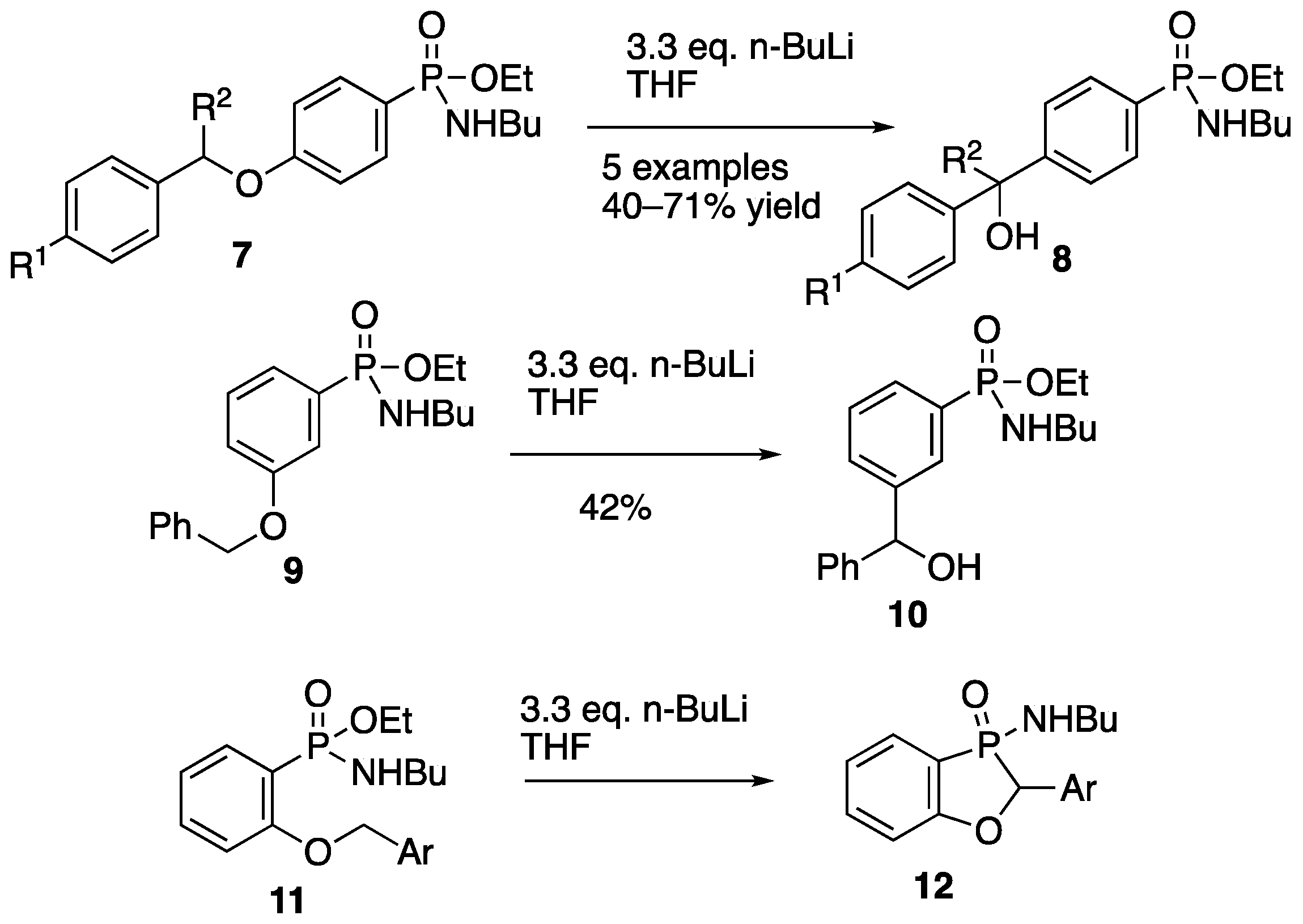
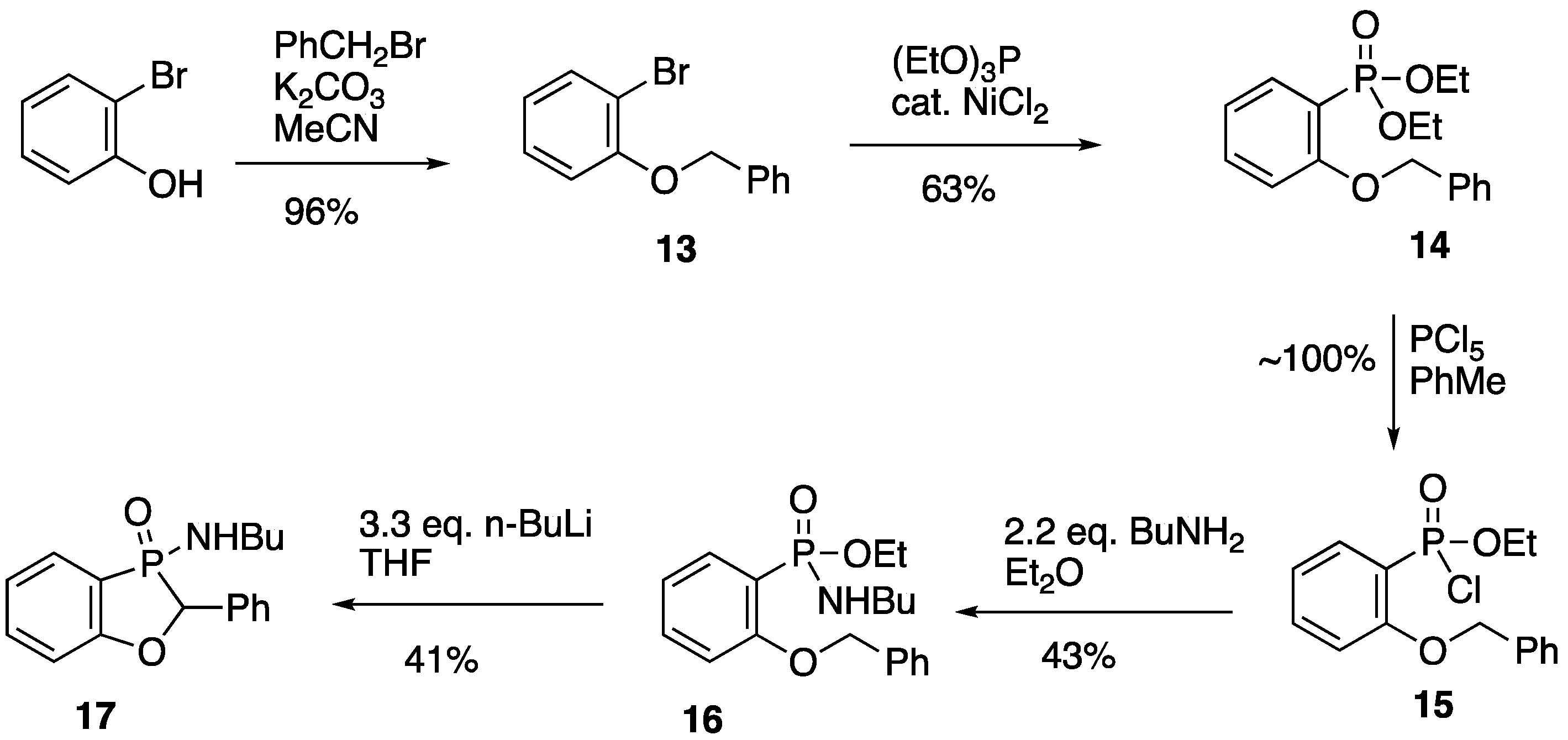
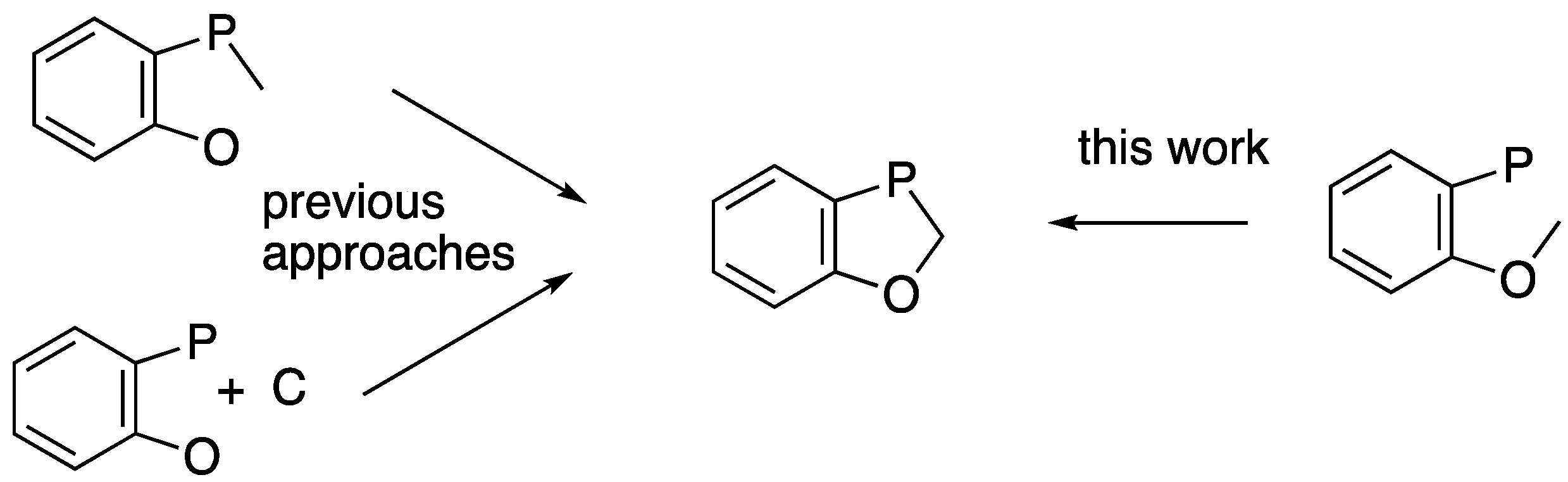
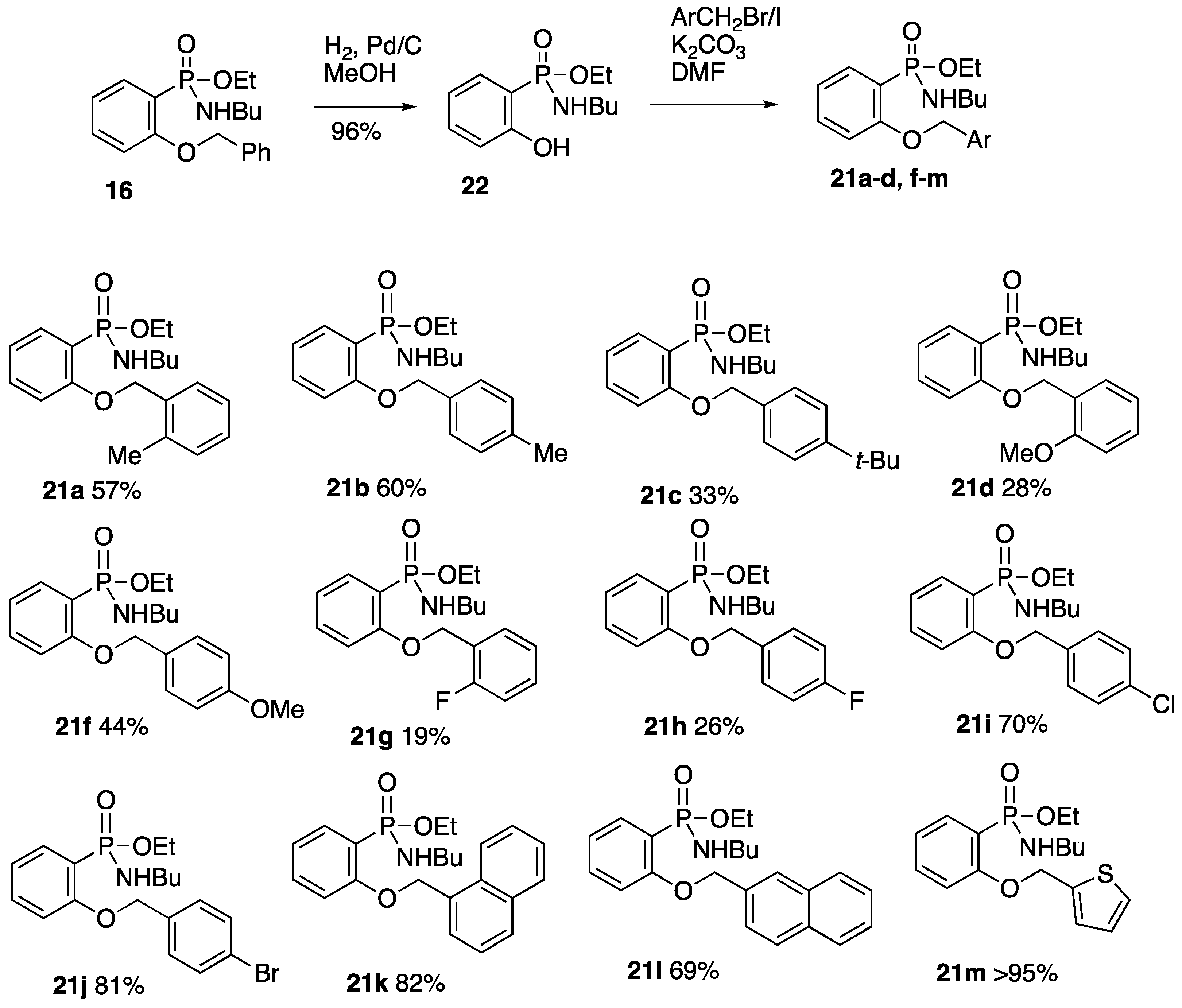
2.2. Synthesis and Rearrangement of Ethyl P-(4-Benzyloxyphenyl)-N-butylphosphonamidate 16
2.2.1. 1-(Benzyloxy)-2-bromobenzene 13
To a stirred solution of 2-bromophenol (4.36 g, 25.2 mmol) in MeCN (60 mL) at rt was added K
2CO
3 (4.74 g, 34.3 mmol) and benzyl bromide (3.0 mL, 4.32 g, 25.2 mmol), and the mixture was stirred at rt overnight. The reaction was diluted with H
2O (75 mL), the layers separated, and the aqueous layer extracted with EtOAc (3 × 75 mL). The combined organic layers were dried over MgSO
4 and concentrated to give
13 (6.35 g, 96%) as a pale-yellow oil which was used without further purification;
1H NMR (400 MHz): 7.55 (1H, dd,
J = 7.8, 1.6 Hz, ArH), 7.50–7.44 (2H, m, ArH), 7.42–7.34 (2H, m, ArH), 7.34–7.27 (1H, m, ArH), 7.22 (1H, ddd,
J = 8.2, 7.4, 1.6 Hz, ArH), 6.92 (1H, dd,
J = 8.2, 1.4 Hz, ArH), 6.83 (1H, ddd,
J = 7.8, 7.4, 1.4 Hz, ArH) and 5.14 (2H, s, OCH
2);
13C NMR (100 MHz) 154.9 (C-O), 136.5 (C), 133.4 (CH), 128.5 (2CH), 128.3 (CH), 127.9 (CH), 126.9 (2CH), 122.1 (CH), 113.8 (CH), 112.4 (C-Br) and 70.7 (OCH
2). The
1H and
13C spectral data were in accordance with those previously reported [
14] (
Supplementary Materials).
