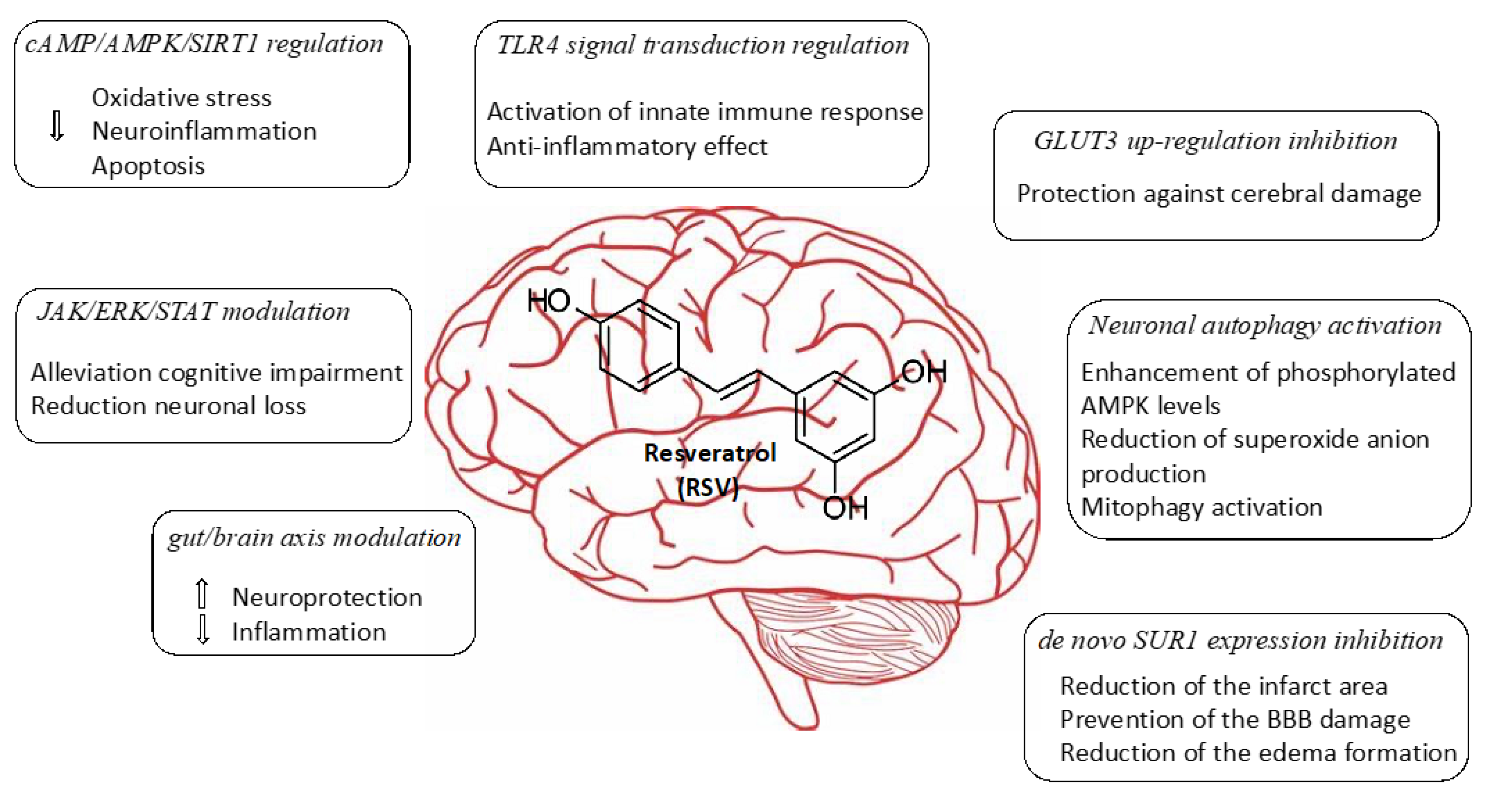
2.1. Regulation of cAMP/AMPK/SIRT1 Pathway
Recently, dysfunction of energy metabolism has been considered as a possible important pathophysiological basis for cerebrovascular accidents. In this way, Wan et al. evaluated the neuroprotector action of RSV in cerebral ischemic injury [
28]. Besides, the authors proposed a mechanism of action based on the regulation of energy metabolism, identifying potential targets of RSV. Results showed that treatment with RSV induced activation of AMPK and SIRT1 by inhibiting cyclic nucleotide PDEs.
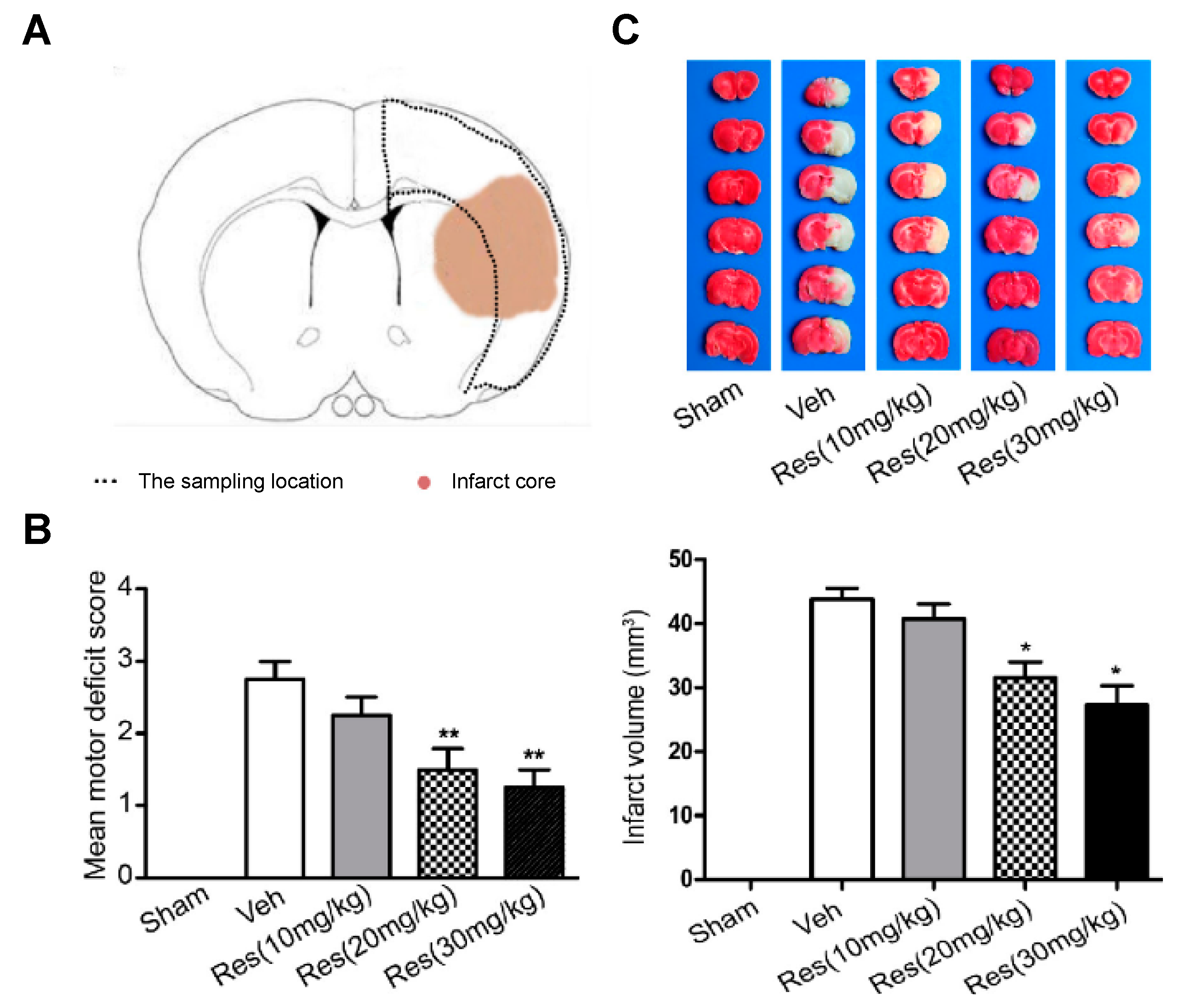
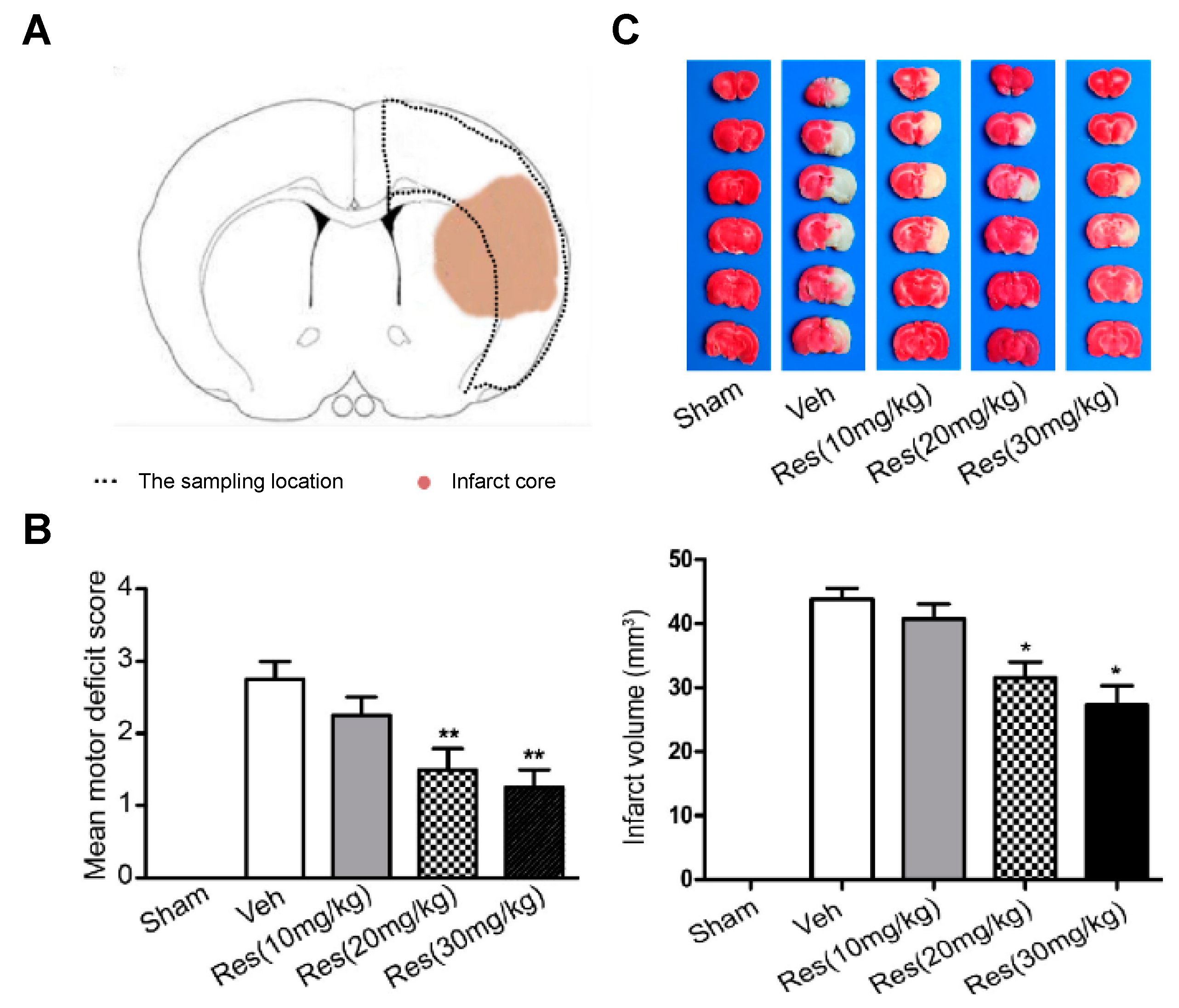
For the development of the studies, in vivo cerebral ischemia injury was induced in 7–8-week-old male Sprague Dawley rats by middle cerebral artery occlusion (MCAO). After 2 h, the cerebral obstruction was removed, and rats were reperfused. RSV dissolved in 5% dimethyl sulfoxide was repeatedly injected via intraperitoneal at 20 mg/kg once a day for 5 days before the occlusion procedure and for the final time before surgery. Results suggested a neuroprotective effect of RSV in MCAO rats since the degree of neurological deficits (based on a 5-point scale) significantly decreased from 2.75 for the control group to 1.67 after RSV treatment (
Figure 2). Besides, these values further decreased with an increase in dose (20 mg/kg to 30 mg/kg). On the other hand, infarct volume measurements showed that RSV protected the rat brain since the volume measured was significantly lower than for the control group (31.5 mm
3 vs. 43.8 mm
3, respectively). Regarding the mechanism, the authors hypothesized that RSV might exert its neuroprotection by increasing SIRT1 and p-AMPK protein levels. In fact, results revealed that the protein expression of SIRT1 and p-AMPK were increased 1.8-fold and 1.4-fold, respectively, in comparison with the vehicle group. To establish the relationship between SIRT1 and p-AMPK in the signaling pathways mediated by RSV, authors employed dorsomorphin, an AMPK inhibitor, and sirtinol as SIRT1-specific inhibitors together with RSV. The first inhibitor produced a considerable decrease, not only in p-AMPK levels but also in SIRT1 levels, while the second inhibitor only affected SIRT1 levels without modifying AMPK levels. Based on that, the authors suggested that RSV could increase SIRT1 levels by activation of AMPK.
