
Fluoroquinolones-Associated Disability: It Is Not All in Your Head



Abstract
1. Introduction
2. Overview of Fluoroquinolones Toxicity
3. Effects on the Central Nervous System
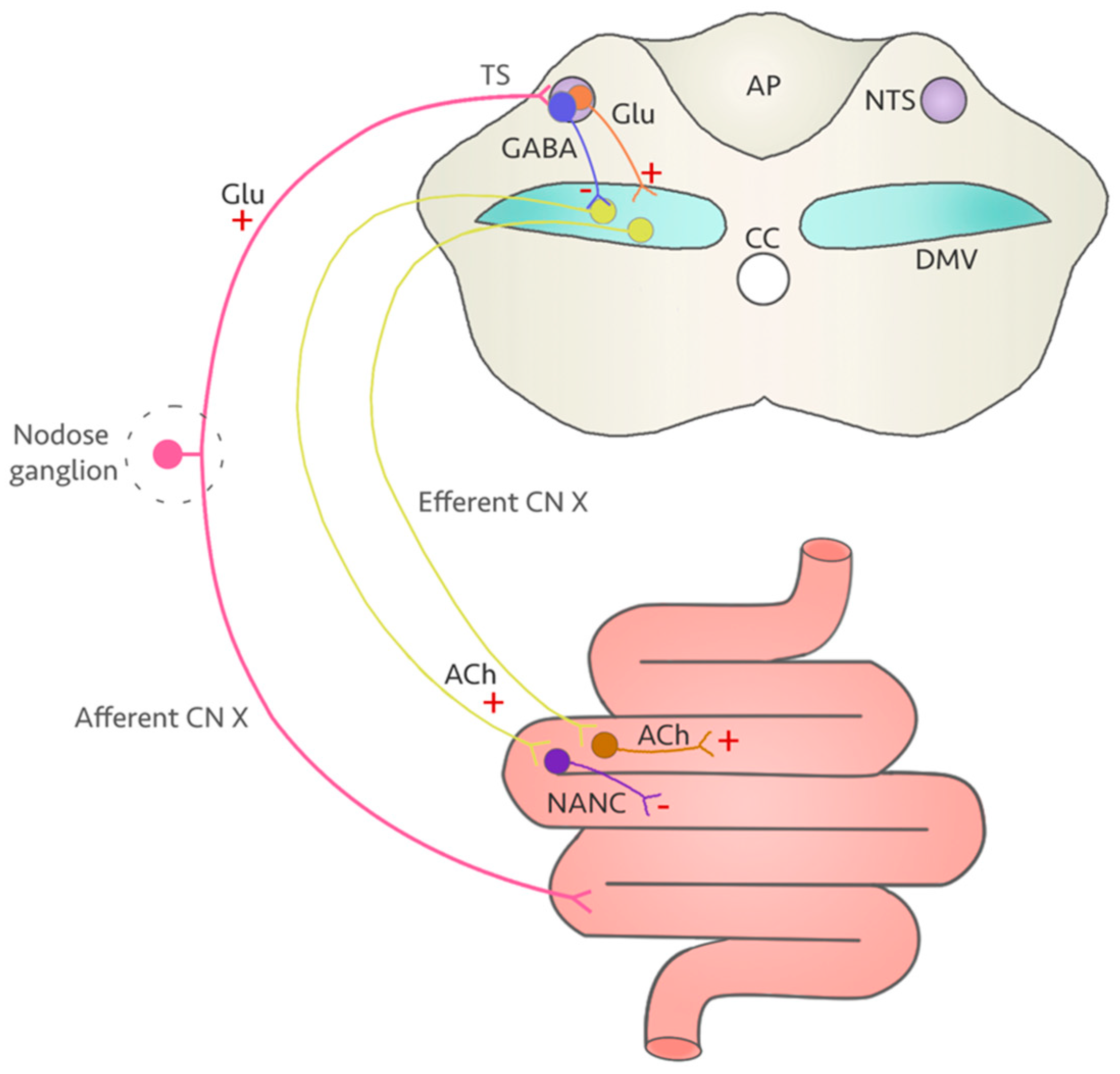
4. A Vulnerable Target: The Vagus Nerve
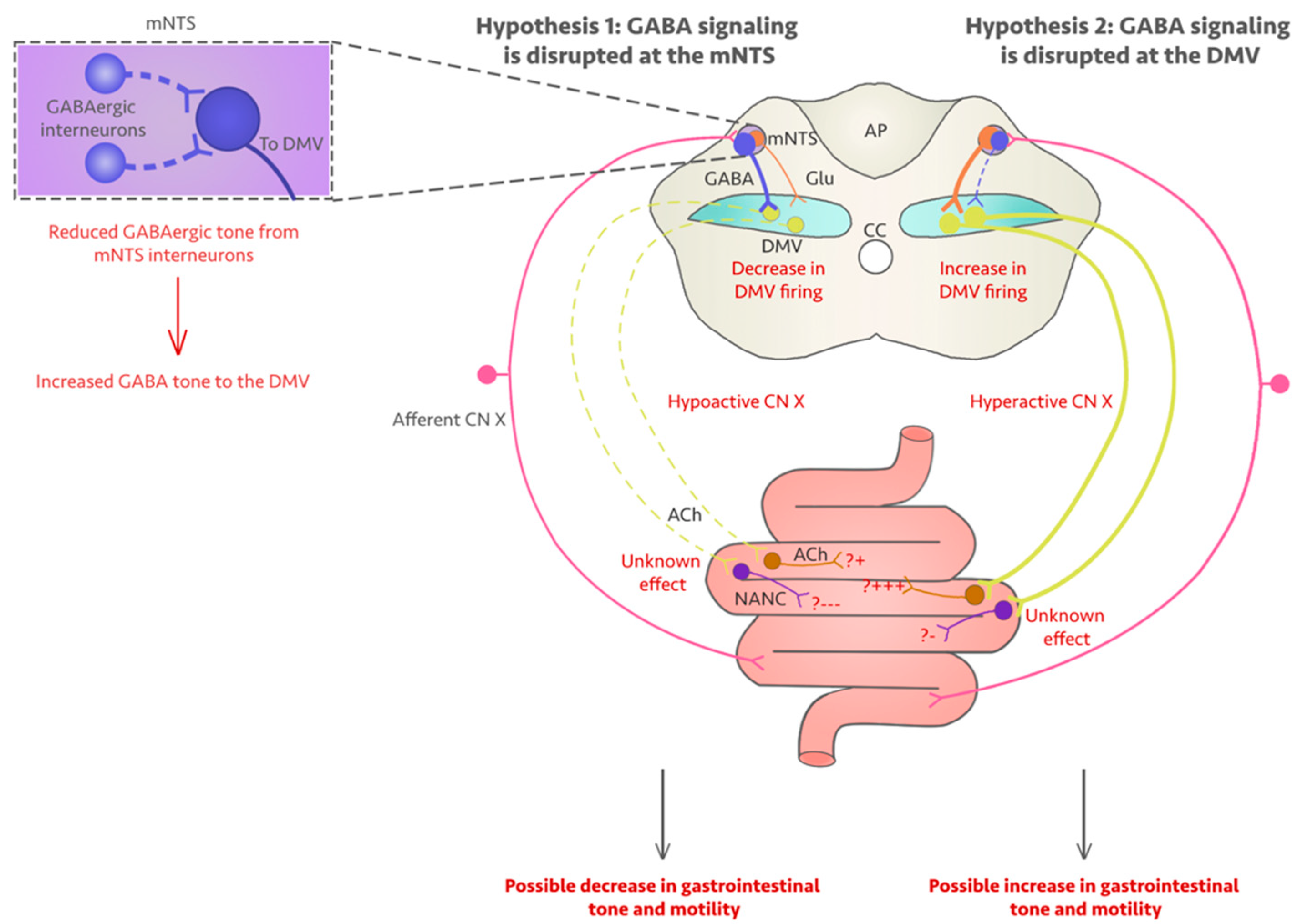
5. Could Fluoroquinolones Compromise Vagus Nerve Function?
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Institute of Diabetes and Digestive and Kidney Diseases. Fluoroquinolones. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. [Google Scholar]
- Kabbani, S.; Hersh, A.L.; Shapiro, D.J.; Fleming-Dutra, K.E.; Pavia, A.T.; Hicks, L.A. Opportunities to Improve Fluoroquinolone Prescribing in the United States for Adult Ambulatory Care Visits. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2018, 67, 134–136. [Google Scholar] [CrossRef]
- Baggs, J.; Fridkin, S.K.; Pollack, L.A.; Srinivasan, A.; Jernigan, J.A. Estimating National Trends in Inpatient Antibiotic Use Among US Hospitals from 2006 to 2012. JAMA Intern. Med. 2016, 176, 1639. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- King, D.E.; Malone, R.; Lilley, S.H. New Classification and Update on the Quinolone Antibiotics. Am. Fam. Physician 2000, 61, 2741–2748. [Google Scholar] [PubMed]
- Stahlmann, R.; Lode, H. Safety Considerations of Fluoroquinolones in the Elderly. Drugs Aging 2010, 27, 193–209. [Google Scholar] [CrossRef]
- Norrby, S.R. Side-Effects of Quinolones: Comparisons between Quinolones and Other Antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 1991, 10, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Stahlmann, R.; Riecke, K. Well tolerated or risky? Adverse effect of quinolones. Pharm. Unserer Zeit 2001, 30, 412–417. [Google Scholar] [CrossRef]
- Chui, C.S.L.; Chan, E.W.; Wong, A.Y.S.; Root, A.; Douglas, I.J.; Wong, I.C.K. Association between Oral Fluoroquinolones and Seizures. Neurology 2016, 86, 1708–1715. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jiang, Z.; Zhao, Q.; Ding, Y. Adverse Reactions of Fluoroquinolones to Central Nervous System and Rational Drug Use in Nursing Care. Pak. J. Pharm. Sci. 2019, 32, 427–432. [Google Scholar]
- Etminan, M.; Brophy, J.M.; Samii, A. Oral Fluoroquinolone Use and Risk of Peripheral Neuropathy: A Pharmacoepidemiologic Study. Neurology 2014, 83, 1261–1263. [Google Scholar] [CrossRef]
- Lewis, T.; Cook, J. Fluoroquinolones and Tendinopathy: A Guide for Athletes and Sports Clinicians and a Systematic Review of the Literature. J. Athl. Train. 2014, 49, 422–427. [Google Scholar] [CrossRef]
- Stephenson, A.L.; Wu, W.; Cortes, D.; Rochon, P.A. Tendon Injury and Fluoroquinolone Use: A Systematic Review. Drug Saf. 2013, 36, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Yarrington, M.E.; Anderson, D.J.; Dodds Ashley, E.; Jones, T.; Davis, A.; Johnson, M.; Lokhnygina, Y.; Sexton, D.J.; Moehring, R.W. Impact of FDA Black Box Warning on Fluoroquinolone and Alternative Antibiotic Use in Southeastern US Hospitals. Infect. Control Hosp. Epidemiol. 2019, 40, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Hooton, T.M.; Naber, K.G.; Wullt, B.; Colgan, R.; Miller, L.G.; Moran, G.J.; Nicolle, L.E.; Raz, R.; Schaeffer, A.J.; et al. International Clinical Practice Guidelines for the Treatment of Acute Uncomplicated Cystitis and Pyelonephritis in Women: A 2010 Update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin. Infect. Dis. 2011, 52, e103–e120. [Google Scholar] [CrossRef] [PubMed]
- Ganjizadeh-Zavareh, S.; Sodhi, M.; Spangehl, T.; Carleton, B.; Etminan, M. Oral Fluoroquinolones and Risk of Fibromyalgia. Br. J. Clin. Pharmacol. 2019, 85, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.K. Peripheral Neuropathy and Guillain-Barré Syndrome Risks Associated with Exposure to Systemic Fluoroquinolones: A Pharmacovigilance Analysis. Ann. Epidemiol. 2014, 24, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Stahlmann, R. Safety Profile of the Quinolones. J. Antimicrob. Chemother. 1990, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, B.A.; Baker, C.A. Fluoroquinolone Toxicity Profiles: A Review Focusing on Newer Agents. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1999, 28, 352–364. [Google Scholar] [CrossRef]
- Shimoda, K. Mechanisms of Quinolone Phototoxicity. Toxicol. Lett. 1998, 102, 369–373. [Google Scholar] [CrossRef]
- Stahlmann, R.; Lode, H. Toxicity of Quinolones. Drugs 1999, 58 (Suppl. 2), 37–42. [Google Scholar] [CrossRef]
- Mehrzad, R.; Barza, M. Weighing the Adverse Cardiac Effects of Fluoroquinolones: A Risk Perspective. J. Clin. Pharmacol. 2015, 55, 1198–1206. [Google Scholar] [CrossRef]