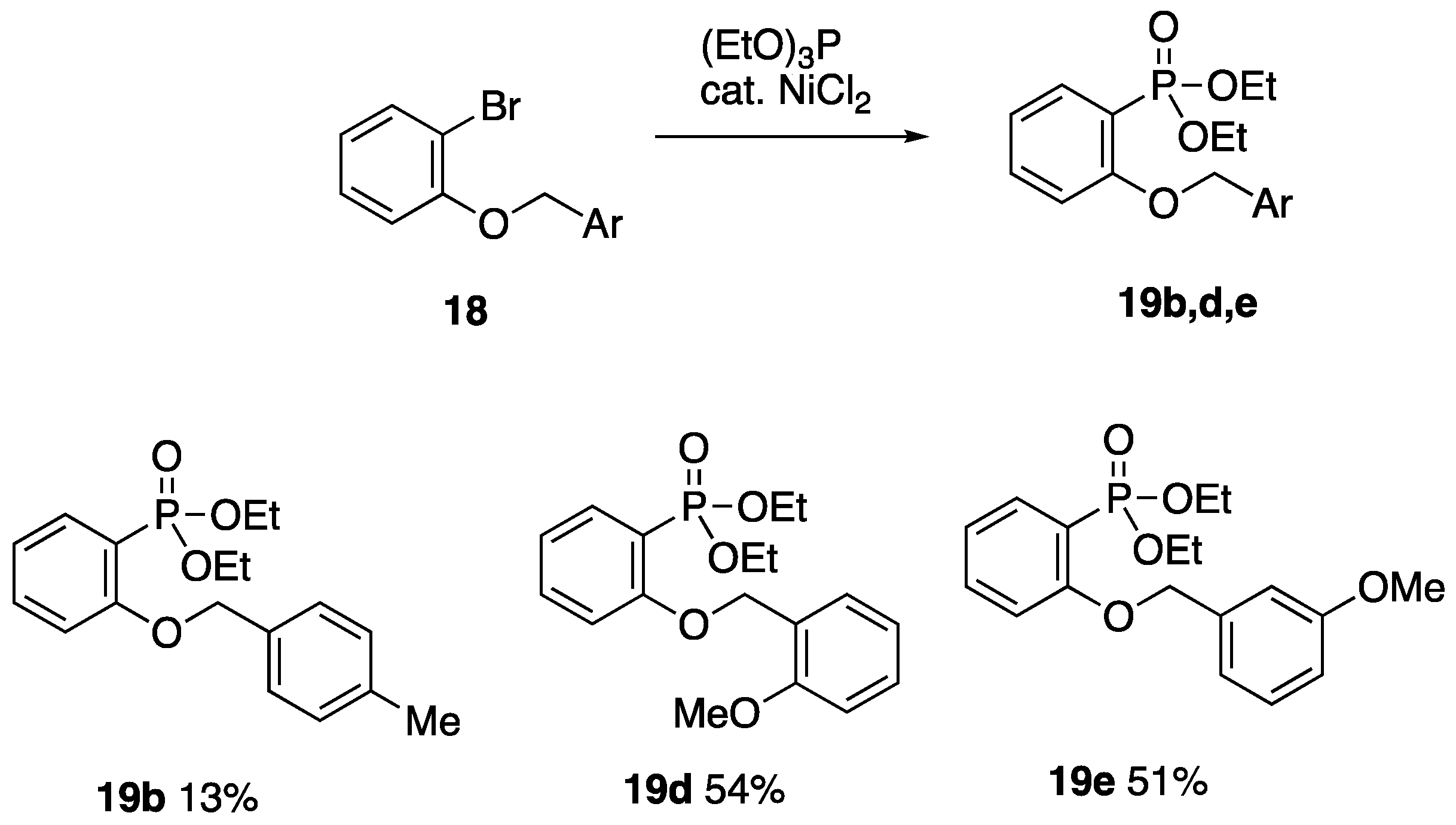
2.2.2. Diethyl (2-Benzyloxyphenyl)phosphonate 14
Following a modified literature procedure [
15], 1-(benzyloxy)-2-bromobenzene
13 (3.77 g, 14.2 mmol) and anhydrous NiCl
2 (0.92 g, 7.1 mmol) were placed in a flask set up for distillation, and a dropping funnel containing triethyl phosphite (3.0 mL, 17.2 mmol) was connected to the still head. The mixture was heated at 150 °C while the phosphite was added dropwise until the mixture was dark red. When the initial dark red colour changed to blue, more phosphite was added until the red colour returned. This was repeated until all the phosphite was added; the mixture was then heated for a further 30 min and cooled to rt. The mixture was taken up in CH
2Cl
2 (50 mL), which was washed with dil. HCl (25 mL), dried, and evaporated. Purification via flash column chromatography (gradient elution hexane/EtOAc 9:1 to 100% ethyl acetate), followed by the removal of triethyl phosphate by Kugelrohr distillation, gave
8 (2.88 g, 63%) as a pale-yellow oil; ν
max/cm
−1 1593, 1477, 1443, 1279, 1242, 1020, 959, 756, 733, 696, 573, 536, and 507;
1H NMR (400 MHz): 7.87 (1H, ddd,
J = 14.9, 7.4, 1.8 Hz, ArH), 7.55–7.50 (2H, m, ArH), 7.47 (1H, dddd,
J = 8.3, 7.4, 1.8, 0.9 Hz, ArH), 7.41–7.35 (2H, m, ArH), 7.33–7.28 (1H, m, ArH), 7.05–6.96 (2H, m, ArH), 5.19 (2H, s, OCH
2Ph), 4.18–4.05 (4H, m, 2 OC
H2CH
3) and 1.28 (6H, t,
J = 7.1 Hz, 2 OCH
2C
H3);
13C NMR (100 MHz): 160.1 (d,
J = 2.7 Hz, C-O), 136.4 (C), 135.1 (d,
J = 7.2 Hz, CH), 134.2 (d,
J = 2.1 Hz, CH), 128.4 (2CH), 127.7 (CH), 126.9 (2CH), 120.5 (d,
J = 14.6 Hz, CH), 117.0 (d,
J = 187 Hz, ArC-P), 112.3 (d,
J = 9.3 Hz, Ar CH), 70.0 (OCH
2Ph), 62.0 (d,
J = 5.6 Hz, 2 O
CH
2CH
3) and 16.2 (d,
J = 6.5 Hz, 2 OCH
2CH
3);
31P NMR (162 MHz): +17.1; HRMS (ESI
+): found 343.1058. C
17H
21NaO
4P (M + Na) requires 343.1075.
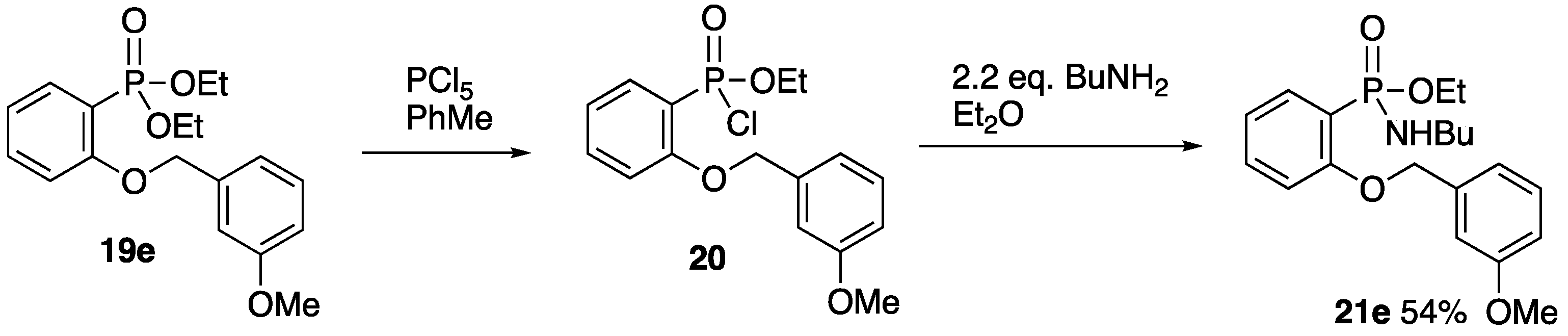
2.2.3. Ethyl (2-Benzyloxyphenyl)phosphonochloridate 15
A solution of diethyl (2-benzyloxyphenyl)phosphonate 14 (1.00 g, 3.1 mmol) in dry toluene (15 mL) was stirred at 0 °C while PCl5 (1.30 g, 6.2 mmol) was added. The mixture was then stirred at rt for 30 min, filtered, and evaporated to give 15 (0.99 g, ~100%) as a yellow oil which was used without further purification; 1H NMR (400 MHz): 7.94 (1H, ddd, J = 16.9, 7.7, 1.8 Hz, ArH), 7.55 (1H, tdd, J = 8.4, 1.8, 1.0 Hz, ArH), 7.54–7.46 (2H, m, ArH), 7.41–7.36 (2H, m, ArH), 7.35–7.32 (1H, m, ArH), 7.09–6.99 (2H, m, ArH), 5.22 (2H, s, OCH2Ph), 4.42–4.26 (2H, m, OCH2CH3) and 1.35 (3 H, td, J = 7.0, 0.5 Hz, OCH2CH3); 13C NMR (125 MHz): 159.8 (4ry, d, J = 2.9 Hz, ArC-O), 136.0 (C), 135.6 (d, J = 2.0 Hz, CH), 134.2 (d, J = 8.6 Hz, CH), 128.5 (2CH), 128.0 (CH), 127.0 (2CH), 120.5 (d, J = 16.4 Hz, CH), 118.5 (d, J = 179.5 Hz, C-P), 112.7 (d, J = 9.8 Hz, CH), 70.5 (OCH2Ph), 63.7 (d, J = 7.7 Hz, OCH2CH3) and 15.8 (d, J = 7.7 Hz, OCH2CH3); 31P NMR (162 MHz): +26.5.