The authors also used rolipram as a PDE inhibitor, and its neuroprotective activity was evaluated and compared with that of RSV. Results showed that both significantly increased ATP levels in MCAO rats compared with sham rats (0.30 µmol/g vs. 0.21 µmol/g). Additionally, rolipram increased the expression and levels of SIRT1 and AMPK proteins. Finally, RSV and rolipram inhibited PDE 4A expression, a representative PDE. Hence, the last results confirmed that RSV inhibited PDEs, specifically PDE4A, and as a result, it increases cAMP levels, and thus, the neuroprotective effect is promoted.
The beneficial effect of RSV in induced cerebral ischemia and reperfusion was demonstrated. A new insight into the molecular mechanism based on the regulation of neuronal metabolism was proposed. This is the first study that attributes RSV direct inhibition of PDEs and may provide a potential therapeutic target for ischemic stroke treatment. Nevertheless, further studies are needed to investigate the fundamental mechanism of SIRT1 and AMPK activation and the cAMP signaling.
2.2. Modulating JAK/ERK/STAT Signaling Pathway
To investigate the protective effect of RSV on hippocampus neurons from cerebral ischemia, Chang et al. administered RSV after an induced stroke. The authors proposed that RSV inhibited not only phosphorylation of Janus kinase (JAK)/extracellular signal-regulated kinases (ERK)/signal traducers and activators of transcription (STAT), exerting a neuroprotective effect, but also promoted downregulation of inflammatory cytokines, reduced neuronal loss, and alleviated cognitive impairment [
30].
The authors carried out in vivo studies inducing a cerebral ischemia–reperfusion injury in Sprague Dawley rats. The occlusion was maintained for 2 h, and treatment started for 7 consecutive days using a concentration of 20 mg/kg of RSV dissolved in 50% propylene glycol. The locomotive activity of rats was evaluated using open-field and closed-field tests after 7 days. One week later, the authors studied both memory and learning abilities using the Morris water maze (MWM) test. Then, 3 weeks later after injury, rats were sacrificed to conduct histology, Western blot assay, lipid peroxidation, and antioxidant enzyme assays. This study demonstrated that RSV was able to protect the neuronal loss in the hippocampus from ischemia/reperfusion injury via modulating the JAK/ERK/STAT signaling pathway. This was confirmed using hematoxylin-eosin (HE) staining by counting the number of necrotic neurons in the hippocampus. In addition, the authors showed that RSV prevented memory deficits in rats and effectively reduced lipid peroxidation involved in programmed cell death cascades and inflammation. Therefore, these preliminary results have served to underscore the potential of RSV in treating ischemia–reperfusion injury in the brain, but further experiments should be properly designed to launch preclinical studies to confirm this capability.