- Rubinstein, E.; Camm, J. Cardiotoxicity of Fluoroquinolones. J. Antimicrob. Chemother. 2002, 49, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.C. Risk Assessment for Antimicrobial Agent-Induced QTc Interval Prolongation and Torsades de Pointes. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2001, 21, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Lubasch, A.; Keller, I.; Borner, K.; Koeppe, P.; Lode, H. Comparative Pharmacokinetics of Ciprofloxacin, Gatifloxacin, Grepafloxacin, Levofloxacin, Trovafloxacin, and Moxifloxacin after Single Oral Administration in Healthy Volunteers. Antimicrob. Agents Chemother. 2000, 44, 2600–2603. [Google Scholar] [CrossRef] [PubMed]
- Sprandel, K.A.; Rodvold, K.A. Safety and Tolerability of Fluoroquinolones. Clin. Cornerstone 2003, 5, S29–S36. [Google Scholar] [CrossRef]
- Liguori, M.J.; Anderson, M.G.; Bukofzer, S.; McKim, J.; Pregenzer, J.F.; Retief, J.; Spear, B.B.; Waring, J.F. Microarray Analysis in Human Hepatocytes Suggests a Mechanism for Hepatotoxicity Induced by Trovafloxacin. Hepatology 2005, 41, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Paterson, J.M.; Mamdani, M.M.; Manno, M.; Juurlink, D.N. Fluoroquinolone Therapy and Idiosyncratic Acute Liver Injury: A Population-Based Study. CMAJ Can. Med. Assoc. J. 2012, 184, 1565–1570. [Google Scholar] [CrossRef]
- Yu, C.; Giuffre, B. Achilles Tendinopathy after Treatment with Fluoroquinolone. Australas. Radiol. 2005, 49, 407–410. [Google Scholar] [CrossRef]
- Kim, G.K. The Risk of Fluoroquinolone-Induced Tendinopathy and Tendon Rupture. J. Clin. Aesthetic Dermatol. 2010, 3, 49–54. [Google Scholar]
- Williams, R.J.; Attia, E.; Wickiewicz, T.L.; Hannafin, J.A. The Effect of Ciprofloxacin on Tendon, Paratenon, and Capsular Fibroblast Metabolism. Am. J. Sports Med. 2000, 28, 364–369. [Google Scholar] [CrossRef]
- Le Huec, J.C.; Schaeverbeke, T.; Chauveaux, D.; Rivel, J.; Dehais, J.; Le Rebeller, A. Epicondylitis after Treatment with Fluoroquinolone Antibiotics. J. Bone Joint Surg. Br. 1995, 77, 293–295. [Google Scholar] [CrossRef]
- Förster, C.; Kociok, K.; Shakibaei, M.; Merker, H.J.; Stahlmann, R. Quinolone-Induced Cartilage Lesions Are Not Reversible in Rats. Arch. Toxicol. 1996, 70, 474–481. [Google Scholar] [CrossRef]
- Fish, D.N. Fluoroquinolone Adverse Effects and Drug Interactions. Pharmacotherapy 2001, 21, 253S–272S. [Google Scholar] [CrossRef]
- Mizuki, Y.; Fujiwara, I.; Yamaguchi, T. Pharmacokinetic Interactions Related to the Chemical Structures of Fluoroquinolones. J. Antimicrob. Chemother. 1996, 37 (Suppl. A), 41–55. [Google Scholar] [CrossRef]
- Beckmann, J.; Elsässer, W.; Gundert-Remy, U.; Hertrampf, R. Enoxacin—A Potent Inhibitor of Theophylline Metabolism. Eur. J. Clin. Pharmacol. 1987, 33, 227–230. [Google Scholar] [CrossRef]
- Efthymiopoulos, C.; Bramer, S.L.; Maroli, A.; Blum, B. Theophylline and Warfarin Interaction Studies with Grepafloxacin. Clin. Pharmacokinet. 1997, 33 (Suppl. 1), 39–46. [Google Scholar] [CrossRef]
- Marchbanks, C. Drug-Drug Interactions with Fluoroquinolones. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1993, 13, 23S–28S. [Google Scholar]
- Davis, R.; Markham, A.; Balfour, J.A. Ciprofloxacin. An Updated Review of Its Pharmacology, Therapeutic Efficacy and Tolerability. Drugs 1996, 51, 1019–1074. [Google Scholar] [CrossRef] [PubMed]
- Stille, W.; Harder, S.; Mieke, S.; Beer, C.; Shah, P.M.; Frech, K.; Staib, A.H. Decrease of Caffeine Elimination in Man during Co-Administration of 4-Quinolones. J. Antimicrob. Chemother. 1987, 20, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, N.; Niki, Y.; Soejima, R. Effect of Levofloxacin on Serum Concentration of Theophylline. Chemotherapy 1992, 40 (Suppl. 3), 68–74. [Google Scholar]
- Christ, W. Central Nervous System Toxicity of Quinolones: Human and Animal Findings. J. Antimicrob. Chemother. 1990, 26 (Suppl. B), 219–225. [Google Scholar] [CrossRef]
- Radandt, J.M.; Marchbanks, C.R.; Dudley, M.N. Interactions of Fluoroquinolones with Other Drugs: Mechanisms, Variability, Clinical Significance, and Management. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1992, 14, 272–284. [Google Scholar] [CrossRef]
- Domagala, J.M. Structure-Activity and Structure-Side-Effect Relationships for the Quinolone Antibacterials. J. Antimicrob. Chemother. 1994, 33, 685–706. [Google Scholar] [CrossRef] [PubMed]
- Smolders, I.; Gousseau, C.; Marchand, S.; Couet, W.; Ebinger, G.; Michotte, Y. Convulsant and Subconvulsant Doses of Norfloxacin in the Presence and Absence of Biphenylacetic Acid Alter Extracellular Hippocampal Glutamate but Not Gamma-Aminobutyric Acid Levels in Conscious Rats. Antimicrob. Agents Chemother. 2002, 46, 471–477. [Google Scholar] [CrossRef]
