
Systematic Investigation of the Solvation Structure in THF-Based Localized High-Concentration Electrolytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Electrolyte Preparation
2.2. Electrolyte Characterizations
3. Results and Discussion
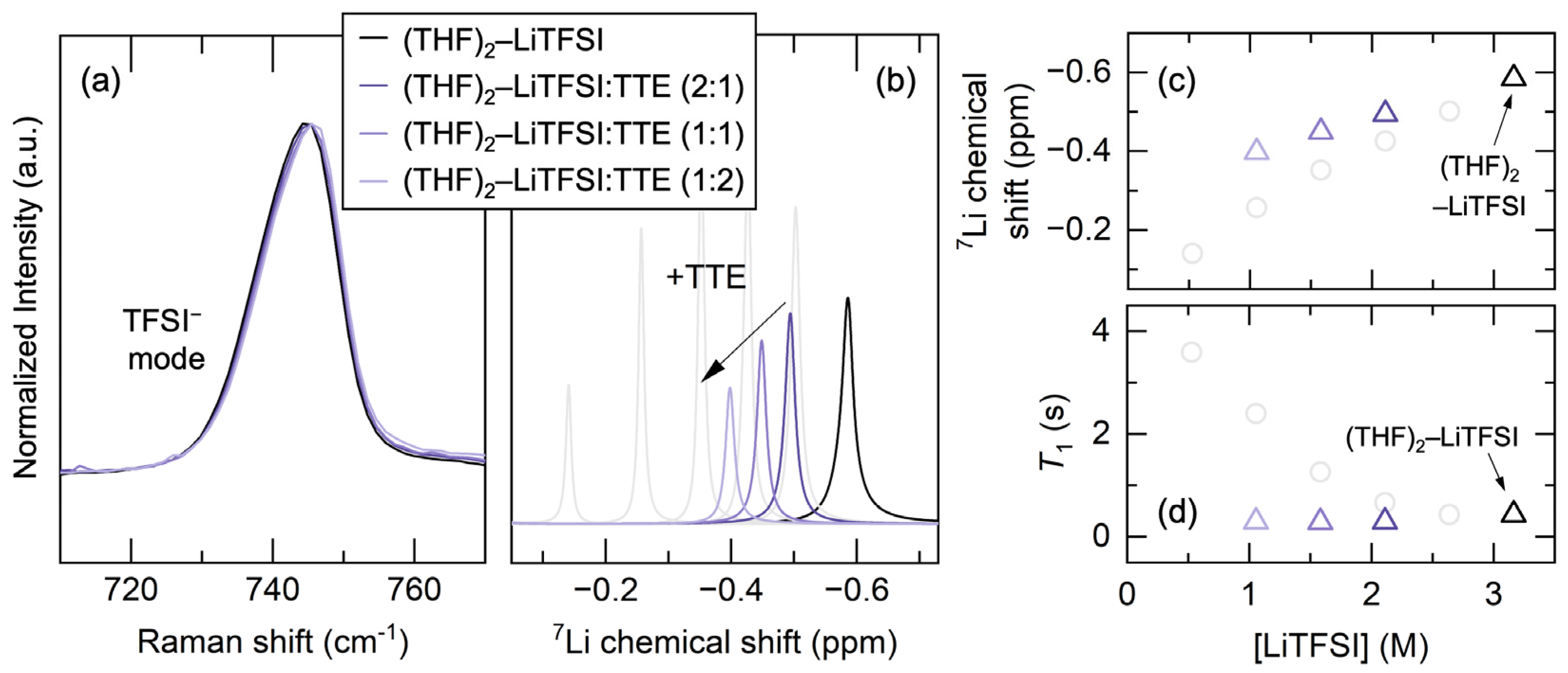
3.1. Raman Spectroscopy
3.2. NMR Spectroscopy
3.3. Effect of Electrolyte Localization via TTE Addition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ren, X.; Zou, L.; Jiao, S.; Mei, D.; Engelhard, M.H.; Li, Q.; Lee, H.; Niu, C.; Adams, B.D.; Wang, C.; et al. High-concentration ether electrolytes for stable high-voltage lithium metal batteries. ACS Energy Lett. 2019, 4, 896–902. [Google Scholar] [CrossRef]
- Efaw, C.M.; Wu, Q.; Gao, N.; Zhang, Y.; Zhu, H.; Gering, K.; Hurley, M.F.; Xiong, H.; Hu, E.; Cao, X.; et al. Localized high-concentration electrolytes get more localized through micelle-like structures. Nat. Mater. 2023, 22, 1531–1539. [Google Scholar] [CrossRef]
- Cao, X.; Jia, H.; Xu, W.; Zhang, J.-G. Localized high-concentration electrolytes for lithium batteries. J. Electrochem. Soc. 2021, 168, 010522. [Google Scholar] [CrossRef]
- Zheng, Y.; Soto, F.A.; Ponce, V.; Seminario, J.M.; Cao, X.; Zhang, J.-G.; Balbuena, P.B. Localized high concentration electrolyte behavior near a lithium–metal anode surface. J. Mater. Chem. A 2019, 7, 25047–25055. [Google Scholar] [CrossRef]
- Sun, J.; Yao, Y.; Cui, X.; Luo, J.; Zhang, J.; Zhao, Y.; Wang, H.; Zhou, J.; Zhu, J.; Wang, Y.; et al. Improving Low-Temperature Tolerance of a Lithium-Ion Battery by a Localized High-Concentration Electrolyte Based on the Weak Solvation Effect. Batter. Energy 2025, 4, e20240106. [Google Scholar] [CrossRef]
- Hossain, J.; Wu, Q.; Bernardez, E.J.M.; Quilty, C.D.; Marschilok, A.C.; Takeuchi, E.S.; Bock, D.C.; Takeuchi, K.J.; Qi, Y. The relationship between ionic conductivity and solvation structures of localized high-concentration fluorinated electrolytes for lithium-ion batteries. J. Phys. Chem. Lett. 2023, 14, 7718–7731. [Google Scholar] [CrossRef] [PubMed]
- van Ekeren, W.; Hall, A.; Lahtinen, K.; Younesi, R. The solvation structure of localized high concentration electrolytes. ChemElectroChem 2024, 11, e202400050. [Google Scholar] [CrossRef]
- Ren, F.; Li, Z.; Chen, J.; Huguet, P.; Peng, Z.; Deabate, S. Solvent–diluent interaction-mediated solvation structure of localized high-concentration electrolytes. ACS Appl. Mater. Interfaces 2022, 14, 4211–4219. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Park, B.K.; Kang, S.; Yang, S.J.; Choi, S.H.; Yoo, D.; Kim, K.J. Bespoke Dual-Layered Interface Enabled by Cyclic Ether in Localized High-Concentration Electrolytes for Lithium Metal Batteries. Adv. Funct. Mater. 2024, 34, 2408365. [Google Scholar] [CrossRef]
- Seo, D.M.; Boyle, P.D.; Sommer, R.D.; Daubert, J.S.; Borodin, O.; Henderson, W.A. Solvate structures and spectroscopic characterization of LiTFSI electrolytes. J. Phys. Chem. B 2014, 118, 13601–13608. [Google Scholar] [CrossRef]
- Jiang, G.; Li, F.; Wang, H.; Wu, M.; Qi, S.; Liu, X.; Yang, S.; Ma, J. Perspective on high-concentration electrolytes for lithium metal batteries. Small Struct. 2021, 2, 2000122. [Google Scholar] [CrossRef]
