adipogenic differentiation kit (Thermo Fisher Scientific, Braunschweig, Germany, 10105453) as outlined earlier [42]. ORO was quantified according to the manufacturers’ protocol, as described before [21].
4.8. Statistical Analysis
X-ray films were quantified by densitometry using ImageJ software. Statistics were calculated with GraphPad Prism 8.0.1 software. Data are presented as mean ± standard deviation (SD). The unpaired, two-tailed Student’s t-test was used for statistical analysis. p < 0.05 was considered statistically significant.
Author Contributions
Conceptualization, X.H. and W.D.; methodology, X.H. and D.D.; formal analysis, X.H. and W.D.; investigation, X.H. and D.D.; writing—original draft, X.H. and W.D.; writing—review and editing, X.H., D.D. and W.D.; visualization, X.H. and W.D.; supervision, X.H. and W.D.; funding acquisition, W.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grant 31770813 awarded by the Natural Science Foundation of China to W.D.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and following the institutional and ethical guidelines of the Department of Surgery at Charité—Universitätsmedizin Berlin, Campus Virchow Klinikum (approval of the local Ethics Committee: EA1/289/16).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
The authors declare no competing interests.
References
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the american association for the study of liver diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. Nafld, nash and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Osna, N.A.; Kharbanda, K.K. Treatment options for alcoholic and non-alcoholic fatty liver disease: A review. World J. Gastroenterol. 2017, 23, 6549–6570. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Guo, G.L. Role of fxr in liver inflammation during nonalcoholic steatohepatitis. Curr. Pharmacol. Rep. 2017, 3, 92–100. [Google Scholar] [CrossRef]
- Maratos-Flier, E. Fatty liver and fgf21 physiology. Exp. Cell Res. 2017, 360, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (nafld)-pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Ruiz, I.; Medina, M.A.; Martinez-Poveda, B. From food to genes: Transcriptional regulation of metabolism by lipids and carbohydrates. Nutrients 2021, 13, 1513. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Ni, W.; Lin, S.; Bian, S.; Zheng, W.; Qu, L.; Fan, Y.; Lu, C.; Xiao, M.; Zhou, P. Usp7 mediates pathological hepatic de novo lipogenesis through promoting stabilization and transcription of znf638. Cell Death Dis. 2020, 11, 843. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992, 61, 761–807. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. Ring domain e3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Dubiel, W.; Dubiel, D.; Wolf, D.A.; Naumann, M. Cullin 3-based ubiquitin ligases as master regulators of mammalian cell differentiation. Trends Biochem. Sci. 2018, 43, 95–107. [Google Scholar] [CrossRef]
- Duda, D.M.; Borg, L.A.; Scott, D.C.; Hunt, H.W.; Hammel, M.; Schulman, B.A. Structural insights into nedd8 activation of cullin-ring ligases: Conformational control of conjugation. Cell 2008, 134, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Deshaies, R.J. Multimodal activation of the ubiquitin ligase scf by nedd8 conjugation. Mol. Cell 2008, 32, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, W.; Nhan, T.; Toth, J.I.; Petroski, M.D.; Wolf, D.A. Cand1 controls in vivo dynamics of the cullin 1-ring ubiquitin ligase repertoire. Nat. Commun. 2013, 4, 1642. [Google Scholar] [CrossRef]