- Palù, G.; Valisena, S.; Ciarrocchi, G.; Gatto, B.; Palumbo, M. Quinolone Binding to DNA Is Mediated by Magnesium Ions. Proc. Natl. Acad. Sci. USA 1992, 89, 9671–9675. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Matsubayashi, K.; Hakusui, H. Interaction of Quinolones with Metal Cations in Aqueous Solution. Chem. Pharm. Bull. 1996, 44, 1425–1430. [Google Scholar] [CrossRef][Green Version]
- Michalak, K.; Sobolewska-Włodarczyk, A.; Włodarczyk, M.; Sobolewska, J.; Woźniak, P.; Sobolewski, B. Treatment of the Fluoroquinolone-Associated Disability: The Pathobiochemical Implications. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Spivey, J.M.; Cummings, D.M.; Pierson, N.R. Failure of Prostatitis Treatment Secondary to Probable Ciprofloxacin-Sucralfate Drug Interaction. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1996, 16, 314–316. [Google Scholar] [CrossRef]
- Seedher, N.; Agarwal, P. Effect of Metal Ions on Some Pharmacologically Relevant Interactions Involving Fluoroquinolone Antibiotics. Drug Metabol. Drug Interact. 2010, 25, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.H.; Chiu, F.C.; Li, R.C. Mechanistic Investigation of the Reduction in Antimicrobial Activity of Ciprofloxacin by Metal Cations. Pharm. Res. 1997, 14, 366–370. [Google Scholar] [CrossRef]
- Koga, H. High-Performance Liquid Chromatography Measurement of Antimicrobial Concentrations in Polymorphonuclear Leukocytes. Antimicrob. Agents Chemother. 1987, 31, 1904–1908. [Google Scholar] [CrossRef] [PubMed]
- Pascual, A.; García, I.; Ballesta, S.; Perea, E.J. Uptake and Intracellular Activity of Trovafloxacin in Human Phagocytes and Tissue-Cultured Epithelial Cells. Antimicrob. Agents Chemother. 1997, 41, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Andriole, V.T. The Quinolones, 3rd ed.; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Egerbacher, M.; Seiberl, G.; Wolfesberger, B.; Walter, I. Ciprofloxacin Causes Cytoskeletal Changes and Detachment of Human and Rat Chondrocytes in Vitro. Arch. Toxicol. 2000, 73, 557–563. [Google Scholar] [CrossRef]
- Shakibaei, M.; Kociok, K.; Förster, C.; Vormann, J.; Günther, T.; Stahlmann, R.; Merker, H.J. Comparative Evaluation of Ultrastructural Changes in Articular Cartilage of Ofloxacin—Treated and Magnesium—Deficient Immature Rats. Toxicol. Pathol. 1996, 24, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Egerbacher, M.; Wolfesberger, B.; Walter, I.; Seiberl, G. Integrins Mediate the Effects of Quinolones and Magnesium Deficiency on Cultured Rat Chondrocytes. Eur. J. Cell Biol. 1999, 78, 391–397. [Google Scholar] [CrossRef]
- Egerbacher, M.; Edinger, J.; Tschulenk, W. Effects of Enrofloxacin and Ciprofloxacin Hydrochloride on Canine and Equine Chondrocytes in Culture. Am. J. Vet. Res. 2001, 62, 704–708. [Google Scholar] [CrossRef]
- Egerbacher, M.; Wolfesberger, B.; Gabler, C. In Vitro Evidence for Effects of Magnesium Supplementation on Quinolone-Treated Horse and Dog Chondrocytes. Vet. Pathol. 2001, 38, 143–148. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuča, K.; Musílek, K. Redox- and Non-Redox-Metal-Induced Formation of Free Radicals and Their Role in Human Disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef]
- Badal, S.; Her, Y.F.; Maher, L.J. Nonantibiotic Effects of Fluoroquinolones in Mammalian Cells. J. Biol. Chem. 2015, 290, 22287–22297. [Google Scholar] [CrossRef]
- Gootz, T.D.; Barrett, J.F.; Sutcliffe, J.A. Inhibitory Effects of Quinolone Antibacterial Agents on Eucaryotic Topoisomerases and Related Test Systems. Antimicrob. Agents Chemother. 1990, 34, 8–12. [Google Scholar] [CrossRef]
- Fox, A.J.S.; Schär, M.O.; Wanivenhaus, F.; Chen, T.; Attia, E.; Binder, N.B.; Otero, M.; Gilbert, S.L.; Nguyen, J.T.; Chaudhury, S.; et al. Fluoroquinolones Impair Tendon Healing in a Rat Rotator Cuff Repair Model: A Preliminary Study. Am. J. Sports Med. 2014, 42, 2851–2859. [Google Scholar] [CrossRef]
- Aranha, O.; Grignon, R.; Fernandes, N.; McDonnell, T.J.; Wood, D.P.; Sarkar, F.H. Suppression of Human Prostate Cancer Cell Growth by Ciprofloxacin Is Associated with Cell Cycle Arrest and Apoptosis. Int. J. Oncol. 2003, 22, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, L.; Ou, R.; Nie, X.; Yang, Y.; Wang, F.; Li, K. Effects of Norfloxacin on Hepatic Genes Expression of P450 Isoforms (CYP1A and CYP3A), GST and P-Glycoprotein (P-Gp) in Swordtail Fish (Xiphophorus helleri). Ecotoxicol. Lond. Engl. 2015, 24, 1566–1573. [Google Scholar] [CrossRef]