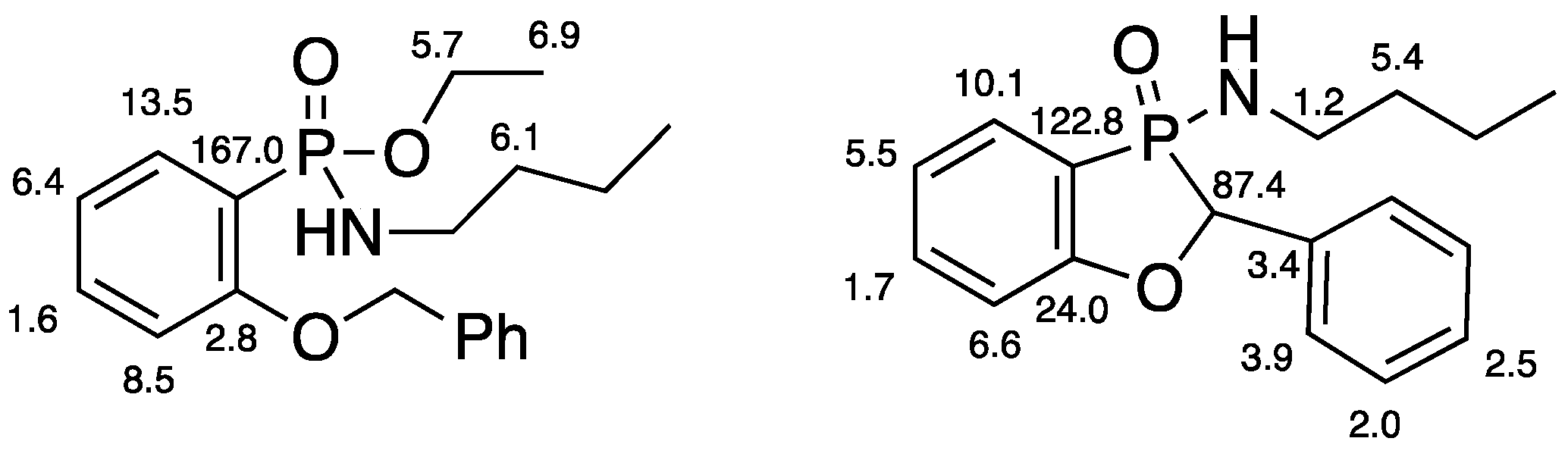
2.2.4. Ethyl P-(2-Benzyloxyphenyl)-N-butylphosphonamidate 16
Following a literature procedure [
16], a solution of
n-butylamine (0.67 mL, 0.50 g, 6.8 mmol) in Et
2O (25 mL) was stirred at 0 °C while a solution of ethyl (2-benzyloxyphenyl)phosphonochloridate
15 (1.00 g, 3.2 mmol) in Et
2O (25 mL) was added dropwise. The mixture was allowed to warm to rt and was stirred for 18 h. Water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with Et
2O (2 × 25 mL), and the combined organic layers were dried and evaporated. Purification by column chromatography (SiO
2, EtOAc/hexane 1:1) gave
16 (480 mg, 43%) as a slightly yellow oil; ν
max/cm
−1 2957, 2930, 2872, 1591, 1440, 1277, 1229, 1086, 1032, 951, 756, 735, 696, 571, and 534;
1H NMR (400 MHz): 7.93 (1H, ddd,
J = 14.2, 7.4, 1.8 Hz, ArH), 7.48–7.43 (3H, m, ArH), 7.42–7.32 (3H, m, ArH), 7.04 (1H, tdd,
J = 7.4, 2.9, 0.9 Hz, ArH), 6.99 (1H, ddd,
J = 8.4, 6.2, 0.9 Hz, ArH), 5.14 (2H, s, OCH
2Ph), 4.09–3.89 (2H, m, OC
H2CH
3), 2.97–2.87 (3H, m, N
HC
H2), 1.34–1.27 (2H, m, NHCH
2C
H2), 1.26 (3H, t,
J = 7.1 Hz, OCH
2C
H3), 1.21–1.09 (2H, m, NHCH
2CH
2C
H2) and 0.79 (3H, t,
