
Stability of the TiO2 Nanuclusters Supported on Fe2O3-Hematite for Application in Electrocatalytic Water Splitting—An Insight from DFT Simulations

 , ,
, ,
Abstract
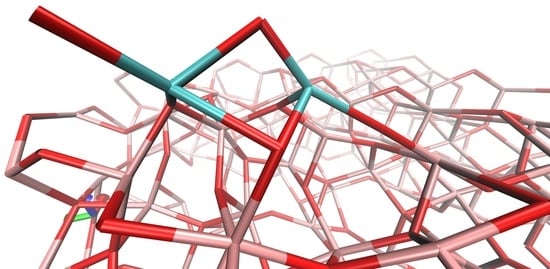
:
1. Introduction
2. Aim and Scope of the Work
3. Model and Computational Details
3.1. Energy of the Systems
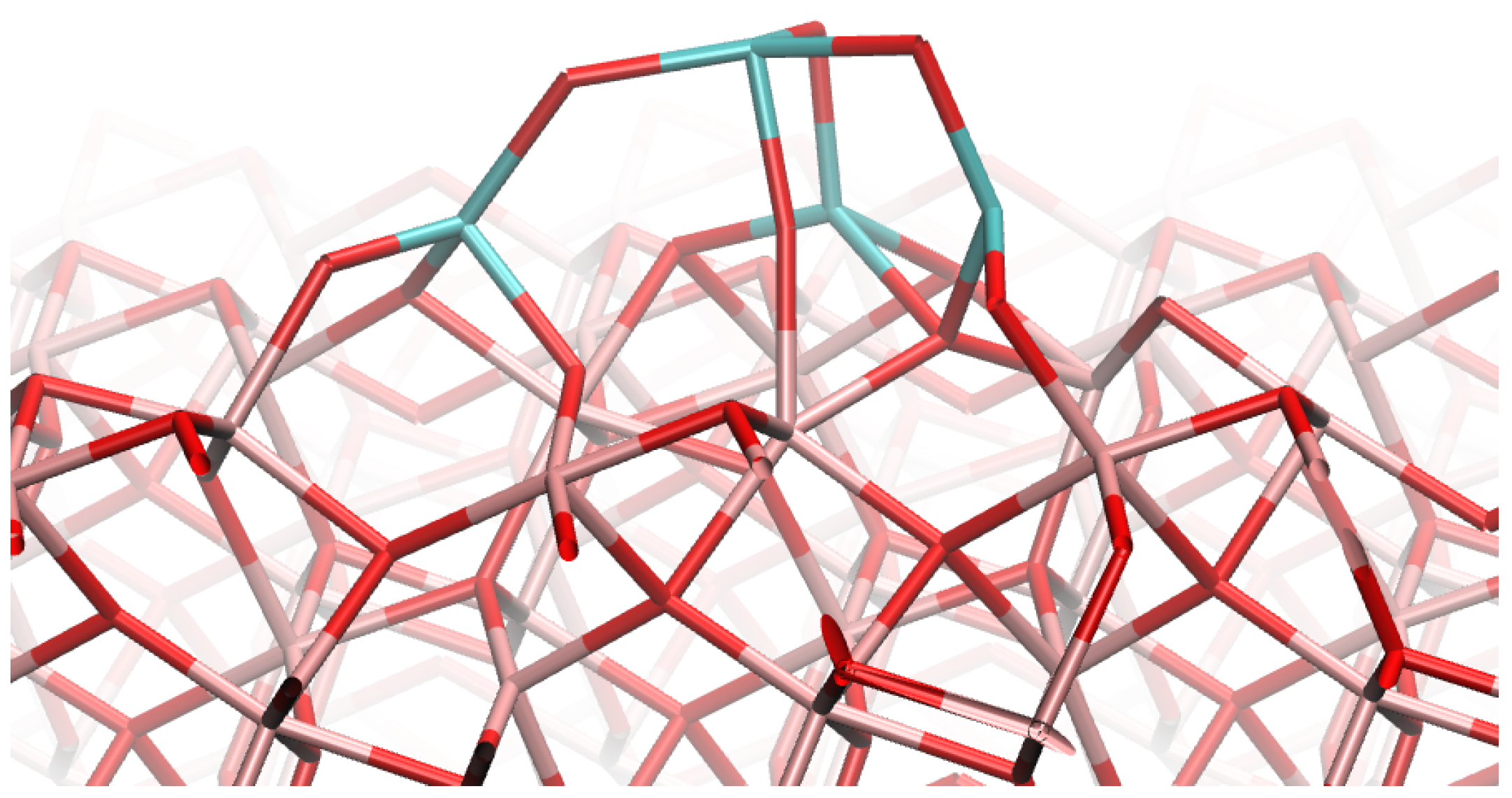
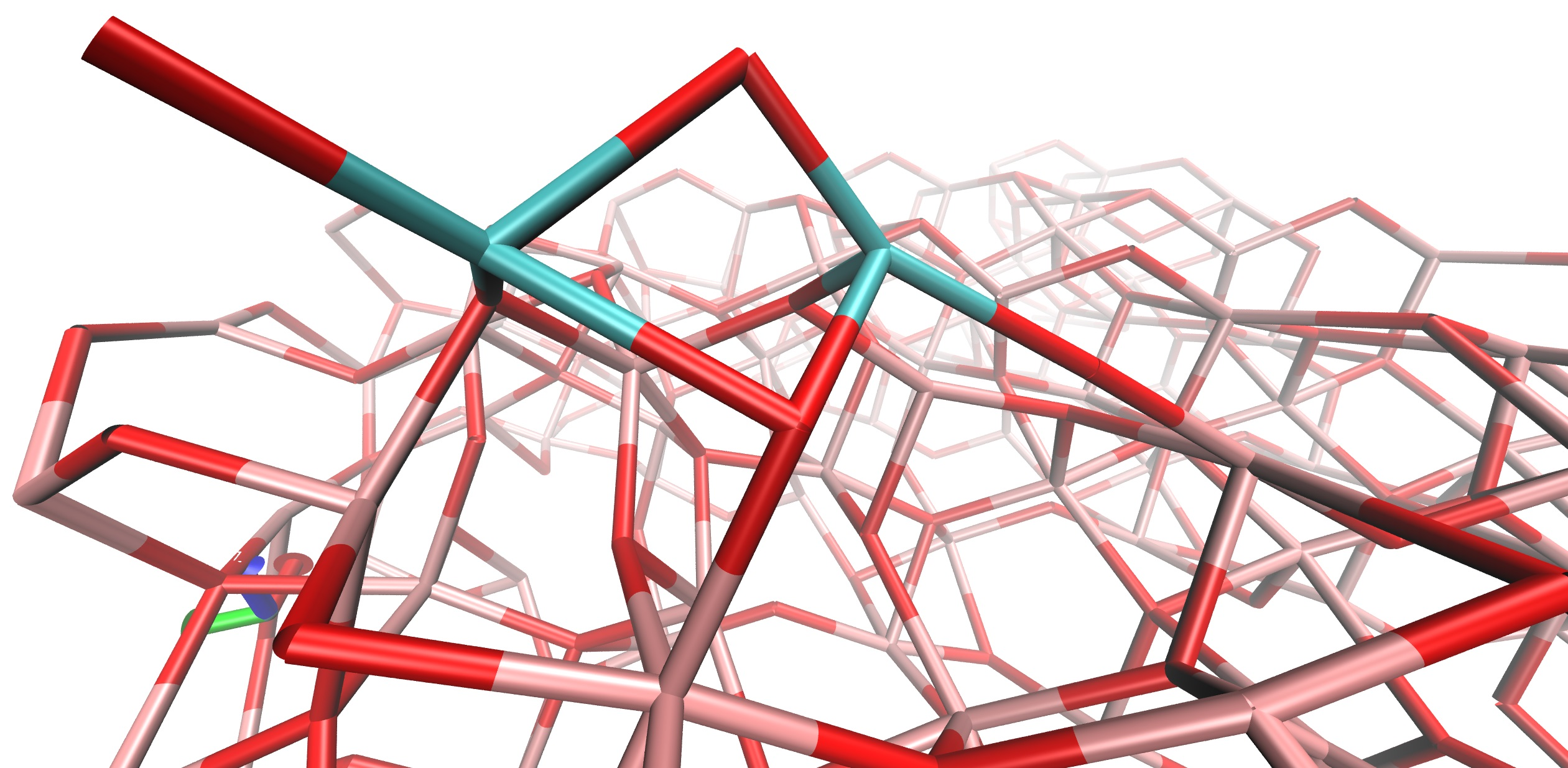
3.2. Base Model
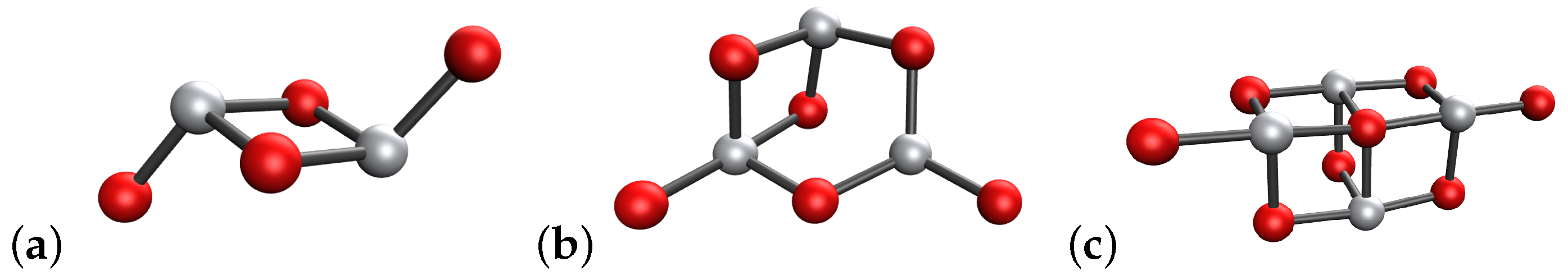
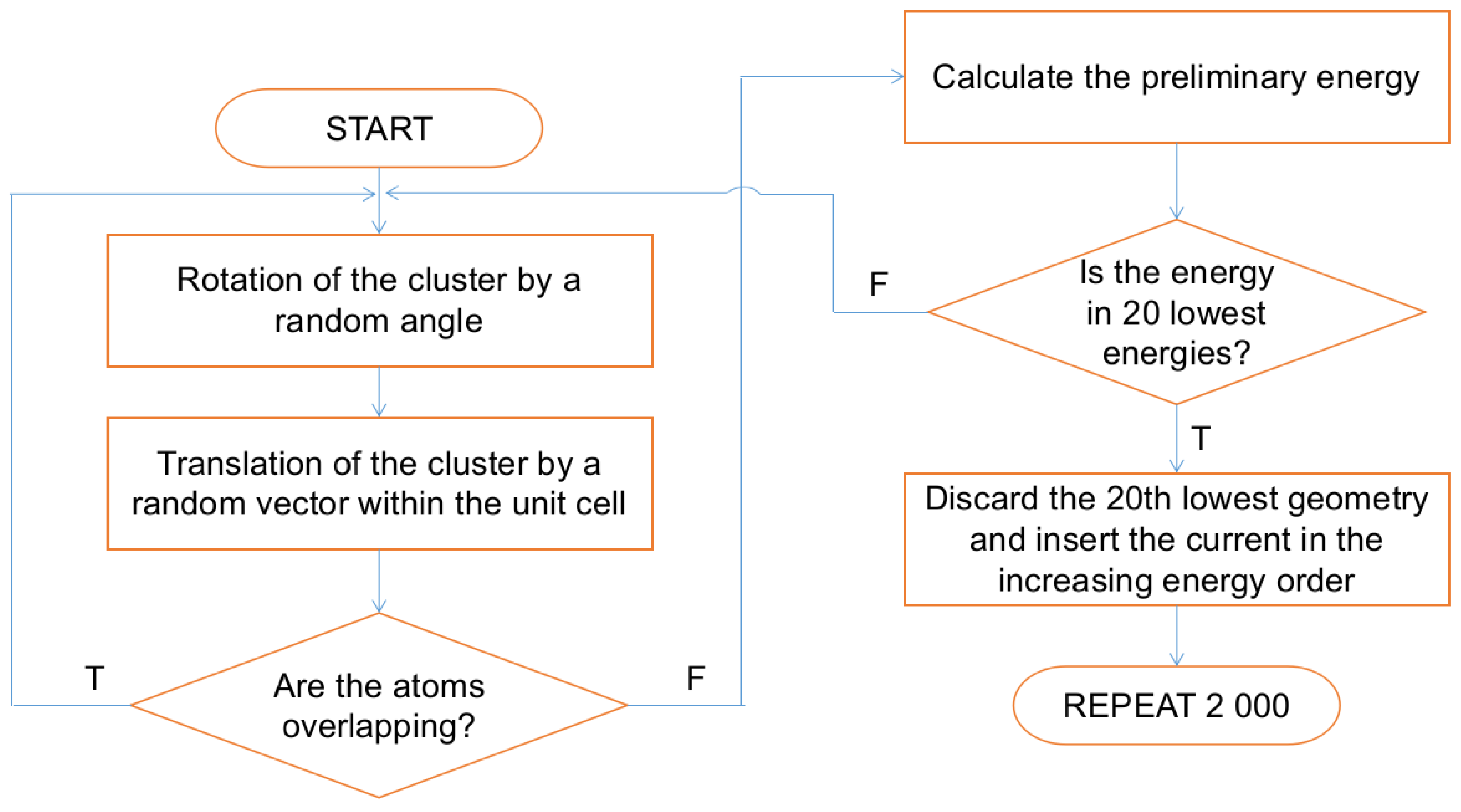
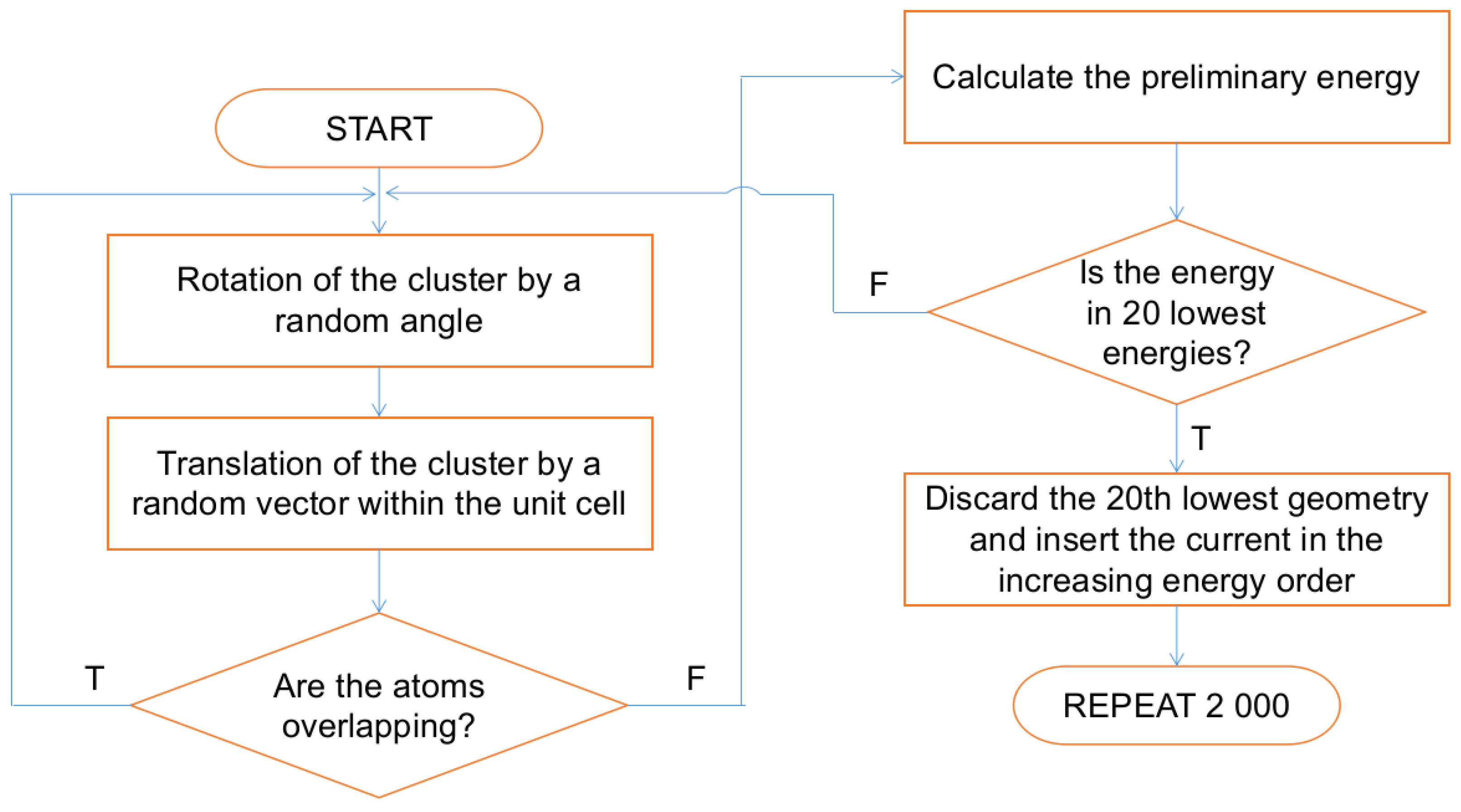
3.3. Initial Geometries
3.4. Molecular Dynamics Simulations
4. Results and Discussion
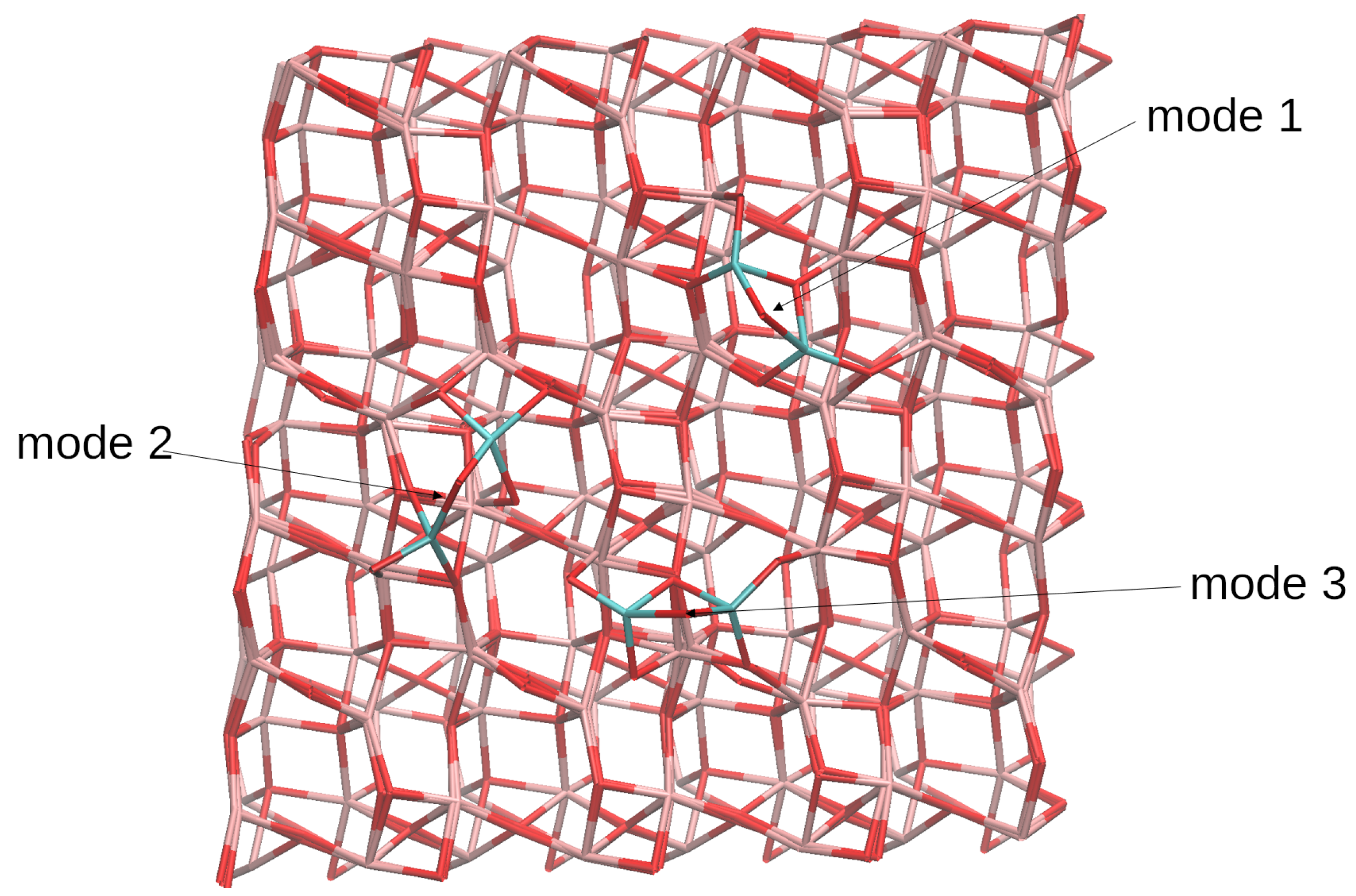
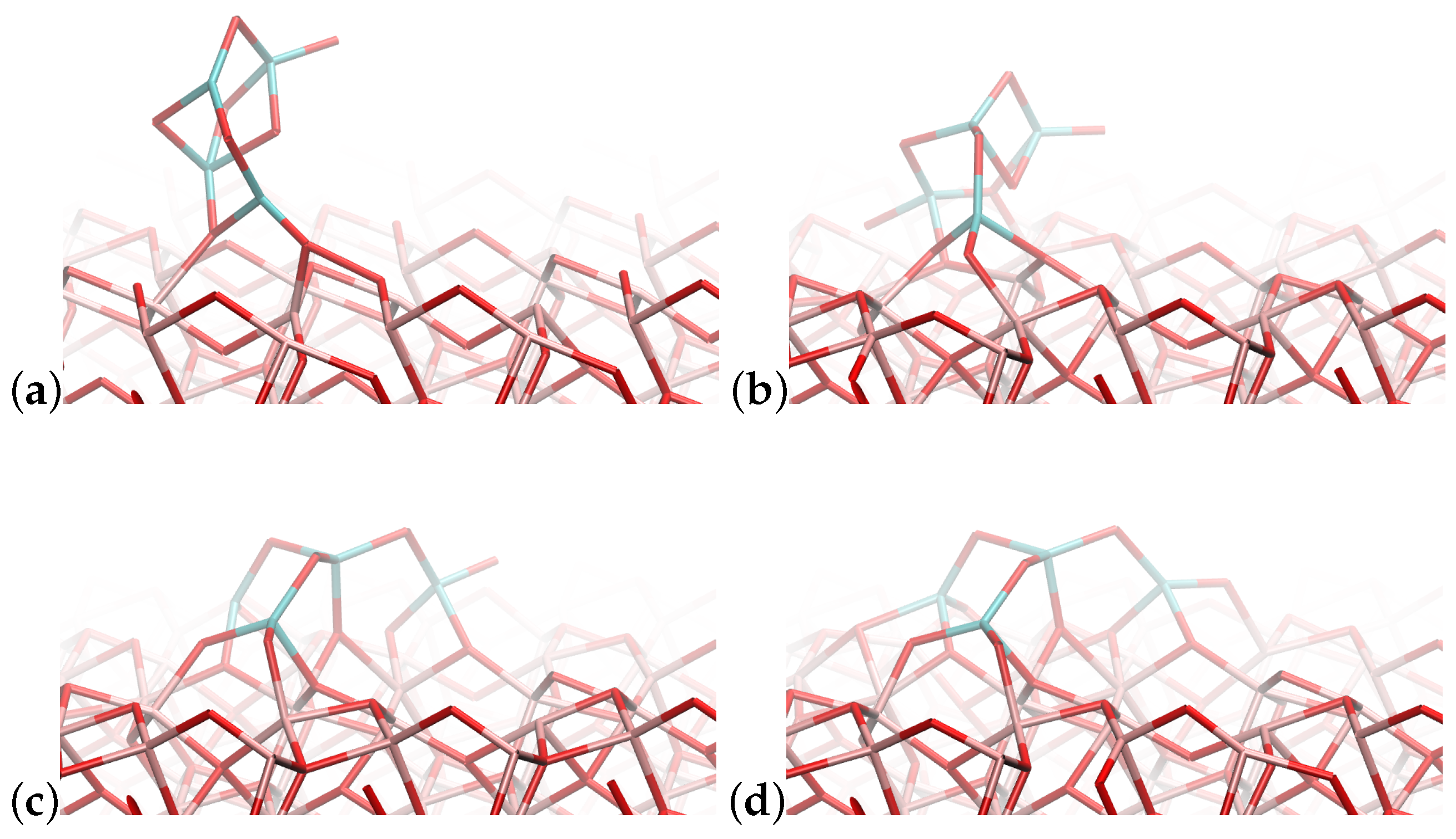
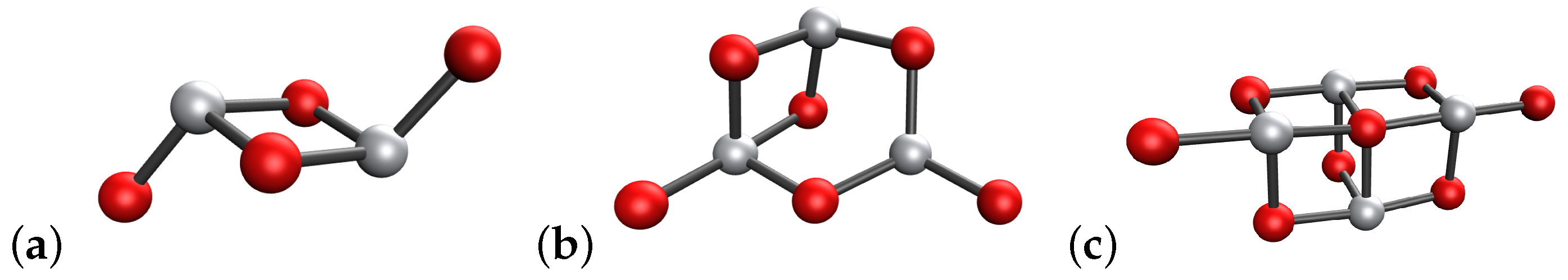
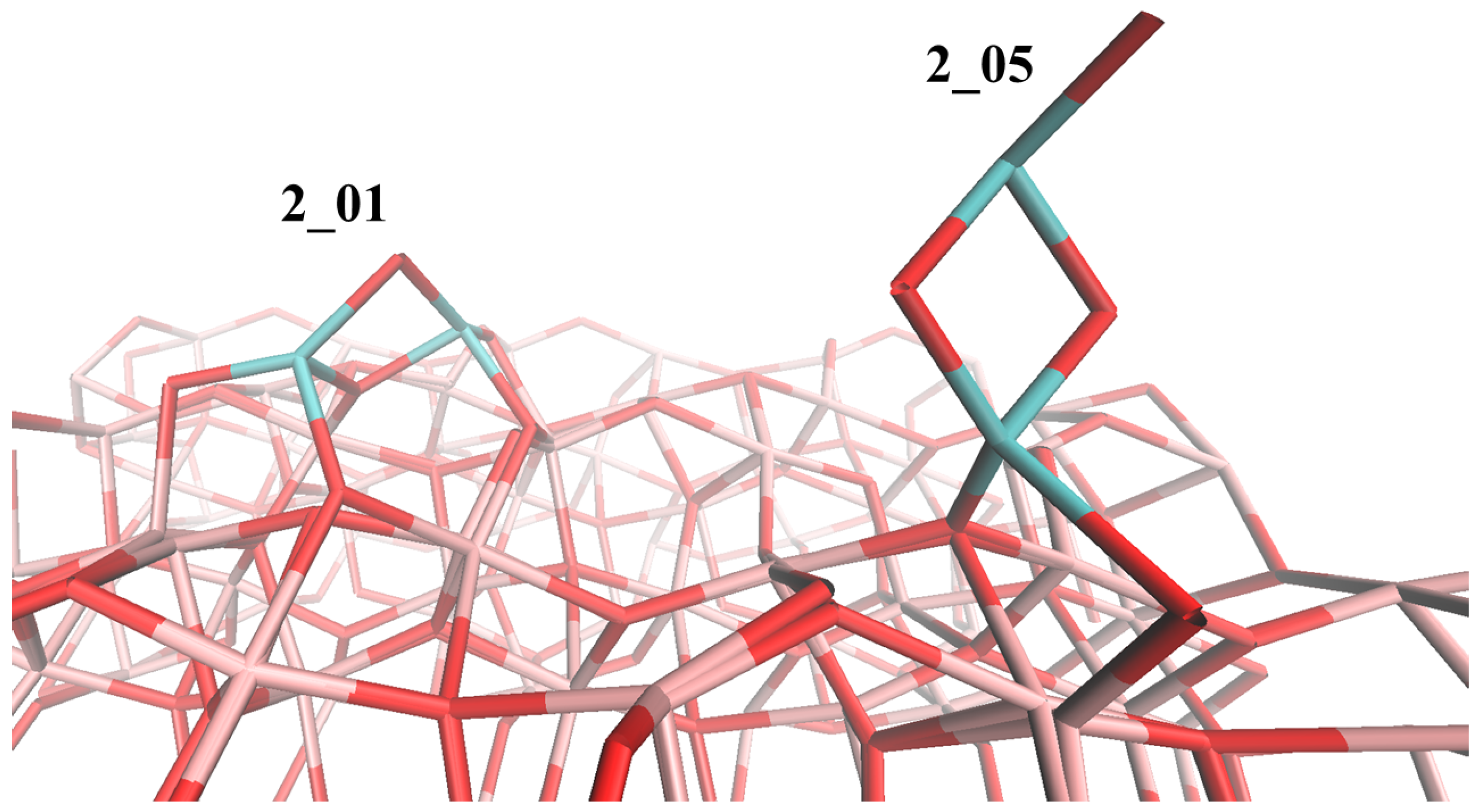
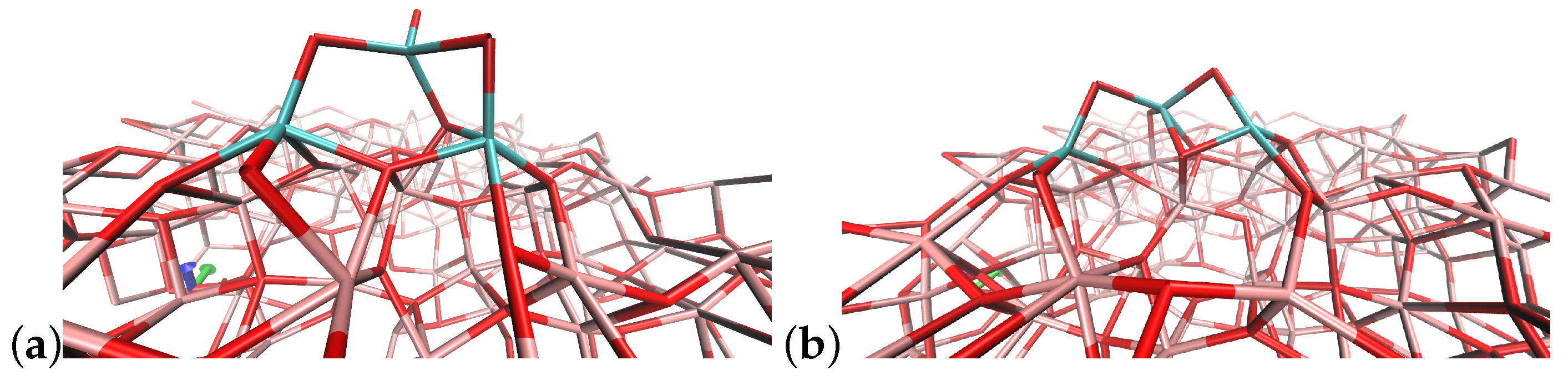
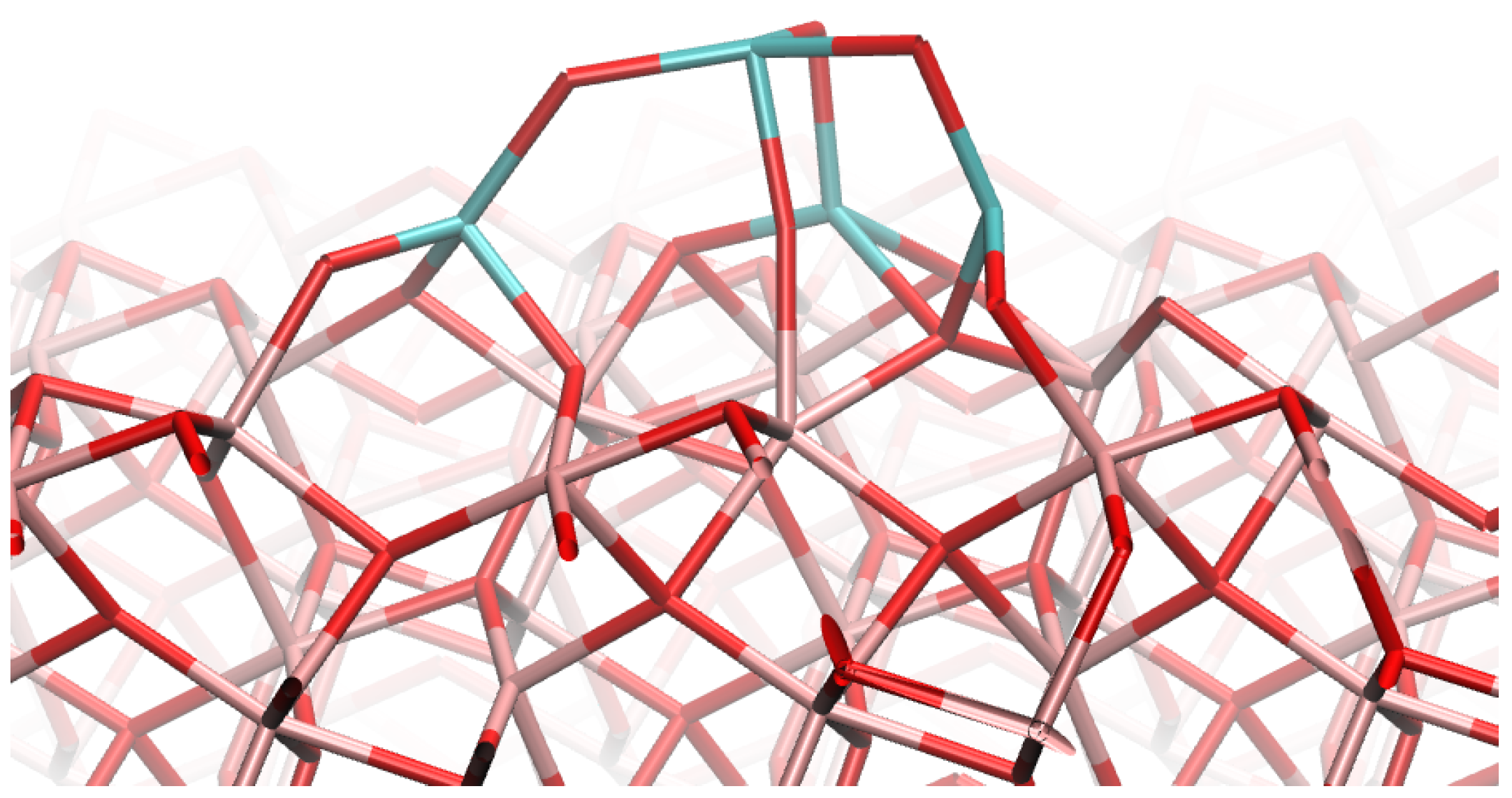
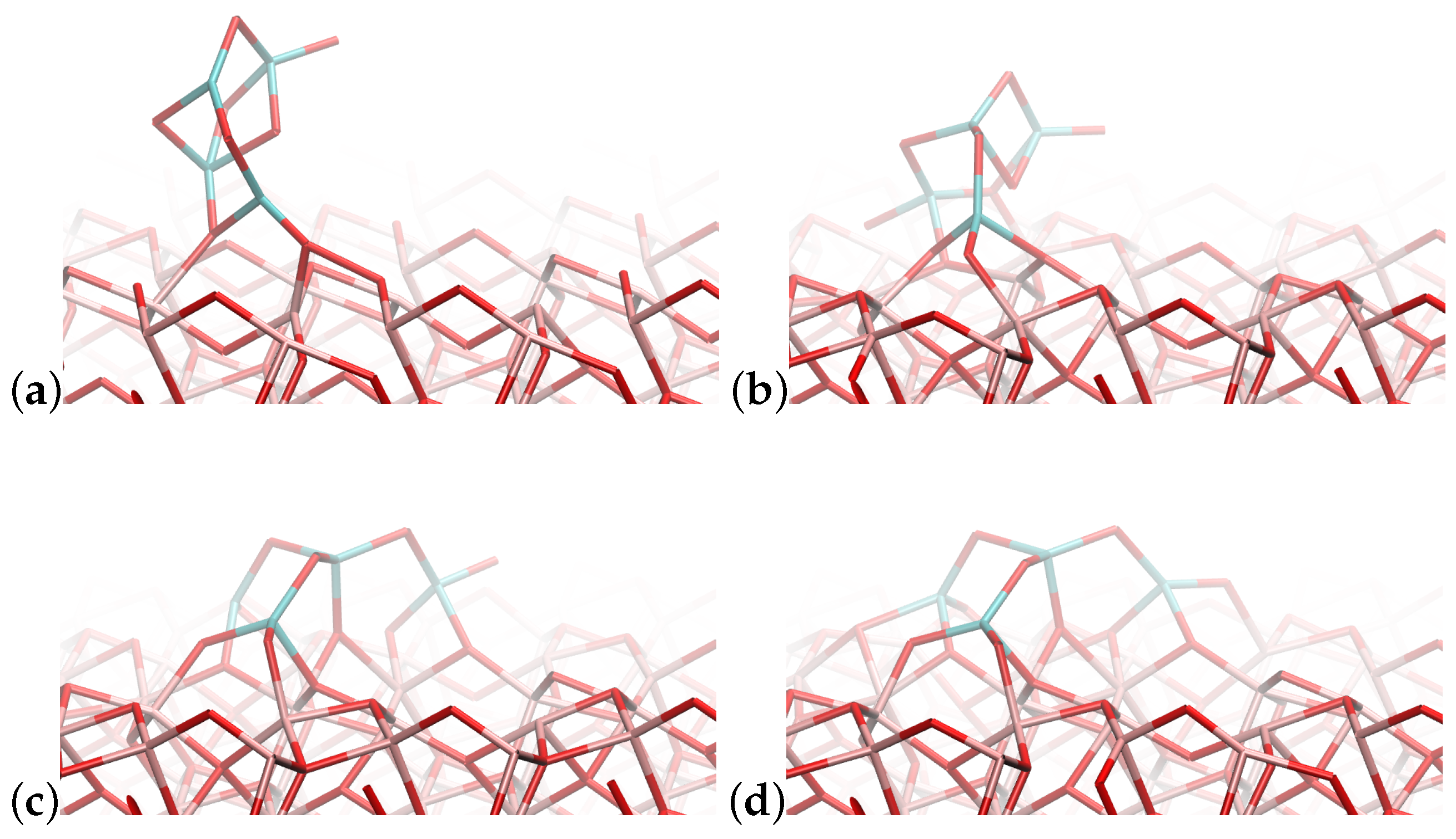
4.1. (TiO2)2 Systems
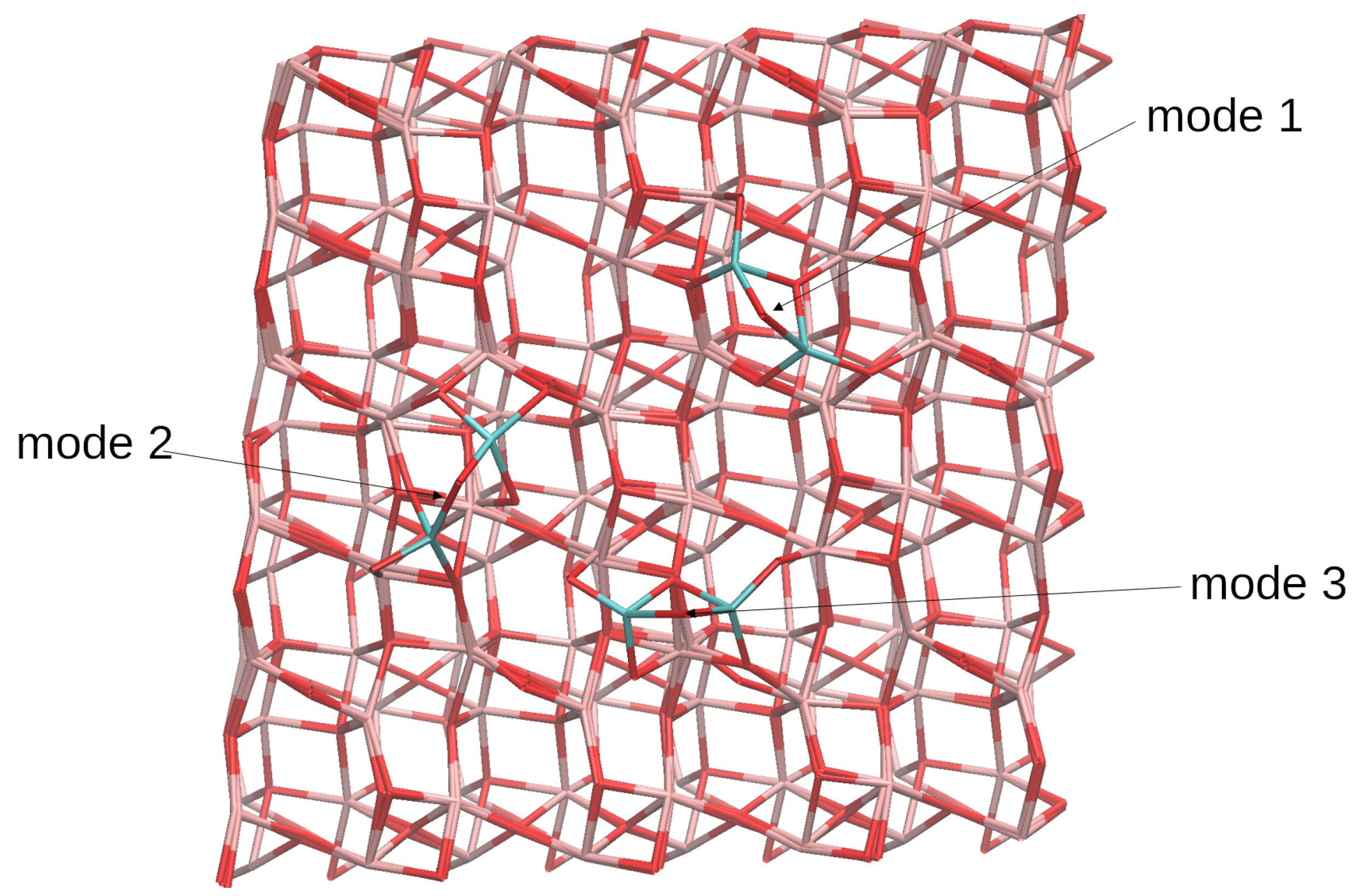
4.2. (TiO2)3 Systems
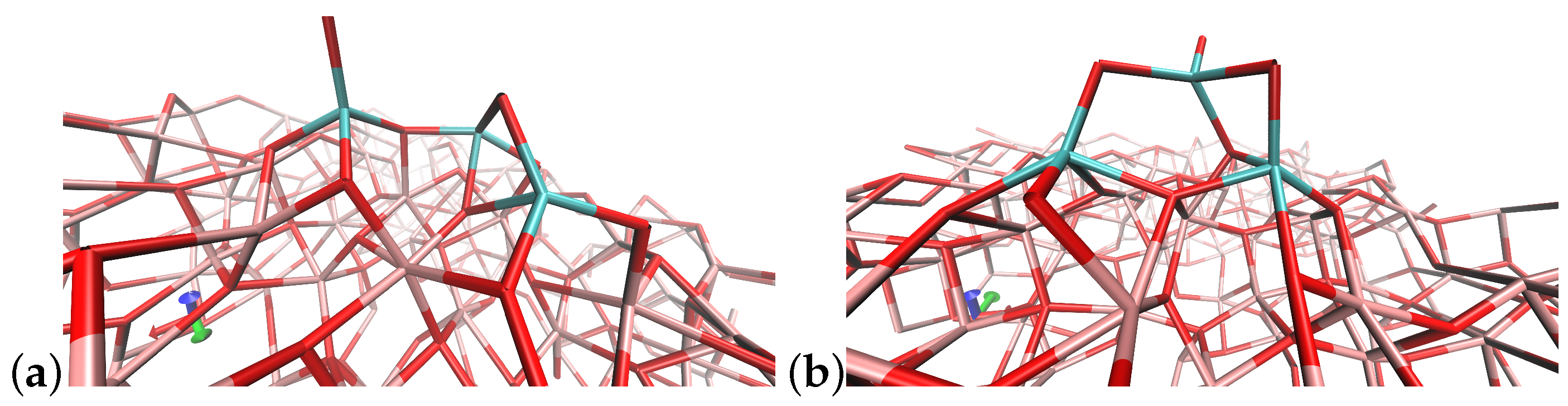
4.3. (TiO2)4 Systems
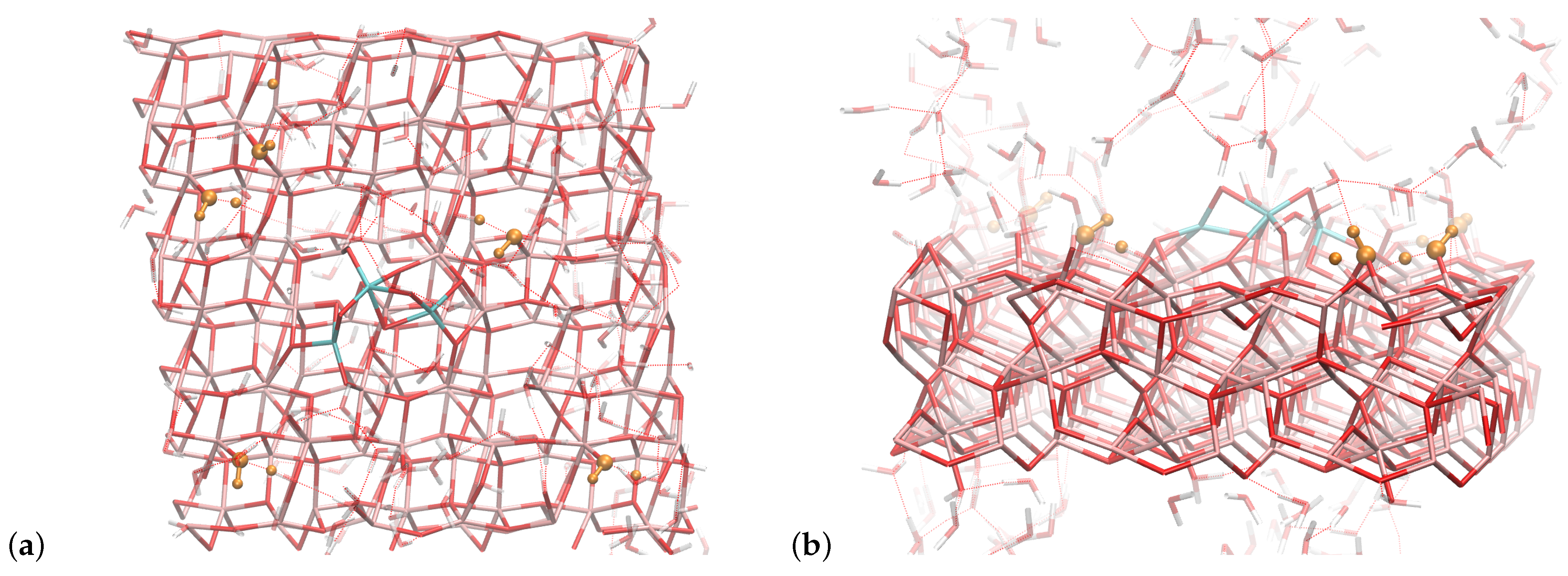
4.4. Solvent Effect
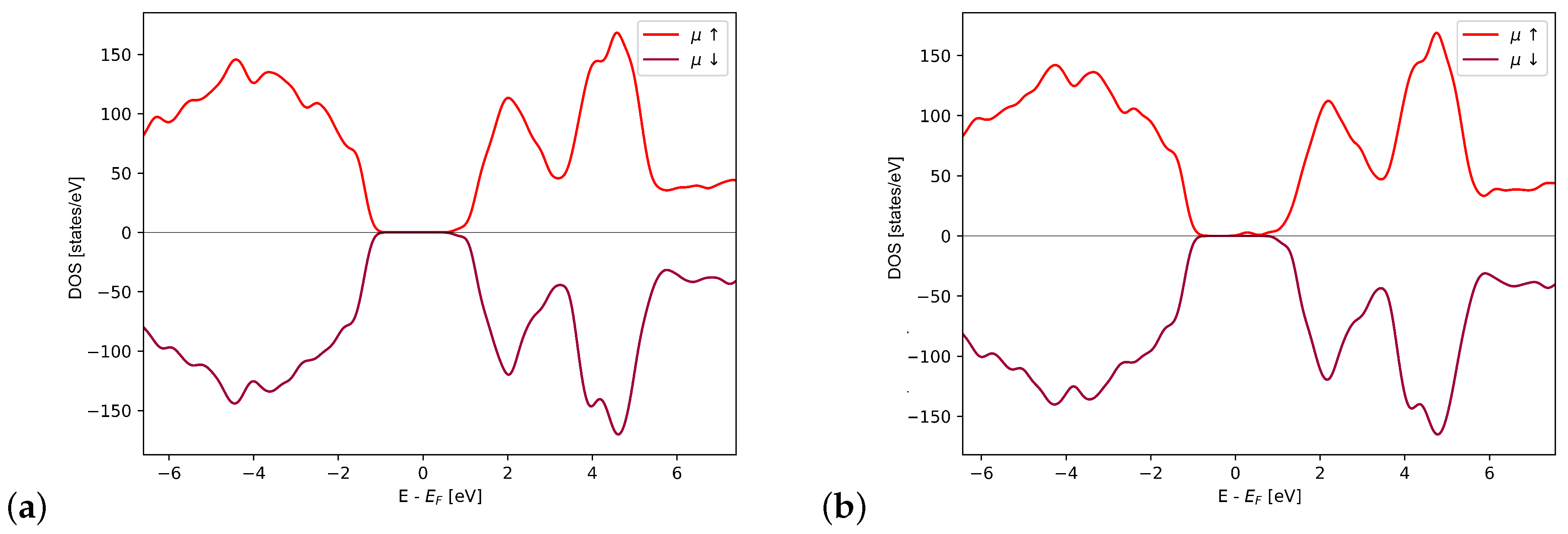
4.5. Density of States
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosa, L.P.; Ribeiro, S.K. The present, past, and future contributions to global warming of CO2 emissions from fuels a key for negotiation in the climate convention. Clim. Chang. 2001, 48, 289–307. [Google Scholar] [CrossRef]