2.3. Regulation of TLR4 Signal Transduction
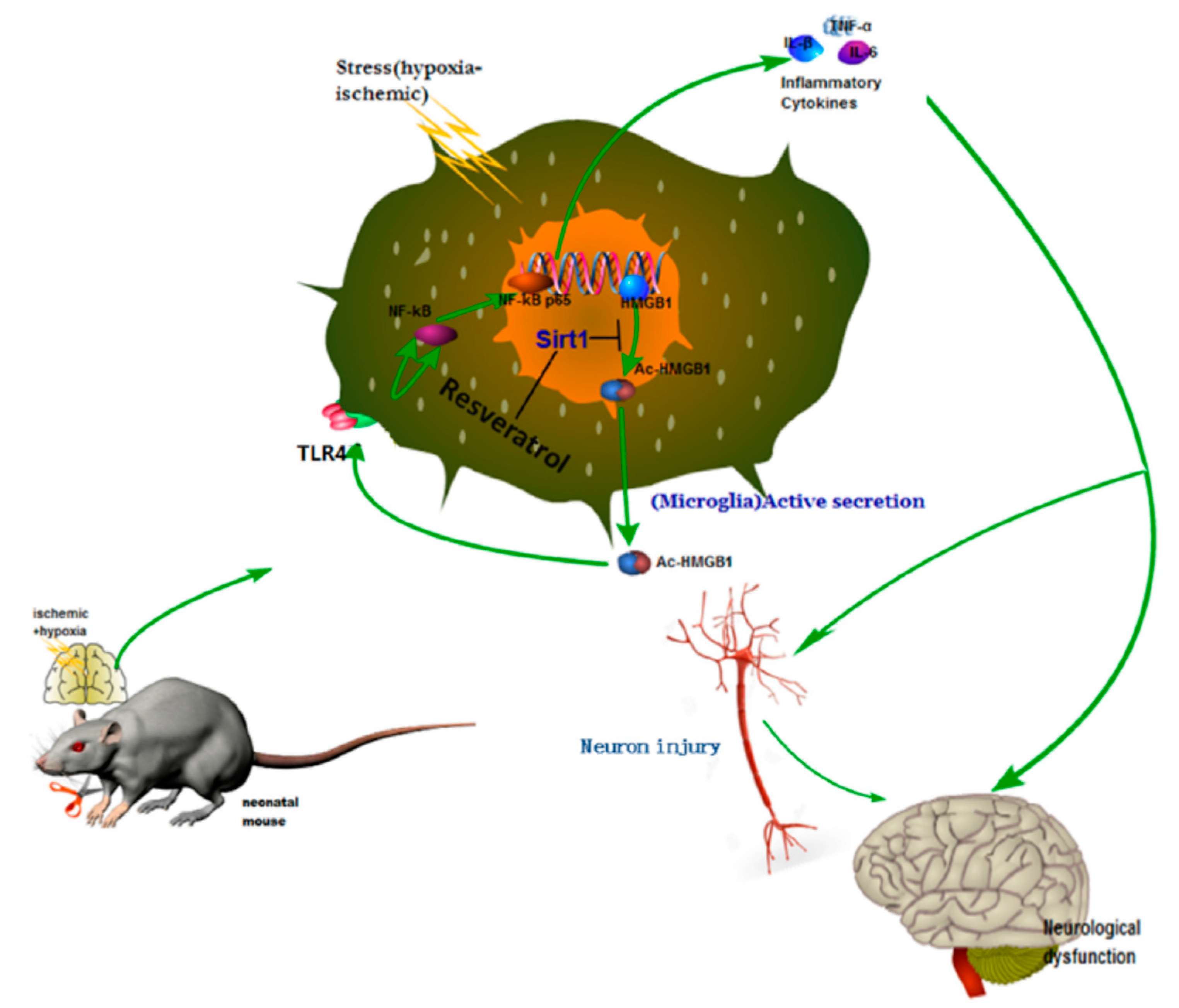
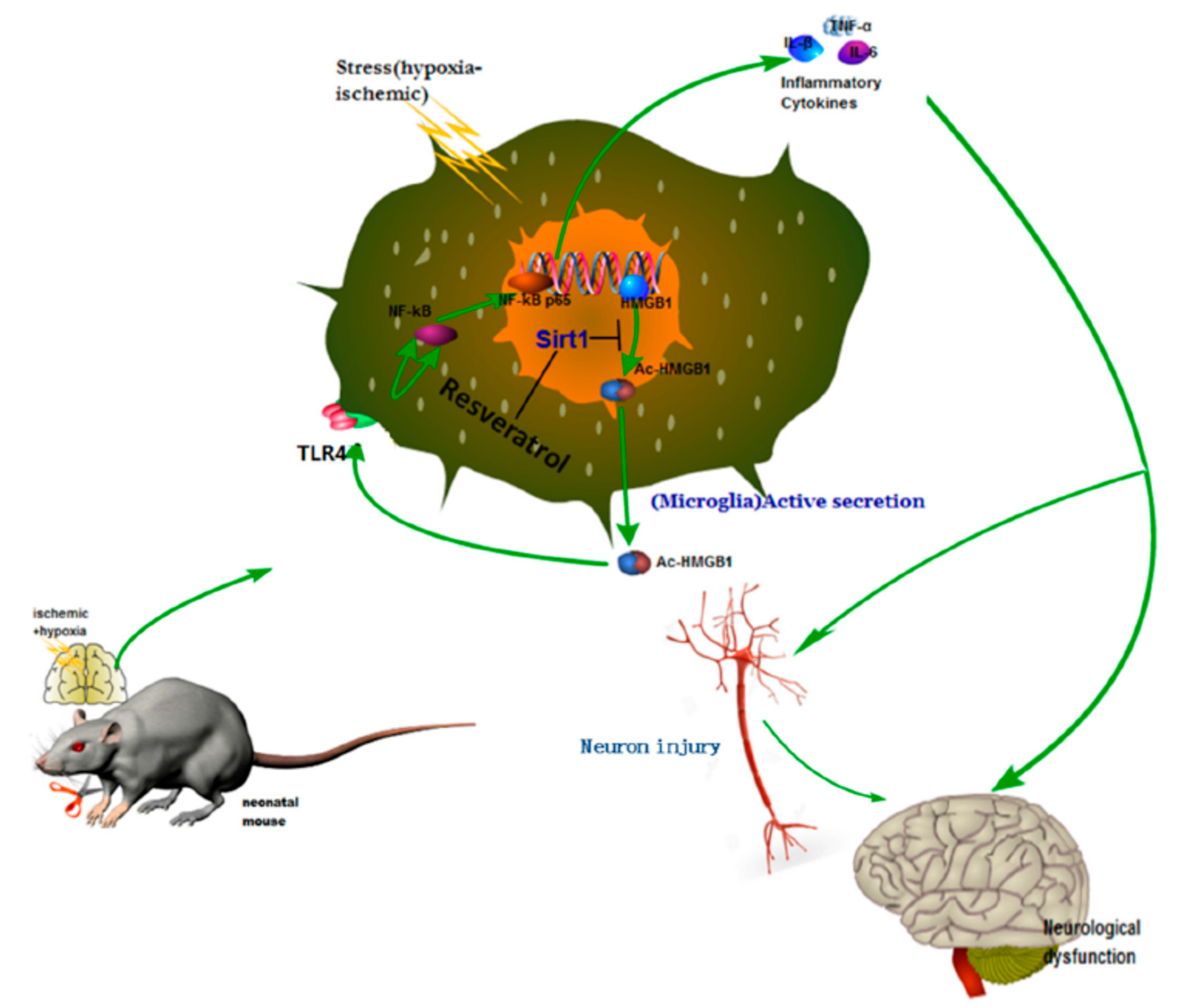
Neonatal hypoxic–ischemic encephalopathy (HIE), or hypoxic–ischemic brain injury (HIBI), is caused by disorders in cerebral blood flow and oxygen supply, being the most common cause of perinatal brain injury, which can lead to neonatal death or cause irreversible or prolonged physical and mental disabilities that lack appropriate treatment. Le et al. evaluated the activity of high mobility group box-1 (HMGB1) through the activation of SIRT1 by RSV in the inhibition of HI insult-induced neuroinflammation [
31]. Using in vivo and in vitro assays, the authors confirmed that RSV inhibited the acetylation level and release of HMGB1, which was actively involved in the activation of the inflammatory cytokines signaling pathways (Toll-Like Receptor 4 (TLR4)/MyD88/Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)), by increasing the expression and activity of SIRT1.