- Hsiao, C.-J.J.; Younis, H.; Boelsterli, U.A. Trovafloxacin, a Fluoroquinolone Antibiotic with Hepatotoxic Potential, Causes Mitochondrial Peroxynitrite Stress in a Mouse Model of Underlying Mitochondrial Dysfunction. Chem. Biol. Interact. 2010, 188, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Liu, R. Oxidative Stress Response of Two Fluoroquinolones with Catalase and Erythrocytes: A Combined Molecular and Cellular Study. J. Hazard. Mater. 2013, 252, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Liu, Z.-Y.; Sun, L.-S.; Li, Y.-J.; Zhang, D.-S.; Pan, R.-T.; Sun, Z.-L. Effect of Danofloxacin on Reactive Oxygen Species Production, Lipid Peroxidation and Antioxidant Enzyme Activities in Kidney Tubular Epithelial Cell Line, LLC-PK1. Basic Clin. Pharmacol. Toxicol. 2013, 113, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Pouzaud, F.; Bernard-Beaubois, K.; Thevenin, M.; Warnet, J.-M.; Hayem, G.; Rat, P. In Vitro Discrimination of Fluoroquinolones Toxicity on Tendon Cells: Involvement of Oxidative Stress. J. Pharmacol. Exp. Ther. 2004, 308, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Kumbhar, G.B.; Khan, A.M.; Rampal, S. Evaluation of Gatifloxacin for Its Potential to Induce Antioxidant Imbalance and Retinopathy in Rabbits. Hum. Exp. Toxicol. 2015, 34, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Talla, V.; Veerareddy, P. Oxidative Stress Induced by Fluoroquinolones on Treatment for Complicated Urinary Tract Infections in Indian Patients. J. Young Pharm. JYP 2011, 3, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Cui, Y.; Brown, P.B.; Ge, X.; Xie, J.; Xu, P. Cytotoxic Effects and Apoptosis Induction of Enrofloxacin in Hepatic Cell Line of Grass Carp (Ctenopharyngodon idellus). Fish Shellfish Immunol. 2015, 47, 639–644. [Google Scholar] [CrossRef]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St. Clair, D.K. Manganese Superoxide Dismutase: Guardian of the Powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, S.S. Fluoroquinolone-Induced Depression. Am. J. Psychiatry 1995, 152, 954–955. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, A.; Srinath, D. Levofloxacin-Induced Acute Anxiety and Insomnia. J. Neurosci. Rural Pract. 2012, 3, 212–214. [Google Scholar] [CrossRef]
- Sarro, A.; Sarro, G. Adverse Reactions to Fluroquinolones. An Overview on Mechanistic Aspects. Curr. Med. Chem. 2001, 8, 371–384. [Google Scholar] [CrossRef]
- Hooper, D.C.; Wolfson, J.S. Fluoroquinolone Antimicrobial Agents. N. Engl. J. Med. 1991, 324, 384–394. [Google Scholar] [CrossRef]
- Green, M.A.; Halliwell, R.F. Selective Antagonism of the GABA(A) Receptor by Ciprofloxacin and Biphenylacetic Acid. Br. J. Pharmacol. 1997, 122, 584–590. [Google Scholar] [CrossRef]
- Halliwell, R.F.; Davey, P.G.; Lambert, J.J. Antagonism of GABAA Receptors by 4-Quinolones. J. Antimicrob. Chemother. 1993, 31, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Ilgin, S.; Can, O.D.; Atli, O.; Ucel, U.I.; Sener, E.; Guven, I. Ciprofloxacin-Induced Neurotoxicity: Evaluation of Possible Underlying Mechanisms. Toxicol. Mech. Methods 2015, 25, 374–381. [Google Scholar] [CrossRef]
- Schmuck, G.; Schürmann, A.; Schlüter, G. Determination of the Excitatory Potencies of Fluoroquinolones in the Central Nervous System by an In Vitro Model. Antimicrob. Agents Chemother. 1998, 42, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Bano, D.; Ankarcrona, M. Beyond the Critical Point: An Overview of Excitotoxicity, Calcium Overload and the Downstream Consequences. Neurosci. Lett. 2018, 663, 79–85. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Lee, J.-M.M.; Zipfel, G.J.; Choi, D.W. The Changing Landscape of Ischaemic Brain Injury Mechanisms. Nature 1999, 399, A7–A14. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and Stroke: Identifying Novel Targets for Neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A.; Wang, J. Superoxide and Non-Ionotropic Signaling in Neuronal Excitotoxicity. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to Various Triggers in Neurodegenerative Disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the Physiology and Pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Rachline, J.; Perin-Dureau, F.; Le Goff, A.; Neyton, J.; Paoletti, P. The Micromolar Zinc-Binding Domain on the NMDA Receptor Subunit NR2B. J. Neurosci. 2005, 25, 308–317. [Google Scholar] [CrossRef]
- Kalappa, B.I.; Anderson, C.T.; Goldberg, J.M.; Lippard, S.J.; Tzounopoulos, T. AMPA Receptor Inhibition by Synaptically Released Zinc. Proc. Natl. Acad. Sci. USA 2015, 112, 15749–15754. [Google Scholar] [CrossRef]
- Granzotto, A.; Canzoniero, L.M.T.; Sensi, S.L. A Neurotoxic Ménage-à-Trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade. Front. Mol. Neurosci. 2020, 13, 225. [Google Scholar] [CrossRef]
- Yeragani, V.K.; Krishnan, S.; Engels, H.J.; Gretebeck, R. Effects of Caffeine on Linear and Nonlinear Measures of Heart Rate Variability before and after Exercise. Depress. Anxiety 2005, 21, 130–134. [Google Scholar] [CrossRef]