- Zemla, A.; Thomas, Y.; Kedziora, S.; Knebel, A.; Wood, N.T.; Rabut, G.; Kurz, T. Csn- and cand1-dependent remodelling of the budding yeast scf complex. Nat. Commun. 2013, 4, 1641. [Google Scholar] [CrossRef]
- Pierce, N.W.; Lee, J.E.; Liu, X.; Sweredoski, M.J.; Graham, R.L.; Larimore, E.A.; Rome, M.; Zheng, N.; Clurman, B.E.; Hess, S.; et al. Cand1 promotes assembly of new scf complexes through dynamic exchange of f box proteins. Cell 2013, 153, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Dubiel, D.; Gierisch, M.E.; Huang, X.; Dubiel, W.; Naumann, M. Cand1-dependent control of cullin 1-ring ub ligases is essential for adipogenesis. Biochim. Biophys. Acta 2013, 1833, 1078–1084. [Google Scholar] [CrossRef]
- Reitsma, J.M.; Liu, X.; Reichermeier, K.M.; Moradian, A.; Sweredoski, M.J.; Hess, S.; Deshaies, R.J. Composition and regulation of the cellular repertoire of scf ubiquitin ligases. Cell 2017, 171, 1326–1339 e1314. [Google Scholar] [CrossRef]
- Liu, X.; Reitsma, J.M.; Mamrosh, J.L.; Zhang, Y.; Straube, R.; Deshaies, R.J. Cand1-mediated adaptive exchange mechanism enables variation in f-box protein expression. Mol. Cell 2018, 69, 773–786 e776. [Google Scholar] [CrossRef]
- Dubiel, D.; Ordemann, J.; Pratschke, J.; Dubiel, W.; Naumann, M. Cand1 exchange factor promotes keap1 integration into cullin 3-ring ubiquitin ligase during adipogenesis. Int. J. Biochem. Cell Biol. 2015, 66, 95–100. [Google Scholar] [CrossRef]
- Straube, R.; Shah, M.; Flockerzi, D.; Wolf, D.A. Trade-off and flexibility in the dynamic regulation of the cullin-ring ubiquitin ligase repertoire. PLoS Comput. Biol. 2017, 13, e1005869. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Bucher, P. The pci domain: A common theme in three multiprotein complexes. Trends Biochem. Sci. 1998, 23, 204–205. [Google Scholar] [CrossRef]
- Lingaraju, G.M.; Bunker, R.D.; Cavadini, S.; Hess, D.; Hassiepen, U.; Renatus, M.; Fischer, E.S.; Thoma, N.H. Crystal structure of the human cop9 signalosome. Nature 2014, 512, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Cavadini, S.; Fischer, E.S.; Bunker, R.D.; Potenza, A.; Lingaraju, G.M.; Goldie, K.N.; Mohamed, W.I.; Faty, M.; Petzold, G.; Beckwith, R.E.; et al. Cullin-ring ubiquitin e3 ligase regulation by the cop9 signalosome. Nature 2016, 531, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Cope, G.A.; Suh, G.S.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of predicted metalloprotease motif of jab1/csn5 in cleavage of nedd8 from cul1. Science 2002, 298, 608–611. [Google Scholar] [CrossRef]
- Huang, X.; Ordemann, J.; Pratschke, J.; Dubiel, W. Overexpression of cop9 signalosome subunits, csn7a and csn7b, exerts different effects on adipogenic differentiation. FEBS Open Bio 2016, 6, 1102–1112. [Google Scholar] [CrossRef]
- Wang, J.; Dubiel, D.; Wu, Y.; Cheng, Y.; Wolf, D.A.; Dubiel, W. Csn7b defines a variant cop9 signalosome complex with distinct function in DNA damage response. Cell Rep. 2021, 34, 108662. [Google Scholar] [CrossRef]
- Dubiel, W.; Chaithongyot, S.; Dubiel, D.; Naumann, M. The cop9 signalosome: A multi-dub complex. Biomolecules 2020, 10, 1082. [Google Scholar] [CrossRef]
- Ji, Y.X.; Huang, Z.; Yang, X.; Wang, X.; Zhao, L.P.; Wang, P.X.; Zhang, X.J.; Alves-Bezerra, M.; Cai, L.; Zhang, P.; et al. The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat. Med. 2018, 24, 213–223. [Google Scholar] [CrossRef]