- Chen, X.; Meng, Y.; Xiao, D.; Wu, Y.; Qin, L. Tunning solvation structure in non-flammable, localized high-concentration electrolytes with enhanced stability towards all aluminum substrate-based K batteries. Energy Storage Mater. 2023, 61, 102923. [Google Scholar] [CrossRef]
- Ren, X.; Chen, S.; Lee, H.; Mei, D.; Engelhard, M.H.; Burton, S.D.; Zhao, W.; Zheng, J.; Li, Q.; Ding, M.S.; et al. Localized high-concentration sulfone electrolytes for high-efficiency lithium-metal batteries. Chem 2018, 4, 1877–1892. [Google Scholar] [CrossRef]
- Wang, Z.; He, Z.; Wang, Z.; Yang, J.; Long, K.; Wu, Z.; Zhou, G.; Mei, L.; Chen, L. A nitrile solvent structure induced stable solid electrolyte interphase for wide-temperature lithium-ion batteries. Chem. Sci. 2024, 15, 13768–13778. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Hua, H.; Lai, P.; Zhao, J. A multifunctional dual-salt localized high-concentration electrolyte for fast dynamic high-voltage lithium battery in wide temperature range. Adv. Energy Mater. 2021, 11, 2101775. [Google Scholar] [CrossRef]
- Cai, W.; Deng, Y.; Deng, Z.; Jia, Y.; Li, Z.; Zhang, X.; Xu, C.; Zhang, X.; Zhang, Y.; Zhang, Q. Quasi-localized high-concentration electrolytes for high-voltage lithium metal batteries. Adv. Energy Mater. 2023, 13, 2301396. [Google Scholar] [CrossRef]
- Piao, Z.; Gao, R.; Liu, Y.; Zhou, G.; Cheng, H. A review on regulating Li+ solvation structures in carbonate electrolytes for lithium metal batteries. Adv. Mater. 2023, 35, 2206009. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Jia, Z.; Ruan, D.; Nian, Q.; Fan, J.; Chen, S.; He, Z.; Wang, D.; Jiang, J.; Ma, J.; et al. Molecular anchoring of free solvents for high-voltage and high-safety lithium metal batteries. Nat. Commun. 2024, 15, 2033. [Google Scholar] [CrossRef] [PubMed]
- Amine, R.; Liu, J.; Acznik, I.; Sheng, T.; Lota, K.; Sun, H.; Sun, C.; Fic, K.; Zuo, X.; Ren, Y.; et al. Regulating the hidden solvation-ion-exchange in concentrated electrolytes for stable and safe lithium metal batteries. Adv. Energy Mater. 2020, 10, 2000901. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Cao, X.; Engelhard, M.H.; Liu, W.; Burton, S.D.; Lee, H.; Niu, C.; Matthews, B.E.; Zhu, Z.; et al. Enabling high-voltage lithium-metal batteries under practical conditions. Joule 2019, 3, 1662–1676. [Google Scholar] [CrossRef]
- Pang, Q.; Shyamsunder, A.; Narayanan, B.; Kwok, C.Y.; Curtiss, L.A.; Nazar, L.F. Tuning the electrolyte network structure to invoke quasi-solid state sulfur conversion and suppress lithium dendrite formation in Li–S batteries. Nat. Energy 2018, 3, 783–791. [Google Scholar] [CrossRef]
- Xu, R.; Ding, J.; Ma, X.; Yan, C.; Yao, Y.; Huang, J. Designing and demystifying the lithium metal interface toward highly reversible batteries. Adv. Mater. 2021, 33, 2105962. [Google Scholar] [CrossRef]
- Wang, C.; Zhuang, Y.; Qin, Y.; Dong, Y.; Liu, L.; Zhang, L.; Du, J. Design and eco-efficiency analysis of sustainable extractive distillation process combining preconcentration and solvent recovery functions for separating the tetrahydrofuran/ethanol/water ternary multi-azeotropic mixture. Process. Saf. Environ. Prot. 2022, 159, 795–808. [Google Scholar] [CrossRef]