For this purpose, the in vivo experiments were performed using the Rice–Vannucci method. The hypoxic–ischemic brain injury was induced in postnatal day 7 pups, which were then placed in a hypoxia chamber for 1 h. After the injury and 12 h later, RSV at a concentration of 100 mg/mL in PBS was administered via intraperitoneal injection using a volume of 45–55 µL/mouse. Behavioral tests showed that the performance anomalies were significantly ameliorated after RSV administration. The Western blotting analysis from brain tissues and the conditioned medium of BV2 microglial cell culture indicated that RSV increased levels of SIRT1 and decreased TLR4, MyD88, and nuclear NF-κB. Besides, the authors observed changes in the localization of HMGB1 after injury. First, it was in the nucleus of the microglia, and then it was partially located out of the nucleus. Interestingly, RSV diminished the cytoplasmatic localization of HGMB1. Using mouse microglial BV2 cells after hypoxic–ischemic insult by oxygen–glucose deprivation (OGD), in vitro tests were performed to elucidate the role of TLR4 (the classical receptor of HMBG1). The stress produced by OGD significantly increased the mRNA expression and levels of cytokines (IL-1β, IL-6, TNF-α) and the expression of TLR4 and NF-κB. The in vitro results demonstrated that RSV also inhibited the amplification of inflammation at a concentration of 25 µM. The activation of SIRT1 due to RSV was confirmed using in vitro tests with stressed microglia cells and co-immunoprecipitation. The expression and deacetylase activity of SIRT1 as well as the interaction between SIRT1 and HGMB1 were increased by RSV (
Figure 3). As a consequence, the level of acetylated HGMB1 and nucleocytoplasmatic translocation was inhibited. Finally, confocal imaging of BV2 cells evidenced the neuroprotective effect of RSV. So far, the specific mechanism for the treatment of neuroinflammation induced by hypoxic–ischemic insult in the immature brain was unclear. However, the authors described the action of essential factors such as HMGB1 and TLR4 involved in the neuroinflammation. Similarly, other authors also observed that RSV could inhibit the inflammation as a regulator of microglia/macrophages activation, reducing the production of inflammatory mediators in the ischemic cortex (IL-1β, TNF-α, and ROS). Thus, a neuroprotection effect was evidenced via reduction of glial cell activation and prevention of delayed neuronal cell death [
24].
In this same direction, other reports have reaffirmed the potent anti-inflammatory properties of RSV and also suggest its effect by modulating TLR4 expression in the HMGB1–TLR4 signaling pathway [
26]. Overall, these works represent a start point for the potential treatment of this high-incidence disorder based on the administration of RSV.
2.4. Mechanism Targeting Gut/Brain Axis
Inflammation and immune system activation are two important processes that can affect the brain after acute stroke. Importantly, emerging evidence has proved the role of the microbiota–gut brain axis in the ischemic brain. In this sense, Dou et al. demonstrated for the first time that RSV might inhibit systemic post-stroke inflammations, inducing neuroprotection by modulating intestinal-immune-cell-mediated inflammation [
32]. The authors proposed one of the possible multifactorial mechanisms through which RSV could act is by modulating T helper 17 (Th17)/regulatory T (Treg) and Th1/Th2 cells as well as the expression of their related cytokines in the small intestine.
The authors carried out in vivo studies by inducing focal cerebral ischemia in mice. This was produced by applying a transient occlusion in the middle cerebral artery (MCAO) for 60 min. Then, extensive in vivo experiments were performed by dividing mice into nine experimental groups, including controls. Some groups were pre-treated with vancomycin, clavulanic acid, or amoxicillin during 3 days of previous MCAO. Additionally, fecal microbiota transplantation and RSV were administered intraperitoneally to experimental groups in order to confirm the neuroprotective effect of RSV, which was directly mediated by the intestinal flora. The RSV concentration used in this study was 200 mg/kg. After 3 days of treatment, mice were sacrificed, and a series of quantification tests (e.g., flow cytometry, Western blotting, albumin quantification by ELISA, vascular permeability assay, and RNA extraction) were performed to measure the mRNA levels of two cytokines (IFN-γ and IL-4) and Th17/Treg response. Interestingly, the authors found an important role of the gut–brain axis in acute ischemic stroke, demonstrating the neuroprotective effect of RSV when administered. Therefore, the authors showed that RSV was able to modulate Th1/Th2 balance and reduce small intestinal pro-inflammatory cytokines, including intestinal vascular permeability. The results also confirmed the ability of RSV to protect against systemic post-stroke inflammation in mice in the cortex and striatum.