- Yeragani, V.K.; Pohl, R.; Berger, R.; Balon, R.; Ramesh, C.; Glitz, D.; Srinivasan, K.; Weinberg, P. Decreased Heart Rate Variability in Panic Disorder Patients: A Study of Power-Spectral Analysis of Heart Rate. Psychiatry Res. 1993, 46, 89–103. [Google Scholar] [CrossRef]
- Yeragani, V.K.; Sobolewski, E.; Igel, G.; Johnson, C.; Jampala, V.C.; Kay, J.; Hillman, N.; Yeragani, S.; Vempati, S. Decreased Heart-Period Variability in Patients with Panic Disorder: A Study of Holter ECG Records. Psychiatry Res. 1998, 78, 89–99. [Google Scholar] [CrossRef]
- Stakenborg, N.; Giovangiulio, M.D.; Boeckxstaens, G.E.; Matteoli, G. The Versatile Role of the Vagus Nerve in the Gastrointestinal Tract. EMJ Gastroenterol. 2013, 1, 106–114. [Google Scholar]
- Baker, E.; Lui, F. Neuroanatomy, Vagal Nerve Nuclei. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Altschuler, S.M.; Escardo, J.; Lynn, R.B.; Miselis, R.R. The Central Organization of the Vagus Nerve Innervating the Colon of the Rat. Gastroenterology 1993, 104, 502–509. [Google Scholar] [CrossRef]
- Browning, K.N.; Travagli, R.A. Central Nervous System Control of Gastrointestinal Motility and Secretion and Modulation of Gastrointestinal Functions. Compr. Physiol. 2014, 4, 1339–1368. [Google Scholar] [CrossRef] [PubMed]
- Gillis, R.A.; Dezfuli, G.; Bellusci, L.; Vicini, S.; Sahibzada, N. Brainstem Neuronal Circuitries Controlling Gastric Tonic and Phasic Contractions: A Review. Cell. Mol. Neurobiol. 2021, 1–28. [Google Scholar] [CrossRef]
- Travagli, R.A.; Anselmi, L. Vagal Neurocircuitry and Its Influence on Gastric Motility. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Fornai, M.; Antonioli, L.; Colucci, R.; Tuccori, M.; Blandizzi, C. Pathophysiology of Gastric Ulcer Development and Healing: Molecular Mechanisms and Novel Therapeutic Options; IntechOpen: London, UK, 2011. [Google Scholar]
- Travagli, R.A.; Gillis, R.A.; Rossiter, C.D.; Vicini, S. Glutamate and GABA-Mediated Synaptic Currents in Neurons of the Rat Dorsal Motor Nucleus of the Vagus. Am. J. Physiol. 1991, 260, G531–G536. [Google Scholar] [CrossRef]
- Sivarao, D.V.; Krowicki, Z.K.; Hornby, P.J. Role of GABAA Receptors in Rat Hindbrain Nuclei Controlling Gastric Motor Function. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 1998, 10, 305–313. [Google Scholar] [CrossRef]
- Travagli, R.A.; Gillis, R.A. Hyperpolarization-Activated Currents, IH and IKIR, in Rat Dorsal Motor Nucleus of the Vagus Neurons in Vitro. J. Neurophysiol. 1994, 71, 1308–1317. [Google Scholar] [CrossRef]
- Eglen, R.M. Muscarinic Receptors and Gastrointestinal Tract Smooth Muscle Function. Life Sci. 2001, 68, 2573–2578. [Google Scholar] [CrossRef]
- Sanders, K.M.; Ward, S.M. Nitric Oxide and Its Role as a Non-adrenergic, Non-cholinergic Inhibitory Neurotransmitter in the Gastrointestinal Tract. Br. J. Pharmacol. 2019, 176, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.C.; Hermann, G.E.; Travagli, R.A. Brainstem Pathways Responsible for Oesophageal Control of Gastric Motility and Tone in the Rat. J. Physiol. 1999, 514 Pt 2, 369–383. [Google Scholar] [CrossRef]
- Cruz, M.T.; Murphy, E.C.; Sahibzada, N.; Verbalis, J.G.; Gillis, R.A. A Reevaluation of the Effects of Stimulation of the Dorsal Motor Nucleus of the Vagus on Gastric Motility in the Rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R291–R307. [Google Scholar] [CrossRef] [PubMed]
- Krowicki, Z.K.; Sharkey, K.A.; Serron, S.C.; Nathan, N.A.; Hornby, P.J. Distribution of Nitric Oxide Synthase in Rat Dorsal Vagal Complex and Effects of Microinjection of Nitric Oxide Compounds upon Gastric Motor Function. J. Comp. Neurol. 1997, 377, 49–69. [Google Scholar] [CrossRef]
- Rogers, R.C.; Travagli, R.A.; Hermann, G.E. Noradrenergic Neurons in the Rat Solitary Nucleus Participate in the Esophageal-Gastric Relaxation Reflex. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R479–R489. [Google Scholar] [CrossRef]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and Subsequent Risk of Parkinson’s Disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Babic, T.; Travagli, R.A. Neural Control of the Pancreas. Pancreapedia Exocrine Pancreas Knowl. Base 2016. [Google Scholar] [CrossRef]
- Clyburn, C.; Travagli, R.A.; Browning, K.N. Acute High-Fat Diet Upregulates Glutamatergic Signaling in the Dorsal Motor Nucleus of the Vagus. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G623–G634. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, R.; Fortna, S.R.; Browning, K.N. Exposure to a High Fat Diet during the Perinatal Period Alters Vagal Motoneurone Excitability, Even in the Absence of Obesity. J. Physiol. 2015, 593, 285–303. [Google Scholar] [CrossRef]
- McMenamin, C.A.; Travagli, R.A.; Browning, K.N. Perinatal High Fat Diet Increases Inhibition of Dorsal Motor Nucleus of the Vagus Neurons Regulating Gastric Functions. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2018, 30. [Google Scholar] [CrossRef]