J = 7.3 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (125 MHz): 159.0 (d,
J = 2.8 Hz, ArC-O), 136.1 (C), 134.4 (d,
J = 6.4 Hz, CH), 133.3 (d,
J = 1.6 Hz, Ar CH), 128.6 (2CH), 128.2 (CH), 127.3 (2CH), 120.8 (d,
J = 13.5 Hz, CH), 119.8 (d,
J = 167.0 Hz, ArC-P), 111.7 (d,
J = 8.5 Hz, CH), 70.3 (OCH
2Ph), 60.2 (d,
J = 5.7 Hz, O
CH
2CH
3) 40.3 (NHCH
2), 34.0 (d,
J = 6.1 Hz, NHCH
2CH
2), 19.6 (NHCH
2CH
2CH
2), 16.3 (d,
J = 6.9 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +21.3; HRMS (ESI
+): found 370.1529. C
19H
26NNaO
3P (M + Na) requires 370.1548.
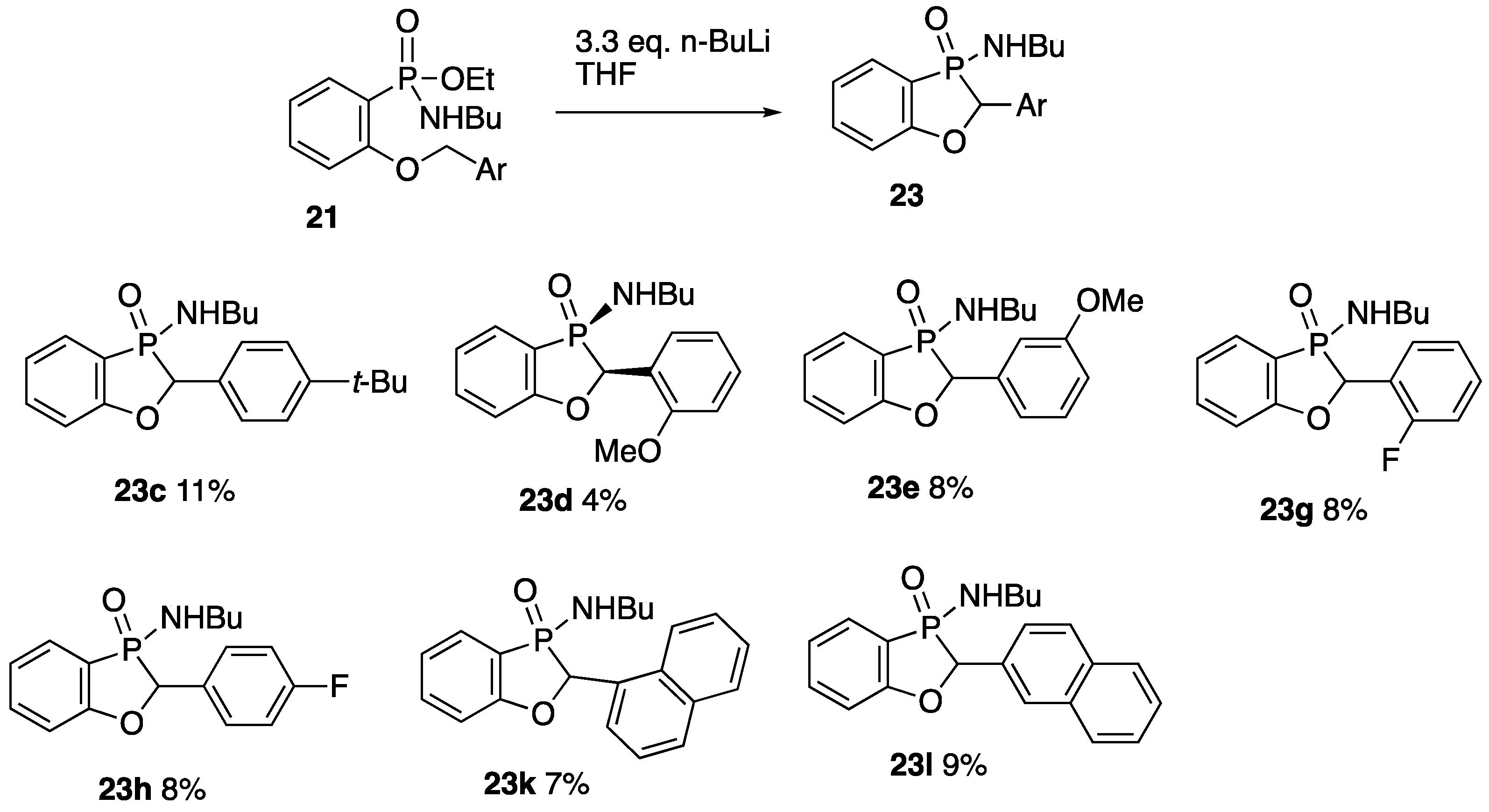
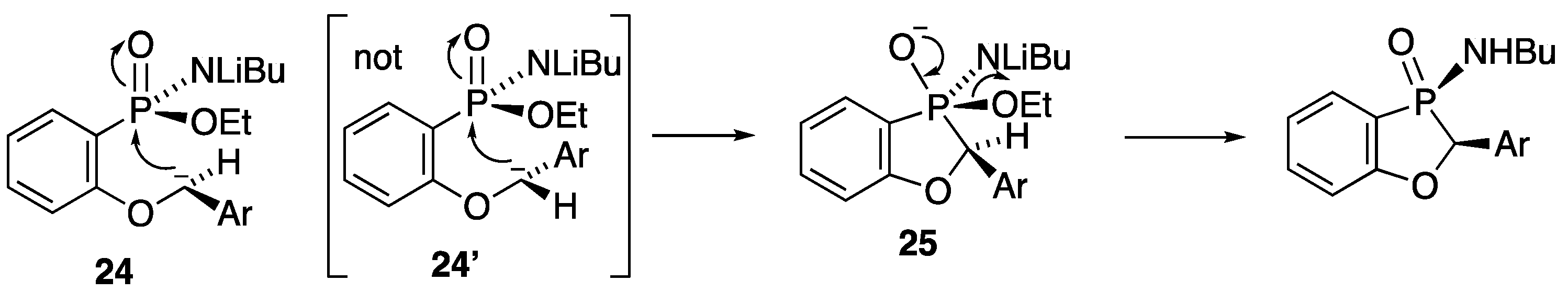
2.2.5. 3-Butylamino-2-phenyl-2H-benzo[d][1,3]oxaphosphole 3-Oxide 17
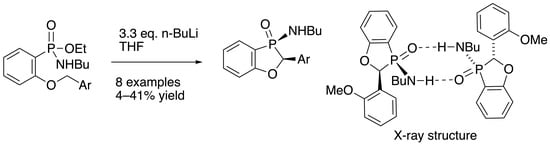
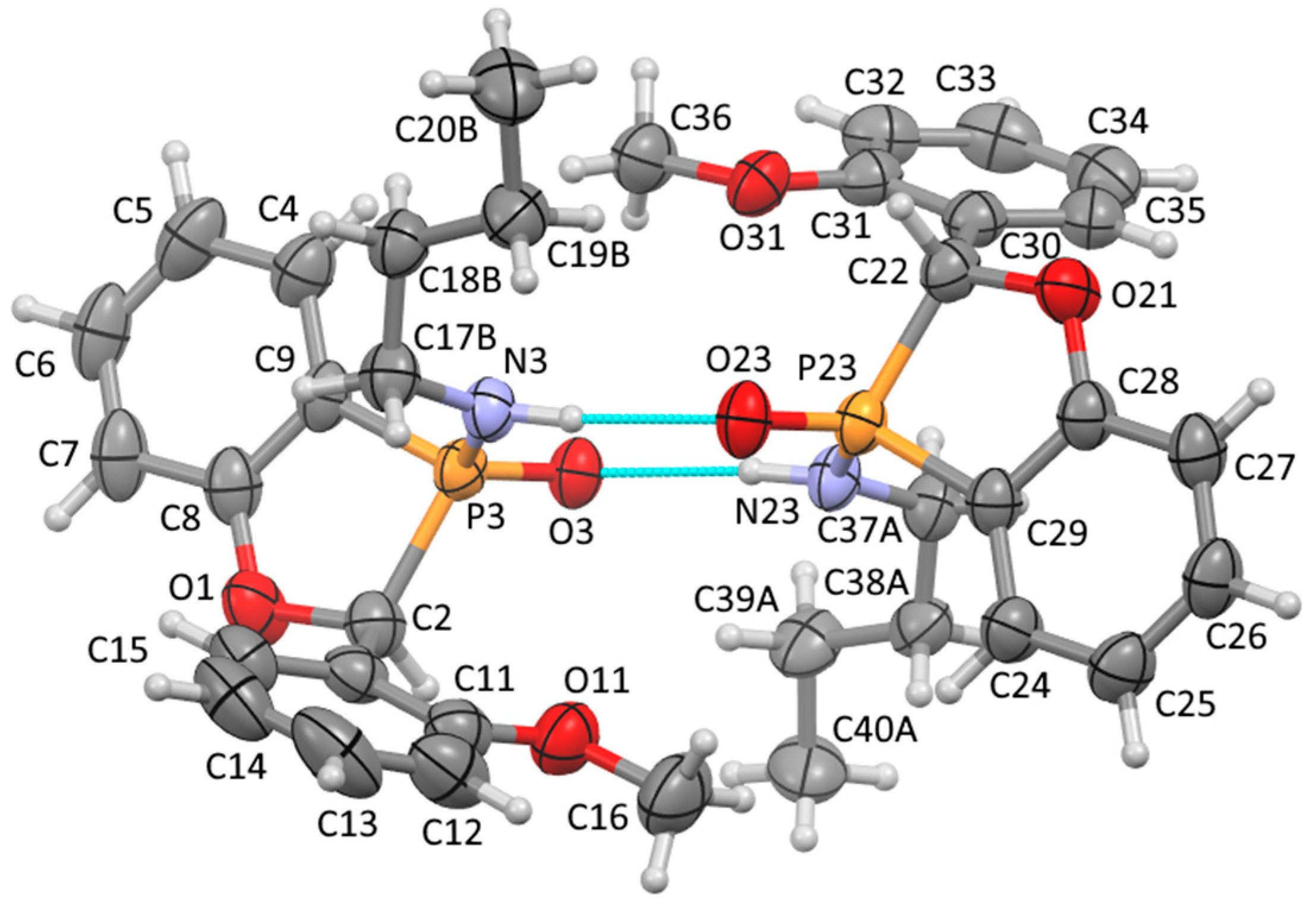
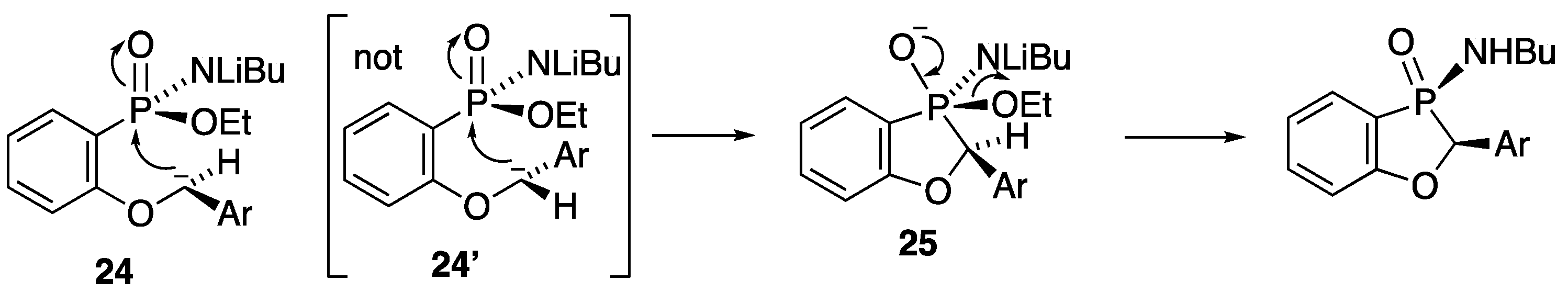
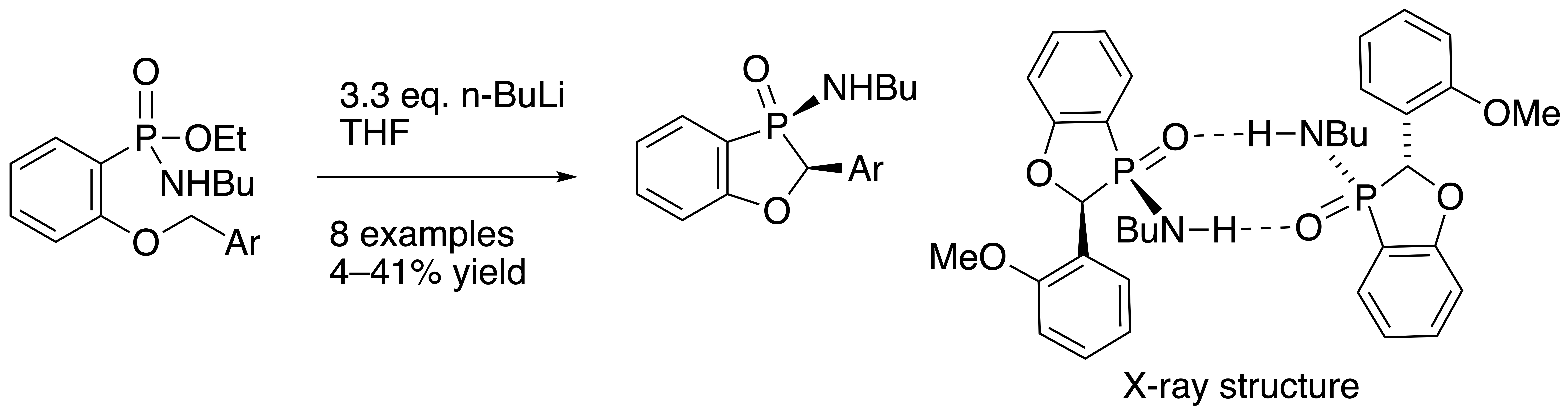
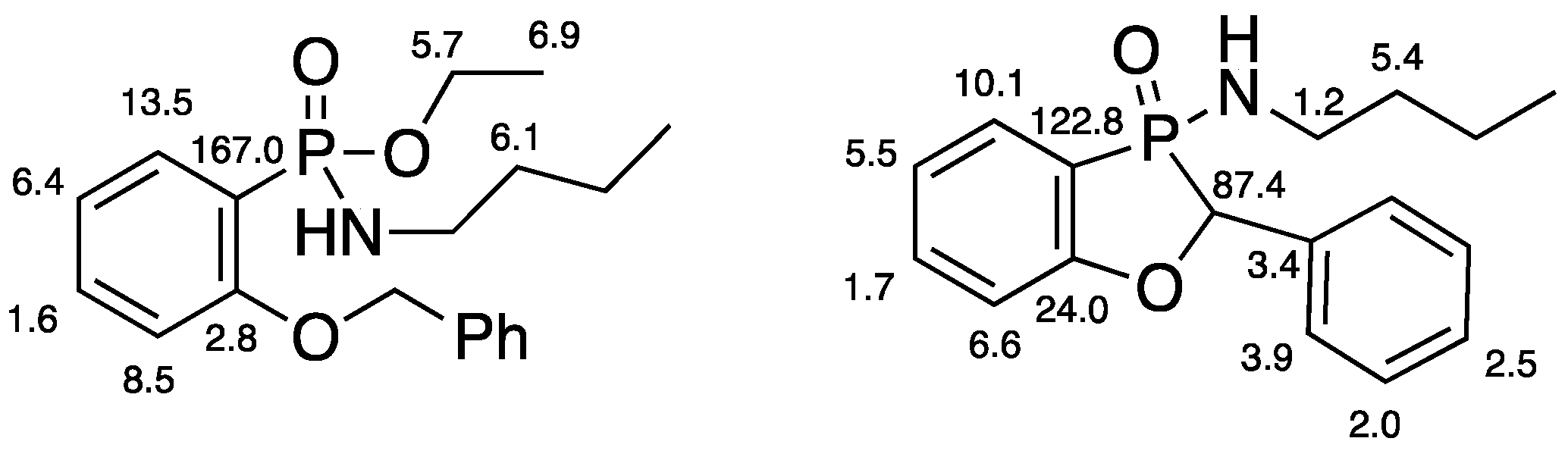
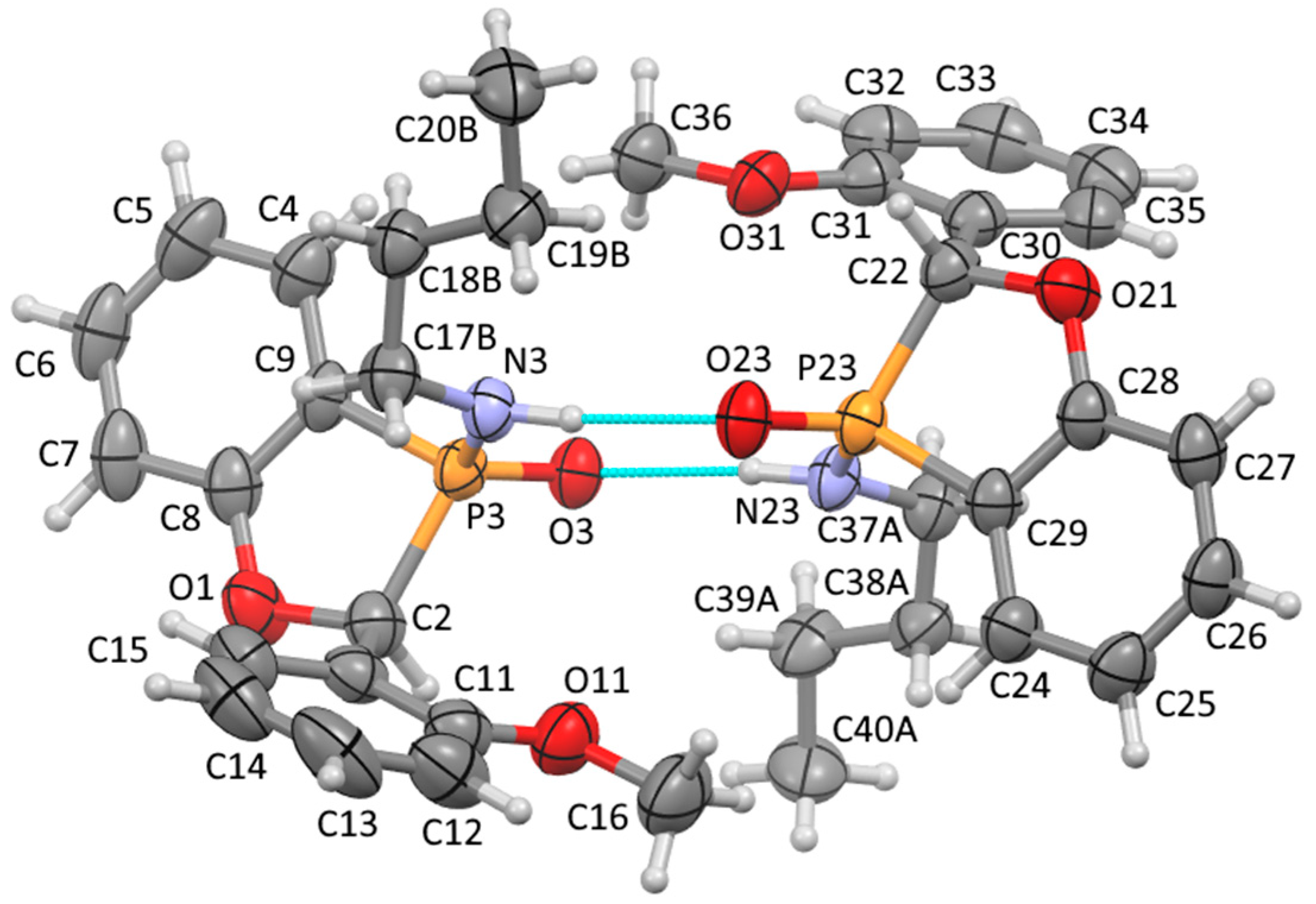
A solution of ethyl P-(2-benzyloxyphenyl)-N-butylphosphonamidate 16 (69.5 mg, 0.2 mmol) in dry THF (2 mL) was stirred at rt under N2 while n-butyllithium (0.37 mL, 0.66 mmol) was added by syringe. After 20 min, the mixture was added to saturated