2.5. Inhibition of GLUT3 Up-Regulation
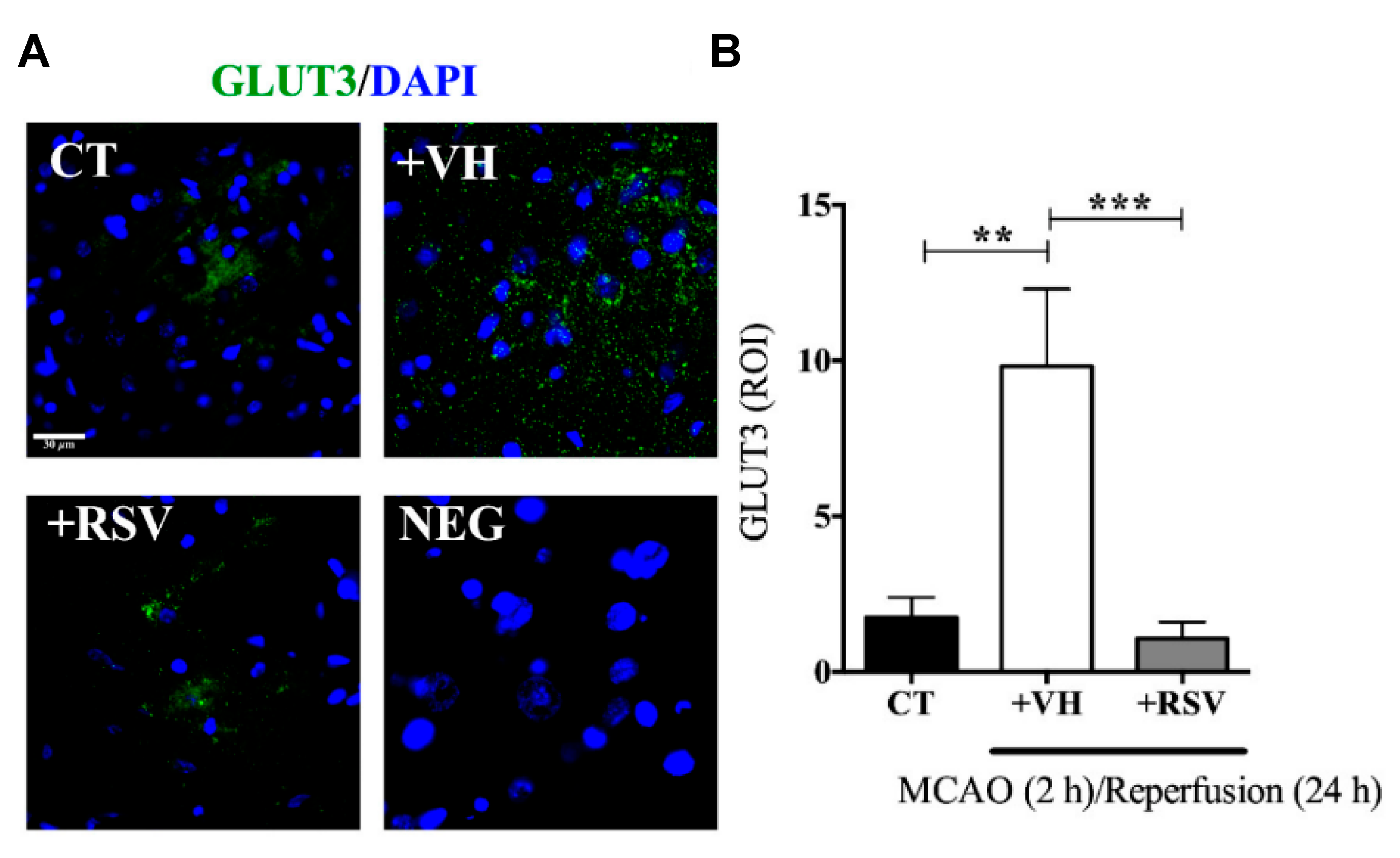
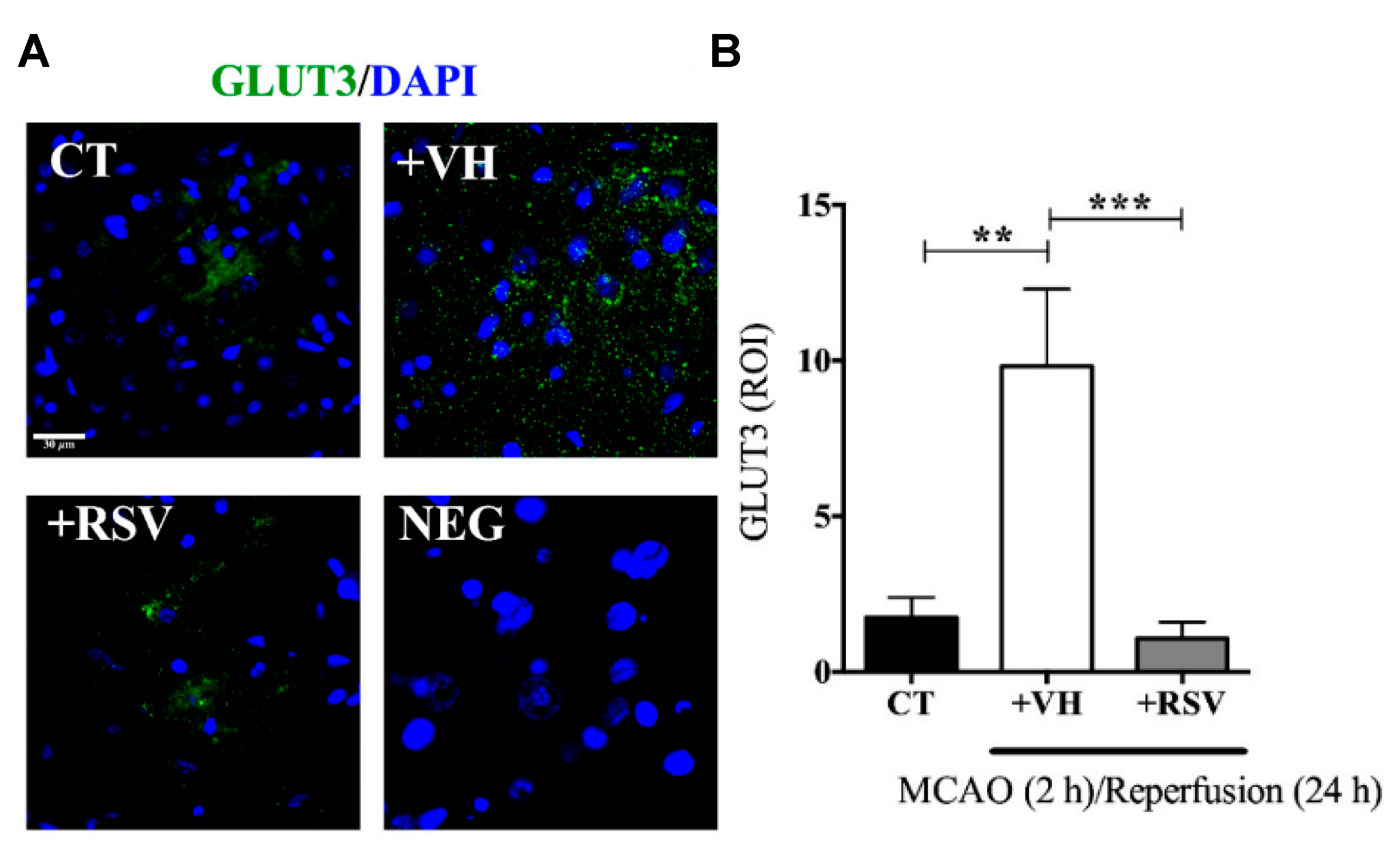
After cerebral ischemia, the brain metabolic energy is reduced. In consequence, an up-regulation of glucose transporter (GLUT3) is an adaptive response for the rapid nutrient supply. Nonetheless, the fluctuation in glucose levels alters mitochondrial functionality, promotes apoptosis, and stimulates pro-inflammatory factors. On this basis, Gutiérrez Aguilar et al. studied the effect of RSV on GLUT3 expression levels after induced ischemia [
33]. Results showed that the polyphenol protects neurons from middle cerebral artery occlusion damage and prevents GLUT3 up-regulation in the injured brain that could depend on AMPK activation.
In this study, male Wistar rats were subjected to MCAO for 2 h, followed by different times of reperfusion (1–24 h). RSV (1.9 mg/kg) was administrated intravenously, diluted in ethanol at the onset of reperfusion. The infarct area measurements demonstrated that RSV reduced MCAO-induced damage since the expression levels of Microtubule-associated protein 2 (MAP2), an indicator of surviving neurons, were conserved after the injury, ensuring the integrity of the fibers. Furthermore, RSV prevented the increment in GLUT3 mRNA stimulated by 2 h of MCAO and 1 h of reperfusion, while those levels greatly increased in the group without treatment (2.5-fold) (
Figure 4). Besides, the same result was obtained for GLUT3 protein levels for 24 h of reperfusion and confirmed by immunofluorescence microscopy. The authors also evaluated the role of RSV in the inactivation of GLUT3 via p-AMPK, specifically through p65-NF-κB, a regulator of GLUT3 expression. For this purpose, glutamate-induced excitotoxicity (model of ischemic stroke) was induced in mixed cultures of neurons and astrocytes. The authors found that RSV increased the phosphorylation of AMPK in both types of cells, indicating that this molecule not only induced an endogenous response but also activated a compensatory mechanism in astrocytes. Based on these results, the authors proposed a mechanism based on RSV-triggered phosphorylation of AMPK. This factor activates SIRT, which subsequently inhibits the transcriptional activity of p65-NF-κB, which is involved in the GLUT3 regulation. However, this is still considered a preliminary study, and further research is essential to improve the comprehension of the process.
2.6. Activation of Neuronal Autophagy
Based on the fact that AMPK preserves the cellular energy levels and may contribute to neuroprotection by activating autophagy after cellular stress such as cerebral ischemia or excitotoxicity, Pineda-Ramírez et al. investigated the neuroprotector effect of resveratrol on this via [
34]. They demonstrated that RSV enhanced AMPK activity and drove autophagy, suggesting an increase of survival neurons. Herein, cerebral ischemia was induced in Wistar rats through the MCAO for 2 h, and then reperfusion was induced. A single dose of RSV (1.8 mg/kg in water–ethanol 50%
v/v) was administrated in the tail vein at the onset of reperfusion. Results showed that p-AMPK levels were significantly increased when RSV was administrated in comparison with the non-treated group. Besides, the infarct area detected was markedly reduced after treatment with RSV than for the control group (20.76 ± 1.67% and 28.08 ± 2.84%, respectively). The employment of an AMPK inhibitor, compound C ((6-[4-(2-piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-ylpyrrazolo [1,5-a]–pyrimidine), showed interference with the protective effect of RSV. For instance, the infarct area was reduced to only 25.7 ± 2.25%; the rat survival after 24 h of reperfusion was 57% and 90.47% for rats treated with RSV. This suggested that the neuroprotector effect of the polyphenol was partially related to AMPK activation.