- Komander, D.; Lord, C.J.; Scheel, H.; Swift, S.; Hofmann, K.; Ashworth, A.; Barford, D. The structure of the cyld usp domain explains its specificity for lys63-linked polyubiquitin and reveals a b box module. Mol. Cell 2008, 29, 451–464. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. Cyld is a deubiquitinating enzyme that negatively regulates nf-kappab activation by tnfr family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Hellerbrand, C.; Massoumi, R. Cylindromatosis--a protective molecule against liver diseases. Med. Res. Rev. 2016, 36, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fukushima, H.; North, B.J.; Nagaoka, Y.; Nagashima, K.; Deng, F.; Okabe, K.; Inuzuka, H.; Wei, W. Scfbeta-trcp regulates osteoclastogenesis via promoting cyld ubiquitination. Oncotarget 2014, 5, 4211–4221. [Google Scholar] [CrossRef] [PubMed]
- Dubiel, D.; Bintig, W.; Kahne, T.; Dubiel, W.; Naumann, M. Cul3 neddylation is crucial for gradual lipid droplet formation during adipogenesis. Biochim. Biophys. Acta 2017, 1864, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.Q.; Li, X.Y.; Wang, L.; Feng, Z.L.; Li, X.F.; Wen, Y.F.; Han, J.X. Palmitate induces fat accumulation by activating c/ebpbeta-mediated g0s2 expression in hepg2 cells. World J. Gastroenterol. 2017, 23, 7705–7715. [Google Scholar] [CrossRef]
- Labrie, M.; Lalonde, S.; Najyb, O.; Thiery, M.; Daneault, C.; Des Rosiers, C.; Rassart, E.; Mounier, C. Apolipoprotein d transgenic mice develop hepatic steatosis through activation of ppargamma and fatty acid uptake. PLoS ONE 2015, 10, e0130230. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to nash pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef]
- Huang, X.; Ordemann, J.; Muller, J.M.; Dubiel, W. The cop9 signalosome, cullin 3 and keap1 supercomplex regulates chop stability and adipogenesis. Biol. Open 2012, 1, 705–710. [Google Scholar] [CrossRef]
- Schlierf, A.; Altmann, E.; Quancard, J.; Jefferson, A.B.; Assenberg, R.; Renatus, M.; Jones, M.; Hassiepen, U.; Schaefer, M.; Kiffe, M.; et al. Targeted inhibition of the cop9 signalosome for treatment of cancer. Nat. Commun. 2016, 7, 13166. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Sintchak, M.D.; Gavin, J.M.; Liao, H.; Bruzzese, F.J.; Bump, N.J.; Soucy, T.A.; Milhollen, M.A.; Yang, X.; Burkhardt, A.L.; et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: The nedd8 e1 inhibitor mln4924 forms a nedd8-amp mimetic in situ. Mol. Cell 2010, 37, 102–111. [Google Scholar] [CrossRef]
- Jin, D.; Li, B.; Deng, X.W.; Wei, N. Plant cop9 signalosome subunit 5, csn5. Plant Sci. 2014, 224C, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hetfeld, B.K.; Helfrich, A.; Kapelari, B.; Scheel, H.; Hofmann, K.; Guterman, A.; Glickman, M.; Schade, R.; Kloetzel, P.M.; Dubiel, W. The zinc finger of the csn-associated deubiquitinating enzyme usp15 is essential to rescue the e3 ligase rbx1. Curr. Biol. 2005, 15, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Naumann, M. Csn-associated usp48 confers stability to nuclear nf-kappab/rela by trimming k48-linked ub-chains. Biochim. Biophys. Acta 2015, 1853, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Li, H. Innate immune regulatory networks in hepatic lipid metabolism. J. Mol. Med. 2019, 97, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Chen, M.M.; Cai, J.J.; Yu, Y.; She, Z.G.; Li, H. Current and emerging approaches for nonalcoholic steatohepatitis treatment. Gene Expr. 2019, 19, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Horner, R.; Gassner, J.; Kluge, M.; Tang, P.; Lippert, S.; Hillebrandt, K.H.; Moosburner, S.; Reutzel-Selke, A.; Pratschke, J.; Sauer, I.M.; et al. Impact of percoll purification on isolation of primary human hepatocytes. Sci. Rep. 2019, 9, 6542. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).