- Clyburn, C.; Howe, C.A.; Arnold, A.C.; Lang, C.H.; Travagli, R.A.; Browning, K.N. Perinatal High-Fat Diet Alters Development of GABAA Receptor Subunits in Dorsal Motor Nucleus of Vagus. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G40–G50. [Google Scholar] [CrossRef]
- Talley, N.J.; Howell, S.; Poulton, R. Obesity and Chronic Gastrointestinal Tract Symptoms in Young Adults: A Birth Cohort Study. Am. J. Gastroenterol. 2004, 99, 1807–1814. [Google Scholar] [CrossRef]
- Ballanyi, K.; Doutheil, J.; Brockhaus, J. Membrane Potentials and Microenvironment of Rat Dorsal Vagal Cells in Vitro during Energy Depletion. J. Physiol. 1996, 495 Pt 3, 769–784. [Google Scholar] [CrossRef]
- Dean, J.B.; Mulkey, D.K. Continuous Intracellular Recording from Mammalian Neurons Exposed to Hyperbaric Helium, Oxygen, or Air. J. Appl. Physiol. 2000, 89, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Kulik, A.; Trapp, S.; Ballanyi, K. Ischemia but Not Anoxia Evokes Vesicular and Ca(2+)-Independent Glutamate Release in the Dorsal Vagal Complex in Vitro. J. Neurophysiol. 2000, 83, 2905–2915. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trapp, S.; Lückermann, M.; Brooks, P.A.; Ballanyi, K. Acidosis of Rat Dorsal Vagal Neurons in Situ during Spontaneous and Evoked Activity. J. Physiol. 1996, 496 Pt 3, 695–710. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Bennun, A. Characterization of the Norepinephrine-Activation of Adenylate Cyclase Suggests a Role in Memory Affirmation Pathways. Biosystems 2010, 100, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Roosevelt, R.W.; Smith, D.C.; Clough, R.W.; Jensen, R.A.; Browning, R.A. Increased Extracellular Concentrations of Norepinephrine in Cortex and Hippocampus Following Vagus Nerve Stimulation in the Rat. Brain Res. 2006, 1119, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.S.; McCall, J.G.; Charney, D.S.; Murrough, J.W. The Role of the Locus Coeruleus in the Generation of Pathological Anxiety. Brain Neurosci. Adv. 2020, 4, 2398212820930321. [Google Scholar] [CrossRef] [PubMed]
- Ganong, W. Circumventricular Organs: Definition and Role in the Regulation of Endocrine and Autonomic Function. Clin. Exp. Pharmacol. Physiol. 2000, 27, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.; Ballanyi, K. KATP Channel Mediation of Anoxia-Induced Outward Current in Rat Dorsal Vagal Neurons In Vitro. J. Physiol. 1995, 487, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, L.; Bove, C.; Coleman, F.H.; Le, K.; Subramanian, M.P.; Venkiteswaran, K.; Subramanian, T.; Travagli, R.A. Ingestion of Subthreshold Doses of Environmental Toxins Induces Ascending Parkinsonism in the Rat. NPJ Park. Dis. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Hay, M.; Amilhon, B.; Jean, A.; Moyse, E. In Vivo Neurogenesis in the Dorsal Vagal Complex of the Adult Rat Brainstem. Neuroscience 2005, 130, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.L.; Doheny, K.K.; Travagli, R.A. Necrotizing Enterocolitis Attenuates Developmental Heart Rate Variability Increases in Newborn Rats. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2019, 31, e13484. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nozu, T.; Kumei, S.; Ohhira, M.; Okumura, T. IL-1 Receptor Antagonist Blocks the Lipopolysaccharide-Induced Inhibition of Gastric Motility in Freely Moving Conscious Rats. Dig. Dis. Sci. 2012, 57, 2555–2561. [Google Scholar] [CrossRef][Green Version]
- Ammori, J.B.; Zhang, W.-Z.; Li, J.-Y.; Chai, B.-X.; Mulholland, M.W. Effect of Intestinal Inflammation on Neuronal Survival and Function in the Dorsal Motor Nucleus of the Vagus. Surgery 2008, 144, 149–158. [Google Scholar] [CrossRef]
- Hermann, G.E.; Tovar, C.A.; Rogers, R.C. Induction of Endogenous Tumor Necrosis Factor-Alpha: Suppression of Centrally Stimulated Gastric Motility. Am. J. Physiol. 1999, 276, R59–R68. [Google Scholar] [CrossRef]
- Emch, G.S.; Hermann, G.E.; Rogers, R.C. Tumor Necrosis Factor-Alpha Inhibits Physiologically Identified Dorsal Motor Nucleus Neurons in Vivo. Brain Res. 2002, 951, 311–315. [Google Scholar] [CrossRef]
- Suzuki, T.; Takizawa, T.; Kamio, Y.; Qin, T.; Hashimoto, T.; Fujii, Y.; Murayama, Y.; Patel, A.B.; Ayata, C. Noninvasive Vagus Nerve Stimulation Prevents Ruptures and Improves Outcomes in a Model of Intracranial Aneurysm in Mice. Stroke 2019, 50, 1216–1223. [Google Scholar] [CrossRef]
- Norcliffe-Kaufmann, L. The Vagus and Glossopharyngeal Nerves in Two Autonomic Disorders. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 2019, 36, 443–451. [Google Scholar] [CrossRef]