aqueous ammonium chloride (2 mL), and the mixture was extracted with Et2O (3 × 2 mL). Drying and evaporation of the combined extracts gave, after purification via preparative TLC, (EtOAc/hexane 1:1) 17 (24.5 mg, 41%) as a pale-yellow oil; νmax/cm−13184, 2957, 2930, 2872, 1599, 1578, 1449, 1204, 1155, 1126, 1094, 988, 916, 827, 756, 729, 696, and 515; 1H NMR (400 MHz): 7.66–7.52 (2H, m, ArH), 7.42–7.38 (4H, m, ArH), 7.37–7.32 (1H, m, ArH), 7.15–7.07 (2H, m, ArH), 5.57 (1H, d, J = 9.9 Hz, CHP), 2.49–2.37 (1H, m, NHCHH), 2.30–2.21 (1H, m, NHCHH), 1.04–0.94 (4H, m, NHCH2CH2CH2) and 0.70 (3H, t, J = 6.9 Hz, NHCH2CH2CH2CH3); 1H{31P} NMR (400 MHz): 5.57 (1H, s); 13C NMR (100 MHz): 164.6 (d, J = 24.0 Hz, ArC-O), 135.5 (d, J = 1.7 Hz, CH), 134.7 (d, J = 3.4 Hz, C), 129.0 (d, J = 5.5 Hz, CH), 128.8 (d, J = 2.0 Hz, 2CH), 128.0 (d, J = 2.5 Hz, CH), 124.9 (d, J = 3.9 Hz, 2CH), 122.4 (d, J = 10.1 Hz, CH), 114.3 (d, J = 6.6 Hz, CH), 113.9 (d, J = 122.8 Hz, ArC-P), 79.7 (d, J = 87.4 Hz, CHP), 40.2 (d, J = 1.2 Hz, NHCH2), 33.7 (d, J = 5.4 Hz, NHCH2CH2), 19.4 (NHCH2CH2CH2) and 13.5 (NHCH2CH2CH2CH3); 31P NMR (162 MHz): +44.6; HRMS (ESI+): found 302.1295. C17H22NOP (M + H) requires 302.1310.