- Hansen, J.; Sato, M.; Ruedy, R.; Lacis, A.; Oinas, V. Global warming in the twenty-first century: An alternative scenario. Proc. Natl. Acad. Sci. USA 2000, 97, 9875–9880. [Google Scholar] [CrossRef]
- Crabtree, G.W.; Dresselhaus, M.S.; Buchanan, M.V. The hydrogen economy. Phys. Today 2004, 57, 39–44. [Google Scholar] [CrossRef]
- Shiva Kumar, S.; Himabindu, V. Hydrogen production by PEM water electrolysis—A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar] [CrossRef]
- Ajanovic, A.; Sayer, M.; Haas, R. The economics and the environmental benignity of different colors of hydrogen. Int. J. Hydrogen Energy 2022, 47, 24136–24154. [Google Scholar] [CrossRef]
- Arcos, J.M.M.; Santos, D.M.F. The Hydrogen Color Spectrum: Techno-Economic Analysis of the Available Technologies for Hydrogen Production. Gases 2023, 3, 25–46. [Google Scholar] [CrossRef]
- Ali, D.M.; Salman, S.K. A comprehensive review of the fuel cells technology and hydrogen economy. In Proceedings of the 41st International Universities Power Engineering Conference, UPEC 2006, Newcastle upon Tyne, UK, 6–8 September 2006; Volume 1, pp. 98–102. [Google Scholar] [CrossRef]
- Zegers, P. Fuel cell commercialization: The key to a hydrogen economy. J. Power Sources 2006, 154, 497–502. [Google Scholar] [CrossRef]
- McGlashan, N.R. The thermodynamics of chemical looping combustion applied to the hydrogen economy. Int. J. Hydrogen Energy 2010, 35, 6465–6474. [Google Scholar] [CrossRef]