Furthermore, the effect of RSV in autophagy through activation of AMPK was analyzed by the protein’s expression levels involved in different stages of autophagy in neuronal cultures subjected to excitotoxicity. In relation to the first stages of autophagy, RSV increased Beclin 1 protein level, and thus, the microtubule-associated protein light chain 3 (LC3)-II/LC3-I ratio was significantly increased. Those proteins are involved in the formation of autophagosomes. In the final autophagy stage, p62 protein and lysosomal-associated membrane protein 1 (LAMP1) are important. In this case, RSV considerably reduced the level of LAMP1 without affecting p62 levels. The administration of compound C to prevent the AMPK activation from RSV produced the opposite effect. Thus, the authors demonstrated that RSV induced autophagy through AMPK activation. Specifically, mitochondrial degradation was studied through mitochondrial matrix protein HSP60 expression. The expression of this protein was significant decreased after RSV treatment, and this effect was partially reversed by compound C. Based on this result, the authors proposed that the neuroprotector effect of RSV is due to the activation of AMPK, which leads to degradation of mitochondria by autophagy.
Looking for effective therapeutic agents for ischemic stroke treatment, Liu and co-workers studied the effect of combined therapy using RSV and rosuvastatin [
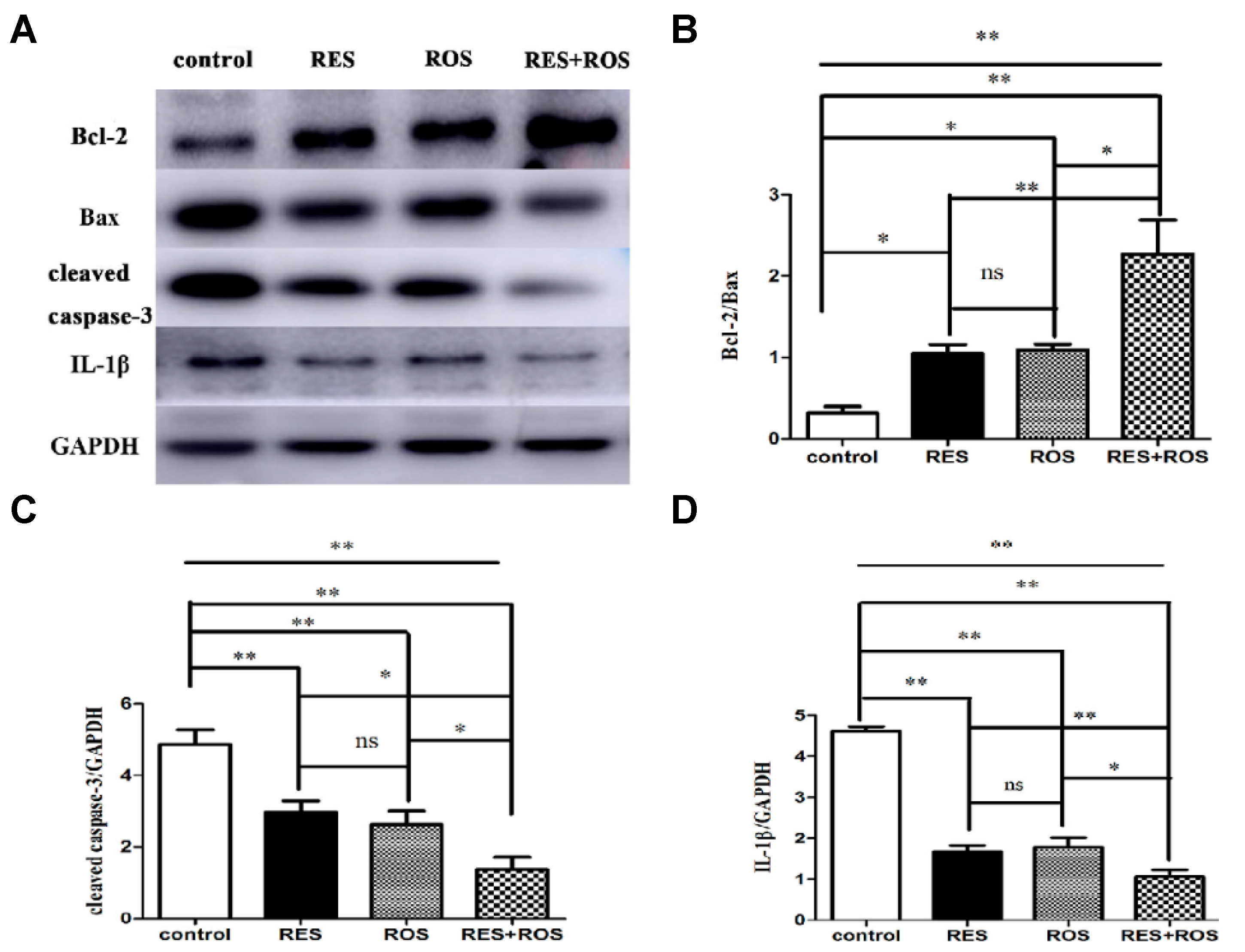
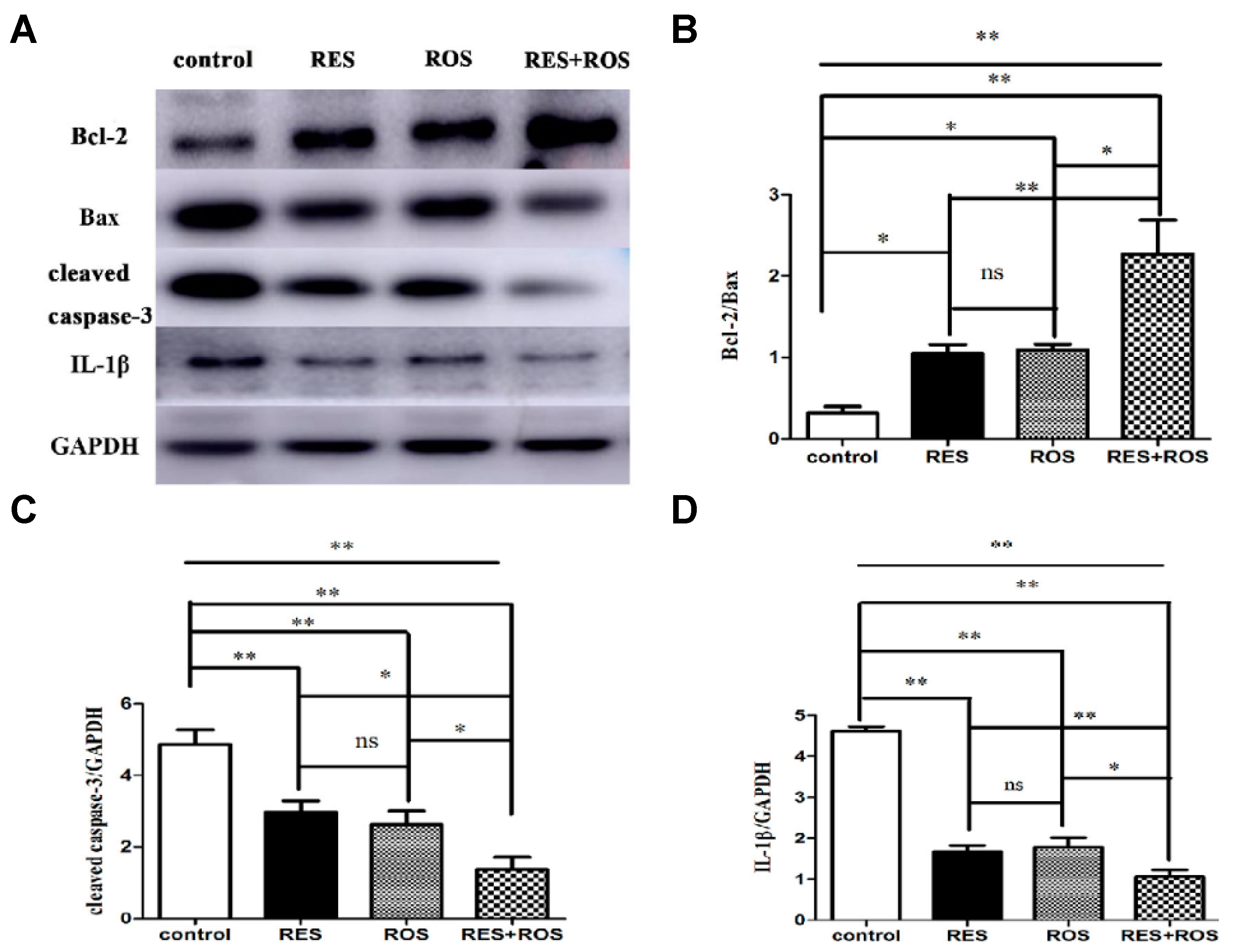
35]. The combined pretreatment synergistically improved induced ischemic stroke. Besides, the authors proposed a possible in vivo mechanism based on the enhancement of anti-apoptosis, anti-inflammation, and autophagy. In the study, MCAO was induced in adult male Sprague Dawley rats for 90 min. Then, the rats were reperfused. The administrations of drugs were carried out via intraperitoneal for RSV at a dose of 50 mg/kg and through intragastric gavage for rosuvastatin at a dose of 10 mg/kg. The doses were given once a day for 7 repeated days before ischemia surgery. Interestingly, the pretreatment with a combination of drugs resulted in a stronger neuroprotective effect evidenced by cerebral infarct measurements and modified neurological severity scores (mNSS) after 24 h of arterial insult. The cerebral infarct volume significantly decreased in comparison with the control group from 0.3 to 0.1, and those values were smaller than for separate therapy (for example, 0.2 for RSV only). Besides, the neurological score decreased considerably from 7 for the control group to 5 due to the combination of drugs. This result showed an improvement in motor, sensory, reflex, and balance of rats and, thus, less neurological damage. In relation to the mechanism, the anti-apoptosis activity was studied, measuring the apoptotic-associated proteins levels, such as Bax, Bcl-2, and caspase-3. The combination of drugs significantly decreased the expression of Bax and caspase 3, both apoptotic markers, compared to the control group (
Figure 5). In contrast, the expression of Bcl-2, an anti-apoptotic factor, increased considerably. Thus, a more enhanced anti-apoptosis was observed by the synergic effect. The inflammatory response was also affected using the combined therapy. In fact, the expression of IL-1β, an inflammatory factor, was remarkably decreased when compared to the separate pretreatment. Thus far, the strong neuroprotective effect was not only related to enhanced anti-apoptosis but also with a greater anti-inflammation. Lastly, the level of autophagy caused by ischemic brain injury was investigated, measuring the levels of LC3II, LC3I, and Beclin-1, all autophagy markers. The combination of drugs produced a considerable increase in Beclin-1 expression and the ratio of LC3-II/LC3-I in contrast with the drugs alone. Thus, neuroprotection was also associated with the upregulation of autophagy levels. Finally, a synergistic effect was clearly observed by the co-administration of a statin and the phytoalexin RSV. This neuroprotective effect was associated with a complex activation that included anti-apoptosis, anti-inflammation, and autophagy.