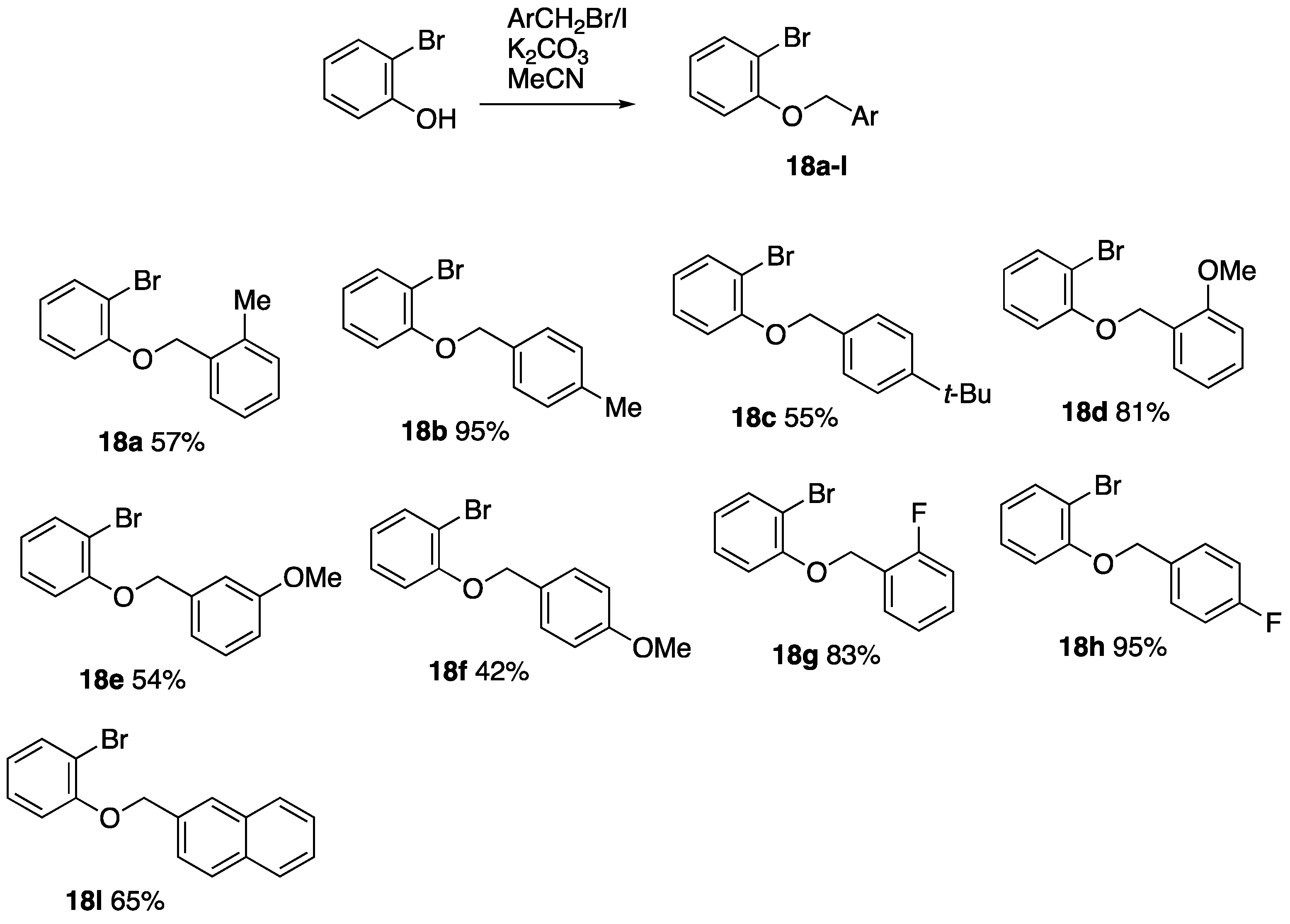
2.3. Synthesis of Substituted 2-Bromophenyl Benzyl Ethers 18
2.3.1. 2-Bromophenyl 2-Methylbenzyl Ether 18a
To a stirred solution of 2-bromophenol (2.9 mL, 4.33 g, 25.0 mmol) and K2CO3 (4.70 g, 34.0 mmol) in MeCN (60 mL) at rt, 2-methylbenzyl bromide (3.4 mL, 4.63 g, 25.0 mmol) was added, and the solution was stirred for 18 h. The reaction mixture was poured into H2O and extracted with EtOAc (2 × 50 mL), and the combined organic fractions were dried over MgSO4 and concentrated to afford, after recrystallisation from hexane, 18a (3.92 g, 57%) as colourless crystals, mp 42–44 °C; νmax/cm−1 3063, 3032, 2972, 2913, 2855, 1585, 1479, 1439, 1275, 1246, 1049, 1028, 737, and 665; 1H NMR (300 MHz): 7.56 (1H, dd, J = 7.9, 1.6 Hz, ArH), 7.53–7.45 (1H, m, ArH), 7.32–7.16 (4 H, m, ArH), 6.98 (1H, dd, J = 8.3, 1.3 Hz, ArH), 6.85 (1 H, td, J = 7.5, 1.2 Hz, ArH), 5.11 (2H, s, OCH2) and 2.40 (3H, s, ArCH3); 13C NMR (75 MHz): 155.1 (C), 136.3 (C), 134.3 (C), 133.5 (CH), 130.3 (CH), 128.4 (CH), 128.2 (2CH), 126.0 (CH), 122.1 (CH), 113.7 (CH), 112.5 (C), 69.4 (CH2) and 19.0 (CH3).
2.3.2. 2-Bromophenyl 4-Methylbenzyl Ether 18b
A solution of sodium iodide (5.34 g, 35.6 mmol) in dry acetone (25 mL) was stirred while 4-methylbenzyl chloride (5.00 g, 35.6 mmol) was added dropwise, and the mixture was stirred for 30 min. The mixture was added to H2O (50 mL) and extracted with Et2O (2 × 50 mL). Drying and evaporation of the extracts gave 4-methylbenzyl iodide 6.52 g, 79%) as a pale-yellow liquid.
To a stirred solution of 2-bromophenol (3.3 mL, 4.87 g, 28.0 mmol) and K2CO3 (7.16 g, 52.0 mmol) in MeCN (90 mL) at rt, 4-methylbenzyl iodide (6.52 g, 28.0 mmol) was added, and the solution was stirred for 18 h. The reaction mixture was poured into H2O and extracted with EtOAc (2 × 50 mL), and the combined organic fractions were dried over MgSO4 and concentrated to afford, after column chromatography (SiO2, hexane/EtOAc 9:1), 18b (6.30 g, 81%) as colourless crystals, mp 54–56 °C; νmax/cm−1 1479, 1454, 1441, 1292, 1285, 1275, 1246, 1213, 1180, 1158, 1055, 1028, 1020, 983, 949, 922, 808, 742, and 664; 1H NMR (300 MHz): 7.55 (1H, dd, J = 7.8, 1.8 Hz, ArH), 7.36 (2H, d, J = 8.1 Hz, ArH), 7.26–7.19 (3 H, m, ArH), 6.93 (1H, dd, J = 8.1, 1.5 Hz, ArH), 6.83 (1H, td, J = 8.1, 7.5, 1.5 Hz, ArH), 5.12 (2H, s, OCH2) and 2.36 (3H, s, CH3); 13C NMR (125 MHz): 155.0 (ArC-O), 137.6 (C), 133.5 (C), 133.4 (CH), 129.2 (2CH), 128.3 (CH), 127.1 (2CH), 122.0 (CH), 113.9 (CH), 112.5 (C-Br), 70.7 (OCH2) and 21.2 (CH3); HRMS (ESI+): found 299.0042. C14H1379BrNaO (M + Na) requires 299.0047.
2.3.3. 2-Bromophenyl 4-tert-Butylbenzyl Ether 18c
The same procedure as in 2.3.1 using 2-bromophenol (5.79 g, 33 mmol), K
2CO
3 (7.20 g, 52 mmol), and 4-
tert-butylbenzyl bromide [
17] (7.63 g, 33 mmol) gave
18c (6.20 g, 55%) as a brown oil; ν
max/cm
−1 2963, 1477, 1462, 1443, 1634, 1294, 1277, 1246, 1233, 1109, 1051, 1030, 1015, 837, 818, 745, 691, 656, and 638;
1H NMR (300 MHz): 7.75 (1H, dd
J = 8.1, 1.5 Hz, ArH), 7.41 (4H, s, ArH), 7.23 (1H, m, ArH), 6.95 (1H, dd
J = 8.4, 1.2 Hz, ArH), 6.83 (1H, ddd
J = 8.1, 7.8, 1.5 Hz, ArH), 5.11 (2H, s, OCH
2) and 1.32 (9H, m, 3 CH
3);
13C NMR (125 MHz): 155.1 (ArC-O), 150.9 (C), 133.5 (C), 133.4 (CH), 128.4 (CH), 126.8 (2CH), 125.5 (2CH), 122.0 (CH), 113.8 (CH), 112.5 (C-Br), 70.6 (CH
2O), 34.5 (
CMe
3) and 31.3 (3CH
3); HRMS (ESI
+) found 341.0512. C
17H
19BrNaO (M + Na) requires 341.0517.