- Christensen, C.H.; Johannessen, T.; Sørensen, R.Z.; Nørskov, J.K. Towards an ammonia-mediated hydrogen economy? Catal. Today 2006, 111, 140–144. [Google Scholar] [CrossRef]
- Zhao, C.; Kou, Y.; Lemonidou, A.; Li, X.; Lercher, J. Highly Selective Catalytic Conversion of Phenolic Bio-Oil to Alkanes. Angew. Chem. 2009, 121, 4047–4050. [Google Scholar] [CrossRef]
- Hellinger, M.; Baier, S.; Mortensen, P.M.; Kleist, W.; Jensen, A.D.; Grunwaldt, J.D. Continuous Catalytic Hydrodeoxygenation of Guaiacol over Pt/SiO2 and Pt/H-MFI-90. Catalysts 2015, 5, 1152–1166. [Google Scholar] [CrossRef]
- Cui, K.; Yang, L.; Ma, Z.; Yan, F.; Wu, K.; Sang, Y.; Chen, H.; Li, Y. Selective conversion of guaiacol to substituted alkylphenols in supercritical ethanol over MoO3. Appl. Catal. B Environ. 2017, 219, 592–602. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, Q.; Xia, R.; Meyer, T.J. CO2 Reduction: From Homogeneous to Heterogeneous Electrocatalysis. Accounts Chem. Res. 2020, 53, 255–264. [Google Scholar] [CrossRef]
- Nabgan, W.; Nabgan, B.; Jalil, A.A.; Ikram, M.; Hussain, I.; Bahari, M.B.; Tran, T.V.; Alhassan, M.; Owgi, A.H.; Parashuram, L.; et al. A bibliometric examination and state-of-the-art overview of hydrogen generation from photoelectrochemical water splitting. Int. J. Hydrogen Energy, 2023; in press. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, J.; Xu, T.; Ji, W.; Zong, X. Recent Advances on Small Band Gap Semiconductor Materials (≤2.1 eV) for Solar Water Splitting. Catalysts 2023, 13, 728. [Google Scholar] [CrossRef]