Other authors also suggested that RSV could induce autophagy as a mechanism of neuroprotection. He et al. hypothesized that RSV treatment could relieve cerebral ischemic injury by inhibiting NLRP3 inflammasome activation through induction of SIRT1-dependent autophagy [
36]. The NLRP3 is a NOD-like receptor belonging to the family of pyrin domain-containing 3 inflammasome which is involved in innate immunity and inflammation. After their stimulation, inflammasomes are activated and lead to caspase-1 activation with the release of bioactive cytokines (IL-1β and IL-18). The induction of autophagy could reduce the inflammasome-derived inflammation and exert the neuroprotective effect. The authors employed healthy male Sprague Dawley rats for the MCAO procedure during 1 h followed by 24 h reperfusion. RSV was injected at a dose of 100 mg/kg (i.p.) at the onset of reperfusion. The results suggested that RSV can ameliorate cerebral ischemic injury in rats since the infarct volume of the RSV-treated group was significantly decreased, the neurological scores were remarkably improved, and the brain water content after reperfusion was reduced in comparison with the non-treated group. Moreover, the levels of NLRP3 inflammasome-regulated cytokines such as caspase-1, IL-1β, and IL-18 were significant inhibited by treatment with RSV. In addition, the protein levels of SIRT1 were upregulated when RSV was administrated in comparison with the MCAO group. On the other hand, the autophagy markers such as LC3B-II/LC3B-I ratio and expression of p62 were increased in the presence of the ischemic injury. The treatment with RSV increased the levels of LC3B-II/LC3B-I ratio even more and decreased the expression of p62, indicating an enhancement of autophagy activity. Finally, to assess if autophagy activity and NLRP3 inflammasome activation were mediated by SIRT1, the SIRT1 siRNA pretreatment (along with or without RSV) was employed. The results showed a significant reverse of RSV effects such as a decrease in SIRT1 expression and in the LC3B-II/LC3B-I ratio and an increase in the expression of p62, i.e., inhibition of autophagy. Besides, the downregulation of NLRP3 inflammasomes by RSV was markedly inhibited by the SIRT1 knockdown. In this way, the authors confirmed that the effect of RSV on autophagy and the inhibition of NLRP3 inflammasome was regulated by the SIRT1 pathway. Thus, these findings demonstrated that RSV administration results are beneficial as a treatment of cerebral ischemic in rats; however, more details of the pathological mechanisms of this injury are required.
2.7. Via De Novo SUR1 Expression Inhibition
Another potential mechanism in which RSV has shown effectiveness is in reducing the infarct area and cerebral edema, resulting in an improvement of the neurological performers, reduction of the blood–brain barrier (BBB) damage, and increase of the survival acting in the process of inhibiting Abcc8 gene transcription that codes for Sulfonylurea receptor 1 (SUR1).
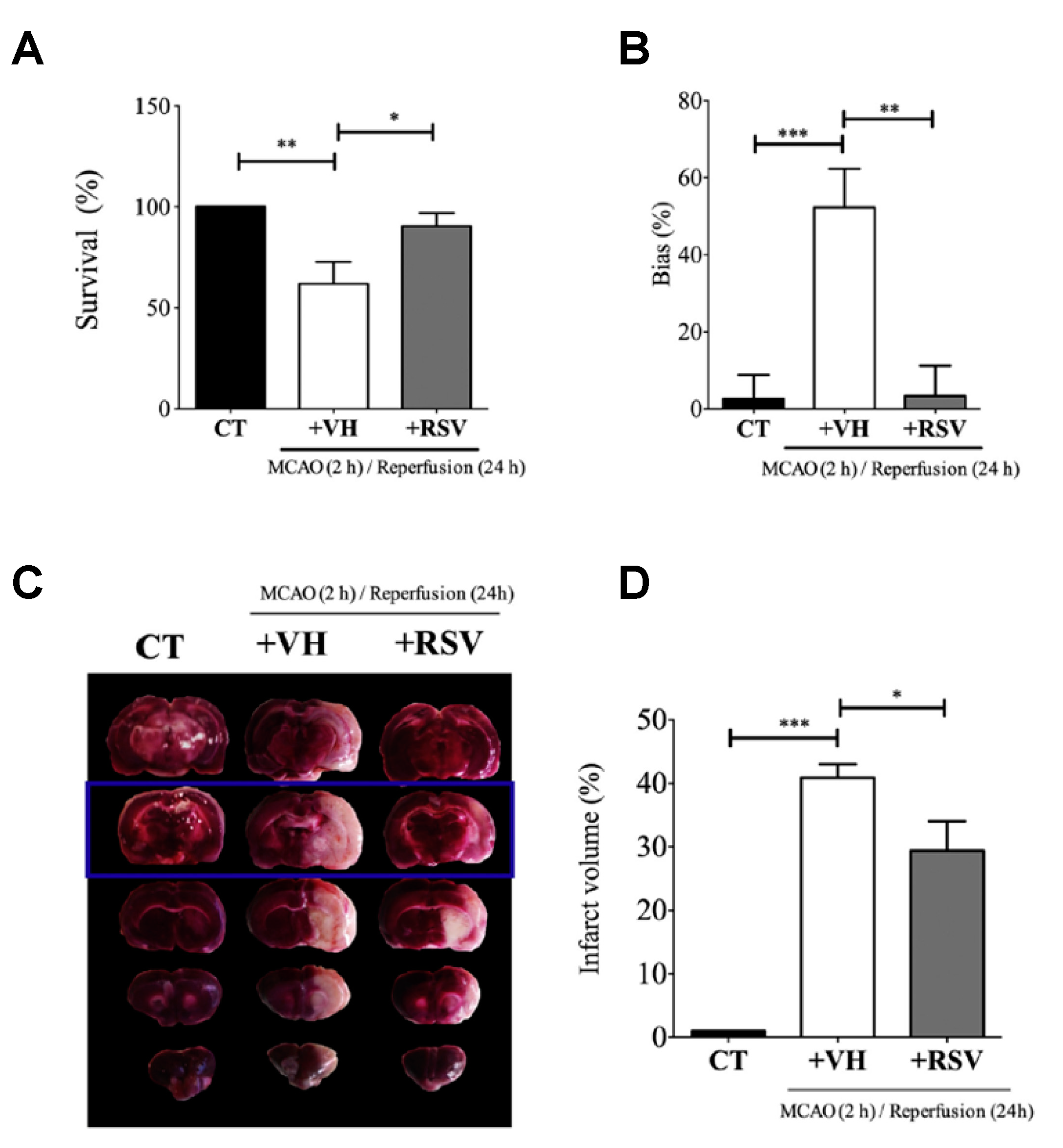
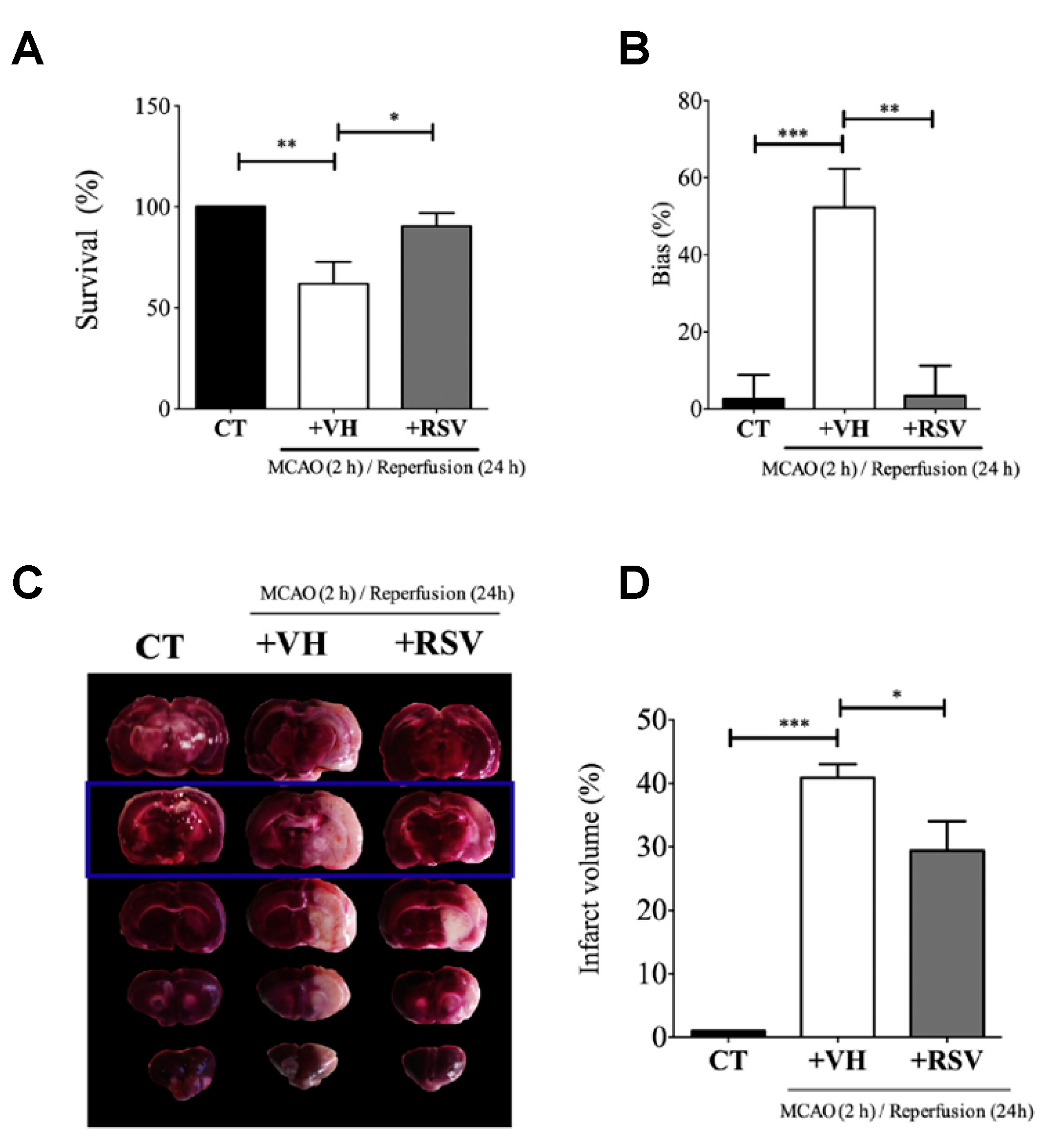