- Newton, E.R.; Akerman, A.W.; Strassle, P.D.; Kibbe, M.R. Association of Fluoroquinolone use With Short-Term Risk of Development of Aortic Aneurysm. JAMA Surg. 2021, 156, 264–272. [Google Scholar] [CrossRef]
- Sanmarco, L.M.; Wheeler, M.A.; Gutiérrez-Vázquez, C.; Polonio, C.M.; Linnerbauer, M.; Pinho-Ribeiro, F.A.; Li, Z.; Giovannoni, F.; Batterman, K.V.; Scalisi, G.; et al. Gut-Licensed IFNγ + NK Cells Drive LAMP1 + TRAIL + Anti-Inflammatory Astrocytes. Nature 2021, 590, 473–479. [Google Scholar] [CrossRef]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and Levofloxacin Attenuate Microglia Inflammatory Response via TLR4/NF-KB Pathway. J. Neuroinflamm. 2019, 16, 148. [Google Scholar] [CrossRef]
- Carreno, F.R.; Frazer, A. Vagal Nerve Stimulation for Treatment-Resistant Depression. Neurother. J. Am. Soc. Exp. Neurother. 2017, 14, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Davey, P.G.; Charter, M.; Kelly, S.; Varma, T.R.; Jacobson, I.; Freeman, A.; Precious, E.; Lambert, J. Ciprofloxacin and Sparfloxacin Penetration into Human Brain Tissue and Their Activity as Antagonists of GABAA Receptor of Rat Vagus Nerve. Antimicrob. Agents Chemother. 1994, 38, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-C.; Chang, S.-S.; Lee, M.G.; Lee, S.-H.; Tsai, Y.-W.; Lin, S.-C.; Chen, S.-T.; Weng, Y.-C.; Porta, L.; Wu, J.-Y.; et al. Risk of Gastrointestinal Perforation in Patients Taking Oral Fluoroquinolone Therapy: An Analysis of Nationally Representative Cohort. PLoS ONE 2017, 12, e0183813. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Smith, B.N. Tonic GABAA Receptor-Mediated Inhibition in the Rat Dorsal Motor Nucleus of the Vagus. J. Neurophysiol. 2010, 103, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Travagli, R.A.; Hermann, G.E.; Browning, K.N.; Rogers, R.C. Musings on the Wanderer: What’s New in Our Understanding of Vago-Vagal Reflexes? III. Activity-Dependent Plasticity in Vago-Vagal Reflexes Controlling the Stomach. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G180–G187. [Google Scholar] [CrossRef]
- Chang, H.Y.; Mashimo, H.; Goyal, R.K. Musings on the Wanderer: What’s New in Our Understanding of Vago-Vagal Reflex? IV. Current Concepts of Vagal Efferent Projections to the Gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G357–G366. [Google Scholar] [CrossRef]
- Herman, M.A.; Cruz, M.T.; Sahibzada, N.; Verbalis, J.; Gillis, R.A. GABA Signaling in the Nucleus Tractus Solitarius Sets the Level of Activity in Dorsal Motor Nucleus of the Vagus Cholinergic Neurons in the Vagovagal Circuit. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 296, G101–G111. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.E.; Travagli, R.A.; Rogers, R.C. Esophageal-Gastric Relaxation Reflex in Rat: Dual Control of Peripheral Nitrergic and Cholinergic Transmission. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1570–R1576. [Google Scholar] [CrossRef] [PubMed]
- Travagli, R.A.; Hermann, G.E.; Browning, K.N.; Rogers, R.C. Brainstem Circuits Regulating Gastric Function. Annu. Rev. Physiol. 2006, 68, 279–305. [Google Scholar] [CrossRef]
- Zhang, X.; Fogel, R. Involvement of Glutamate in Gastrointestinal Vago-Vagal Reflexes Initiated by Gastrointestinal Distention in the Rat. Auton. Neurosci. Basic Clin. 2003, 103, 19–37. [Google Scholar] [CrossRef]
- Zhou, S.-Y.; Lu, Y.-X.; Yao, H.; Owyang, C. Spatial Organization of Neurons in the Dorsal Motor Nucleus of the Vagus Synapsing with Intragastric Cholinergic and Nitric Oxide/VIP Neurons in the Rat. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1201–G1209. [Google Scholar] [CrossRef]
- Fong, A.Y.; Stornetta, R.L.; Foley, C.M.; Potts, J.T. Immunohistochemical Localization of GAD67-Expressing Neurons and Processes in the Rat Brainstem: Subregional Distribution in the Nucleus Tractus Solitarius. J. Comp. Neurol. 2005, 493, 274–290. [Google Scholar] [CrossRef] [PubMed]
- Glatzer, N.R.; Derbenev, A.V.; Banfield, B.W.; Smith, B.N. Endomorphin-1 Modulates Intrinsic Inhibition in the Dorsal Vagal Complex. J. Neurophysiol. 2007, 98, 1591–1599. [Google Scholar] [CrossRef]
- Kawai, Y.; Senba, E. Organization of Excitatory and Inhibitory Local Networks in the Caudal Nucleus of Tractus Solitarius of Rats Revealed in in Vitro Slice Preparation. J. Comp. Neurol. 1996, 373, 309–321. [Google Scholar] [CrossRef]
- Jiang, Y.; Babic, T.; Travagli, R.A. Sex Differences in GABAergic Neurotransmission to Rat DMV Neurons. Am. J. Physiol.-Gastrointest. Liver Physiol. 2019, 317, G476–G483. [Google Scholar] [CrossRef]
- Sugerman, H.J. Increased Intra-Abdominal Pressure and GERD/Barrett’s Esophagus. Gastroenterology 2007, 133, 2075. [Google Scholar] [CrossRef]
- Irritable Bowel Syndrome|Nature Reviews Disease Primers. Available online: https://www.nature.com/articles/nrdp201614 (accessed on 10 June 2021).