- Tamirat, A.G.; Rick, J.; Dubale, A.A.; Su, W.N.; Hwang, B.J. Using hematite for photoelectrochemical water splitting: A review of current progress and challenges. Nanoscale Horizons 2016, 1, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Oh, H.J.; Yun, K.S.; Jung, S.C.; Kang, W.; Kim, S.J. Effect of TiO2 supporting layer on Fe2O3 photoanode for efficient water splitting. Prog. Org. Coatings 2013, 76, 1869–1873. [Google Scholar] [CrossRef]
- Satti, U.Q.; Zaidi, S.J.A.; Riaz, A.; ur Rehman, M.A.; Li, C.X.; Basit, M.A. Simple two-step development of TiO2/Fe2O3 nanocomposite for oxygen evolution reaction (OER) and photo-bio active applications. Colloids Surfaces A Physicochem. Eng. Asp. 2023, 671, 131662. [Google Scholar] [CrossRef]
- Niu, Y.; Li, M.; Jia, X.; Shi, Z.; Liu, H.; Zhang, X. Structures and photocatalytic activity of α-Fe2O3@TiO2 core-shell nanoparticles. Solid State Commun. 2022, 345, 114683. [Google Scholar] [CrossRef]
- Sharma, V.; Dixit, M.; Satsangi, V.R.; Dass, S.; Pal, S.; Shrivastav, R. Photoelectrochemical splitting of water with nanocrystalline Zn1-xMnxO thin films: First-principle DFT computations supporting the systematic experimental endeavor. Int. J. Hydrogen Energy 2014, 39, 3637–3648. [Google Scholar] [CrossRef]
- Mavros, M.G.; Tsuchimochi, T.; Kowalczyk, T.; McIsaac, A.; Wang, L.P.; Voorhis, T.V. What can density functional theory tell us about artificial catalytic water splitting? Inorg. Chem. 2014, 53, 6386–6397. [Google Scholar] [CrossRef]