This study published by Alquisiras-Burgos et al. assessed in vivo studies using male Wistar rats that were divided into three groups: control (CT), vehicle (VH) ischemic group, and the treatment group with RSV [
37]. Then, the authors proceeded to occlude the middle cerebral artery for 2 h. At the beginning of reperfusion, RSV was injected (1.9 mg/kg) in the tail vein. Before rats were sacrificed, the “limb-use asymmetry test” was performed to evaluate the neurological performance of the animals at 24 h of reperfusion. Then, the permeability of BBB was evaluated, and the individual hemisphere water change content (%) was quantified by adding Evans blue (2%) diluted in saline during reperfusion (4 mL/kg) and observing the color change in the tissue. Likewise, the authors quantified the infarct area by TTC staining assay, which showed the highest effect in the somatosensory cortex. Interestingly, the binding activity of specificity protein 1 (SP1) transcription factor was evaluated using electrophoretic mobility shift assays (EMSA) with 5 µg of nuclear extract and 0.2 µg of biotin-labeled oligonucleotides mixed with binding reagents. In addition, immunofluorescence assays with a specific endothelial marker and Western blot assays to quantify SUR1 and Aquaporin-4 (AQP4) expression were assessed (
Figure 6). Despite the fact that a precise mechanism is still unclear, the results in animal models showed that RSV had a tendency to target SP transcription factors and inhibit the expression of SUR1 and AQP4. As a result, the authors found that RSV significantly protected BBB integrity and increased the rate of survival from 61.9% to 90.4%, attenuating infarct volumes. Alternatively, the authors found evidence that the effect of RSV on regulating the balance between matrix metalloproteinase (MMP)-9 and the inhibitor TIMP-1 might prevent the BBB disruption after ischemic stroke.
2.8. Administration of Resveratrol-Loaded Nanoparticles (NPs)
In addition to using RSV itself as a therapeutic molecule to treat ischemic brain injuries, other strategies have been assessed. Lu et al. prepared NPs made up of poly(N-vinylpyrrolidone)-
b-poly(ε-caprolactone) (PVP-PCL) as drug carriers for RSV [
38]. The resulting particles exhibited spherical shape and diameters of 100 nm, according to Scanning Electron Microscopy (SEM) and Dynamic Light Scattering (DLS), respectively. In vitro controlled release experiments were performed. These assays showed an initial burst release of 37% and sustained release of RSV close to 65% after 6 days.
The in vivo effect in model rats was confirmed with different experimental groups. In addition to control groups (saline, empty NPs, and 10 mg/kg unloaded RSV), increasing concentrations of RSV-loaded NPs (1, 5 and 10 mg/kg) were tested. The administration of RSV-loaded NPs was carried out via carotid artery rather than tail vein as this approach showed an increase of the circulating NPs in the cerebral vasculature. Once the parameters were optimized in terms of drug dose (5 mg/kg), the authors investigated the leakage of the BBB with Evan blue (2.5 mL/kg) dissolved in saline (4% w/v). TTC staining assay was also assessed and confirmed the infarct area was reduced by 31.7% and 34.1% after 24 h and 72 h of reperfusion, respectively. In addition, the level of lipid peroxidation was reduced by 56.5%, approximately confirming the reduction of neuronal apoptosis. Finally, this study demonstrated that NPs were able to facilitate the release of RSV in vivo as well as a significant protection against cerebral ischemia/reperfusion, oxidative stress attenuation, and the prevention of brain edema.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}