- Bonaz, B.; Sinniger, V.; Pellissier, S. Vagal Tone: Effects on Sensitivity, Motility, and Inflammation. Neurogastroenterol. Motil. 2016, 28, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Turnidge, J. Pharmacokinetics and Pharmacodynamics of Fluoroquinolones. Drugs 1999, 58 (Suppl. 2), 29–36. [Google Scholar] [CrossRef] [PubMed]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of Drugs through the Blood-Cerebrospinal Fluid/Blood-Brain Barrier for Treatment of Central Nervous System Infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [PubMed]
- Federal Drug Agency Highlights of Prescribing Information, CIPRO©. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/019537s086lbl.pdf (accessed on 14 June 2021).
- Fournel, A.; Marlin, A.; Abot, A.; Pasquio, C.; Cirillo, C.; Cani, P.D.; Knauf, C. Glucosensing in the Gastrointestinal Tract: Impact on Glucose Metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G645–G658. [Google Scholar] [CrossRef]
- Masi, E.B.; Levy, T.; Tsaava, T.; Bouton, C.E.; Tracey, K.J.; Chavan, S.S.; Zanos, T.P. Identification of Hypoglycemia-Specific Neural Signals by Decoding Murine Vagus Nerve Activity. Bioelectron. Med. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Guarino, D.; Nannipieri, M.; Iervasi, G.; Taddei, S.; Bruno, R.M. The Role of the Autonomic Nervous System in the Pathophysiology of Obesity. Front. Physiol. 2017, 8, 665. [Google Scholar] [CrossRef] [PubMed]
- Frontoni, S.; Bracaglia, D.; Gigli, F. Relationship between Autonomic Dysfunction, Insulin Resistance and Hypertension, in Diabetes. Nutr. Metab. Cardiovasc. Dis. NMCD 2005, 15, 441–449. [Google Scholar] [CrossRef]
- Russo, B.; Menduni, M.; Borboni, P.; Picconi, F.; Frontoni, S. Autonomic Nervous System in Obesity and Insulin-Resistance—The Complex Interplay between Leptin and Central Nervous System. Int. J. Mol. Sci. 2021, 22, 5187. [Google Scholar] [CrossRef]
- Muscelli, E.; Emdin, M.; Natali, A.; Pratali, L.; Camastra, S.; Gastaldelli, A.; Baldi, S.; Carpeggiani, C.; Ferrannini, E. Autonomic and Hemodynamic Responses to Insulin in Lean and Obese Humans. J. Clin. Endocrinol. Metab. 1998, 83, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Golomb, B.A.; Koslik, H.J.; Redd, A.J. Fluoroquinolone-Induced Serious, Persistent, Multisymptom Adverse Effects. BMJ Case Rep. 2015, 2015, bcr2015209821. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.L.; Burkholder, C.R.; Doheny, K.K.; Travagli, R.A. Ghrelin Ameliorates the Phenotype of Newborn Rats Induced with Mild Necrotizing Enterocolitis. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2019, 31, e13682. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Interaction | Effect | References |
|---|---|---|
| FQs and cytochrome P450 isozyme | Reduced clearance of theophylline and caffeine | Fish 2001, Pharmacotherapy, [34] Mizuki et al., 1996, J. Antimicrob. Chemother. [35] Beckmann et al., 1987, Eur. J. Clin. Pharmacol. [36] Efthymiopoulos et al., 1997, Clin. Pharmacokinet [37] Marchbanks 1993, Pharmacotherapy [38] Davis et al., 1996, Drugs [39] Stille et al., 1987, J. Antimicrob. Chemother. [40] Okimoto et al., 1992, Chemotherapy [41] |
| FQs and BPAA | Reduced binding of GABA to GABAA receptors | Fish 2001, Pharmacotherapy, [34] Christ 1990, J. Antimicrob. Chemother, [42] Radandt et al., 1992, Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am [43] Domagala 1994, J. Antimicrob. Chemother, [44] Smolders et al., 2002, Antimicrob. Agents Chemother. [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freeman, M.Z.; Cannizzaro, D.N.; Naughton, L.F.; Bove, C. Fluoroquinolones-Associated Disability: It Is Not All in Your Head. NeuroSci 2021, 2, 235-253. https://doi.org/10.3390/neurosci2030017
Freeman MZ, Cannizzaro DN, Naughton LF, Bove C. Fluoroquinolones-Associated Disability: It Is Not All in Your Head. NeuroSci. 2021; 2(3):235-253. https://doi.org/10.3390/neurosci2030017
Chicago/Turabian StyleFreeman, Maya Z., Deanna N. Cannizzaro, Lydia F. Naughton, and Cecilia Bove. 2021. "Fluoroquinolones-Associated Disability: It Is Not All in Your Head" NeuroSci 2, no. 3: 235-253. https://doi.org/10.3390/neurosci2030017
APA StyleFreeman, M. Z., Cannizzaro, D. N., Naughton, L. F., & Bove, C. (2021). Fluoroquinolones-Associated Disability: It Is Not All in Your Head. NeuroSci, 2(3), 235-253. https://doi.org/10.3390/neurosci2030017