- Yatom, N.; Neufeld, O.; Caspary Toroker, M. Toward Settling the Debate on the Role of Fe2O3 Surface States for Water Splitting. J. Phys. Chem. C 2015, 119, 24789–24795. [Google Scholar] [CrossRef]
- Ahn, H.J.; Kment, S.; Naldoni, A.; Zbořil, R.; Schmuki, P. Band gap and Morphology Engineering of Hematite Nanoflakes from an Ex Situ Sn Doping for Enhanced Photoelectrochemical Water Splitting. ACS Omega 2022, 7, 35109–35117. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, F.; Tranca, D.C.; Szyja, B.M.; Van Santen, R.A.; Martínez-Mesa, A.; Uranga-Piña, L.; Seifert, G. Water Splitting on TiO2-Based Electrochemical Cells: A Small Cluster Study. J. Phys. Chem. C 2016, 120, 437–449. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Erratum: Generalized gradient approximation made simple (Physical Review Letters (1996) 77 (3865)). Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]
- Podsiadły-Paszkowska, A.; Tranca, I.; Szyja, B.M. Tuning the Hematite (110) Surface Properties to Enhance Its Efficiency in Photoelectrochemistry. J. Phys. Chem. C 2019, 123, 5401–5410. [Google Scholar] [CrossRef]
- Szyja, B.M.; Podsiadły-Paszkowska, A. Helping Thy Neighbor: How Cobalt Doping Alters the Electrocatalytic Properties of Hematite. J. Phys. Chem. Lett. 2020, 11, 4402–4407. [Google Scholar] [CrossRef] [PubMed]
- Włodarczyk, A.; Uchroński, M.; Podsiadły-Paszkowska, A.; Irek, J.; Szyja, B.M. Mixing ReaxFF parameters for transition metal oxides using force-matching method. J. Mol. Model. 2022, 28. [Google Scholar] [CrossRef] [PubMed]