- Park, H.; Lim, H.-D.; Lim, H.-K.; Seong, W.M.; Moon, S.; Ko, Y.; Lee, B.; Bae, Y.; Kim, H.; Kang, K. High-efficiency and high-power rechargeable lithium–sulfur dioxide batteries exploiting conventional carbonate-based electrolytes. Nat. Commun. 2017, 8, 14989. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, L.; Yu, Y.; He, M.; Zhang, H.; Tang, Y.; Xiong, F.; Gao, S.; Li, A.; Wang, J.; et al. Stabilized Li-S batteries with anti-solvent-tamed quasi-solid-state reaction. Joule 2023, 7, 2074–2091. [Google Scholar] [CrossRef]
- Fawdon, J.; Ihli, J.; La Mantia, F.; Pasta, M. Characterising lithium-ion electrolytes via operando Raman microspectroscopy. Nat. Commun. 2021, 12, 4053. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, V.; Simari, C.; Mangiacapre, E.; Pierini, A.; Gentile, A.; Marchionna, S.; Nicotera, I.; Brutti, S.; Bodo, E. Aprotic Electrolytes Beyond Organic Carbonates: Transport Properties of LiTFSI Solutions in S-Based Solvents. ChemSusChem 2025, 18, e202402273. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Lin, W.; Wei, B.; Chen, D. A concentrated electrolyte of LiTFSI and dimethyl carbonate for high-voltage Li batteries. ACS Appl. Energy Mater. 2023, 6, 9337–9346. [Google Scholar] [CrossRef]
- Alves, C.C.; Campos, T.B.; Alves, W.A. FT-Raman spectroscopic analysis of the most probable structures in aluminum chloride and tetrahydrofuran solutions. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 97, 1085–1088. [Google Scholar] [CrossRef]
- Pham, T.D.; Bin Faheem, A.; Lee, K. Design of a LiF-rich solid electrolyte interphase layer through highly concentrated LiFSI–THF electrolyte for stable lithium metal batteries. Small 2021, 17, 2103375. [Google Scholar] [CrossRef]
- Nanda, R.; Damodaran, K. A review of NMR methods used in the study of the structure and dynamics of ionic liquids. Magn. Reson. Chem. 2018, 56, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.J.; Ciezki, A.A.; Berno, A.; Halalay, I.C.; Goward, G.R. Quantitative operando 7Li NMR investigations of silicon anode evolution during fast charging and extended cycling. J. Am. Chem. Soc. 2023, 145, 21502–21513. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Hwang, Y.; An, Y.J.; Sim, S.; Shin, M. Systematic Investigation of the Solvation Structure in THF-Based Localized High-Concentration Electrolytes. Organics 2026, 7, 10. https://doi.org/10.3390/org7010010
Hwang Y, An YJ, Sim S, Shin M. Systematic Investigation of the Solvation Structure in THF-Based Localized High-Concentration Electrolytes. Organics. 2026; 7(1):10. https://doi.org/10.3390/org7010010
Chicago/Turabian StyleHwang, Yoonha, Yeo Jin An, Soohyun Sim, and Minjeong Shin. 2026. "Systematic Investigation of the Solvation Structure in THF-Based Localized High-Concentration Electrolytes" Organics 7, no. 1: 10. https://doi.org/10.3390/org7010010
APA StyleHwang, Y., An, Y. J., Sim, S., & Shin, M. (2026). Systematic Investigation of the Solvation Structure in THF-Based Localized High-Concentration Electrolytes. Organics, 7(1), 10. https://doi.org/10.3390/org7010010