- Sandratskii, L.M.; Uhl, M.; Kübler, J. Band theory for electronic and magnetic properties of. J. Phys. Condens. Matter 1996, 8, 983. [Google Scholar] [CrossRef]
- Qu, Z.W.; Kroes, G.J. Theoretical study of the electronic structure and stability of titanium dioxide clusters (TiO2)n with n = 1–9. J. Phys. Chem. B 2006, 110, 8998–9007. [Google Scholar] [CrossRef]
- Hjorth Larsen, A.; Jørgen Mortensen, J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The atomic simulation environment—A Python library for working with atoms. J. Phys. Condens. Matter 2017, 29, 27. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G.; Holian, B.L. Kinetic moments method for the canonical ensemble distribution. Phys. Lett. A 1996, 211, 253–257. [Google Scholar] [CrossRef]
- Lo, C.S.; Tanwar, K.S.; Chaka, A.M.; Trainor, T.P. Density functional theory study of the clean and hydrated hematite (1102) surfaces. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 075425. [Google Scholar] [CrossRef]
- Schienbein, P.; Blumberger, J. Nanosecond solvation dynamics of the hematite/liquid water interface at hybrid DFT accuracy using committee neural network potentials. Phys. Chem. Chem. Phys. 2022, 24, 15365–15375. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Liu, C.; Huang, R.; Zhang, H.; Li, X.; García de Arquer, F.P.; Liu, Z.; Li, Y.; Zhang, B. Introducing Brønsted acid sites to accelerate the bridging-oxygen-assisted deprotonation in acidic water oxidation. Nat. Commun. 2022, 13, 4871. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System ID | E (opt) | E (MD) | System ID | E (opt) | E (MD) |
|---|---|---|---|---|---|
| 2_00 | 5.03 | 0.07 | 2_10 | 0.89 | 0.40 |
| 2_01 | 0.00 | 0.15 | 2_11 | 0.57 | 0.72 |
| 2_02 | 6.10 | 0.41 | 2_12 | 0.00 | 0.00 |
| 2_03 | 6.10 | 0.06 | 2_13 | 5.23 | 0.59 |
| 2_04 | 0.32 | 0.49 | 2_14 | 5.24 | 0.62 |
| 2_05 | 6.67 | 1.58 | 2_15 | 5.07 | 0.13 |
| 2_06 | 0.46 | 0.60 | 2_16 | 1.20 | 1.37 |
| 2_07 | 0.33 | 0.50 | 2_17 | 6.88 | 1.79 |
| 2_08 | 0.00 | 0.14 | 2_18 | 1.26 | 1.44 |
| 2_09 | 0.84 | 0.59 | 2_19 | 6.59 | 1.93 |
| System ID | E (opt) | E (MD) | System ID | E (opt) | E (MD) |
|---|---|---|---|---|---|
| 3_00 | 3.22 | 3.75 | 3_10 | 2.07 | 1.77 |
| 3_01 | 1.53 | 1.13 | 3_11 | 3.64 | 4.01 |
| 3_02 | 4.44 | 1.14 | 3_12 | 0.00 | 0.58 |
| 3_03 | 3.59 | 3.75 | 3_13 | 3.81 | 3.07 |
| 3_04 | 1.00 | 1.46 | 3_14 | 3.59 | 4.14 |
| 3_05 | 0.74 | 1.20 | 3_15 | 5.57 | 2.72 |
| 3_06 | 4.23 | – | 3_16 | 2.46 | 0.00 |
| 3_07 | 1.32 | 0.07 | 3_17 | 3.96 | 1.08 |
| 3_08 | 0.61 | 3.75 | 3_18 | 2.22 | 2.72 |
| 3_09 | 4.01 | 4.36 | 3_19 | 2.94 | 1.40 |
| System ID | E (opt) | E (MD) | System ID | E (opt) | E (MD) |
|---|---|---|---|---|---|
| 4_00 | 0.29 | 2.32 | 4_10 | 0.73 | 2.71 |
| 4_01 | 0.69 | 2.69 | 4_11 | 0.50 | 0.90 |
| 4_02 | 3.09 | 5.10 | 4_12 | 3.71 | 2.56 |
| 4_03 | 4.21 | 4.06 | 4_13 | 2.85 | 0.32 |
| 4_04 | 2.85 | 4.05 | 4_14 | 0.00 | 2.00 |
| 4_05 | 3.80 | 3.48 | 4_15 | 4.46 | 2.51 |
| 4_06 | 1.16 | 3.10 | 4_16 | 3.58 | 2.70 |
| 4_07 | 1.12 | 1.68 | 4_17 | 3.48 | 0.86 |
| 4_08 | 2.49 | 0.91 | 4_18 | 0.46 | 2.49 |
| 4_09 | 5.62 | 0.00 | 4_19 | 3.86 | 2.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dymkowski, T.; Żuczkowski, W.; Kołodziejczyk, W.; Hill, G.; Szyja, B.M. Stability of the TiO2 Nanuclusters Supported on Fe2O3-Hematite for Application in Electrocatalytic Water Splitting—An Insight from DFT Simulations. Hydrogen 2023, 4, 573-584. https://doi.org/10.3390/hydrogen4030037
Dymkowski T, Żuczkowski W, Kołodziejczyk W, Hill G, Szyja BM. Stability of the TiO2 Nanuclusters Supported on Fe2O3-Hematite for Application in Electrocatalytic Water Splitting—An Insight from DFT Simulations. Hydrogen. 2023; 4(3):573-584. https://doi.org/10.3390/hydrogen4030037
Chicago/Turabian StyleDymkowski, Tomasz, Wiktor Żuczkowski, Wojciech Kołodziejczyk, Glake Hill, and Bartłomiej M. Szyja. 2023. "Stability of the TiO2 Nanuclusters Supported on Fe2O3-Hematite for Application in Electrocatalytic Water Splitting—An Insight from DFT Simulations" Hydrogen 4, no. 3: 573-584. https://doi.org/10.3390/hydrogen4030037
APA StyleDymkowski, T., Żuczkowski, W., Kołodziejczyk, W., Hill, G., & Szyja, B. M. (2023). Stability of the TiO2 Nanuclusters Supported on Fe2O3-Hematite for Application in Electrocatalytic Water Splitting—An Insight from DFT Simulations. Hydrogen, 4(3), 573-584. https://doi.org/10.3390/hydrogen4030037