2.3.4. 2-Bromophenyl 2-Methoxybenzyl Ether 18d
The same procedure as in 2.3.1 using 2-bromophenol (6.08 g, 35 mmol), K2CO3 (6.64 g, 48 mmol), and 2-methoxybenzyl bromide (6.64 g, 35 mmol) gave 18d (9.10 g, 81%) as a yellow oil; νmax/cm−1 1510, 1310, 1270, 1080, 1060, and 760; 1H NMR (300 MHz): 7.60 (1H, m, ArH), 7.55 (1H, dd J = 8.0, 1.2 Hz, ArH), 7.28 (1H, m, ArH), 7.21 (1H, m, ArH), 7.01–6.95 (2H, m, ArH), 6.87 (1H, m, ArH), 6.82 (1H, ddd J = 8.1, 7.8, 1.5 Hz, ArH), 5.19 (2H, s, OCH2) and 3.86 (3H, s, OCH3); 13C NMR (75 MHz): 156.3 (ArC-O), 155.1 (ArC-O), 133.3 (CH), 128.6 (CH), 128.4 (CH), 127.8 (CH), 124.9 (C), 121.8 (CH), 120.7 (CH), 113.7 (CH), 112.3 (C-Br), 109.9 (CH), 65.8 (OCH2) and 55.3 (OCH3); HRMS (ESI+) found 314.9990. C14H13BrNaO2 (M + Na) requires 314.9997.
2.3.5. 2-Bromophenyl 3-Methoxybenzyl Ether 18e
The same procedure as in 2.3.1 using 2-bromophenol (5.99 g, 34.6 mmol), K2CO3 (6.50 g, 47 mmol), and 3-methoxybenzyl bromide (6.98 g, 34.6 mmol) gave 18e (5.42 g, 54%) as a yellow oil; νmax/cm−1 3063, 3001, 2938, 2835, 1585, 1477, 1277, 1244, 1049, 1023, and 743; 1H NMR (300 MHz): 7.53 (1H, dd, J = 7.9, 1.5 Hz, ArH), 7.32–7.14 (2H, m, ArH), 7.09–6.96 (2H, m, ArH), 6.92–6.74 (3H, m, ArH), 5.09 (2H, s, OCH2) and 3.78 (3H, s, OCH3); 13C NMR (75 MHz): 159.7 (C), 154.8 (C), 138.1 (C), 133.3 (CH), 129.5 (CH), 128.3 (CH), 122.1 (CH), 118.9 (CH), 113.7 (CH), 113.4 (CH), 112.3 (C-Br), 112.2 (CH), 70.4 (CH2) and 55.1 (CH3); HRMS (ESI+) found 314.9989. C14H1379BrNaO2 (M + Na) requires 314.9997.
2.3.6. 2-Bromophenyl 4-Methoxybenzyl Ether 18f
The same procedure as in 2.3.1 using 2-bromophenol (6.28 g, 36.3 mmol), K2CO3 (6.82 g, 49.3 mmol), and 4-methoxybenzyl bromide (7.29 g, 36.3 mmol) gave 18f (4.41 g, 81%) as red crystals, mp 84–87 °C; νmax/cm−1 2999, 2909, 2835, 2361, 1607, 1584, 1510, 1474, 1240, 1171, 1028, 826, 808, and 750; 1H NMR (300 MHz): 7.54 (1H, dd, J = 7.9, 1.6 Hz, ArH), 7.38 (2H, d, J = 9.0 Hz, ArH), 7.26–7.16 (1H, m, ArH), 6.96–6.86 (3H, m, ArH), 6.82 (1H, td, J = 7.7, 1.4 Hz, ArH), 5.07 (2H, s, OCH2) and 3.80 (3H, s, OCH3); 13C NMR (75 MHz): 159.4 (C), 155.1 (C), 133.4 (CH), 128.7 (2CH), 128.5 (C), 128.3 (CH), 122.1 (CH), 114.1 (CH), 113.9 (2CH), 112.6 (C-Br), 70.7 (CH2) and 55.2 (CH3); HRMS (ESI–) found 291.0023. C14H1279BrO2 (M–H) requires 291.0021.
2.3.7. 2-Bromophenyl 2-Fluorobenzyl Ether 18g
The same procedure as in 2.3.2 using 2-bromophenol (3.58 g, 21 mmol), K2CO3 (3.90 g, 28 mmol), and 2-fluorobenzyl iodide (4.89 g, 21 mmol) gave 18g (4.83 g, 83%) as a yellow oil; νmax/cm−1 1585, 1493, 1476, 1456, 1443, 1285, 1273, 1246, 1231, 1053, 1030, 1007, 839, 743, and 665; 1H NMR (300 MHz): 7.64 (1H, m, ArH), 7.56 (1H, dd J = 8.1, 1.8 Hz, ArH), 7.35–7.16 (3H, m, ArH), 7.08 (1H, m, ArH), 6.97 (1H, dd J = 8.1, 1.5 Hz, ArH), 6.86 (1H, td J = 8.1, 1.5 Hz, ArH) and 5.22 (2H, s, OCH2); 13C NMR (100 MHz): 160.0 (d, J = 244.8 Hz, ArC-F), 154.7 (ArC-O), 133.4 (CH), 129.5 (d, J = 8.1 Hz, CH), 129.2 (d, J = 4.2 Hz, CH), 128.5 (CH), 124.3 (d, J = 3.5 Hz, CH), 123.7 (d, J = 13.7 Hz, C), 123.3 (CH), 115.1 (d, J = 20.7 Hz, CH), 113.7 (CH), 112.5 (C-Br) and 64.4 (d, J = 15.0 Hz, OCH2); 19F NMR (376 MHz): –118.9; HRMS (ESI+) found 302.9788. C13H10BrFNaO (M + Na) requires 302.9797.
2.3.8. 2-Bromophenyl 4-Fluorobenzyl Ether 18h
The same procedure as in 2.3.2 using 2-bromophenol (4.36 g, 25 mmol), K2CO3 (4.74 g, 34 mmol), and 4-fluorobenzyl iodide (5.95 g, 25 mmol) gave 18h (6.67 g, 95%) as a yellow oil; νmax/cm−1 1603, 1585, 1572, 1508, 1477, 1464, 1443, 1377, 1294, 1277, 1246, 1223, 1157, 1126, 1053, 1030, 1013, 978, 937, 860, 818, 745, 664, and 600; 1H NMR (300 MHz): 7.65 (1H, dd, J = 7.8, 1.8 Hz, ArH), 7.45 (2H, m, ArH), 7.23 (1H, m, ArH), 7.07 (2H, tt, J = 8.7, 2.1 Hz, ArH), 6.92 (1H, dd J = 8.1, 1.2 Hz, ArH), 6.85 (1H, td J = 7.8, 1.5 Hz, ArH) and 5.10 (2H, s, OCH2); 13C NMR (75 MHz): 162.4 (d, J = 246.4 Hz, ArC-F), 154.8 (ArC-O), 133.5 (CH), 132.2 (d, J = 3.2 Hz, C), 128.8 (d, J = 8.0 Hz, CH), 128.4 (CH), 122.3 (2CH), 115.5 (d, J = 21.6 Hz, 2CH), 113.9 (CH), 112.5 (C-Br) and 70.2 (OCH2); 19F NMR (376 MHz): –114.2; HRMS (ESI+) found 205.0602. C13H10NaO (M + Na − F − Br) requires 205.0629.
2.3.9. 2-Bromophenyl 2-Naphthylmethyl Ether 18l
The same procedure as in 2.3.1 using 2-bromophenol (4.33 g, 25.0 mmol), K2CO3 (4.70 g, 34.0 mmol), and 2-(bromomethyl)naphthalene (5.53 g, 25.0 mmol) gave, after recrystallisation from hexane, 18l (5.05 g, 65%) as light brown crystals, mp 75–77 °C; νmax/cm−1 3067, 3055, 2930, 2878, 1572, 1479, 1442, 1279, 1230, 1030, 1004, 814, and 737; 1H NMR (300 MHz): 7.98–7.75 (4H, m, ArH), 7.66–7.40 (4H, m, ArH), 7.24–7.13 (1H, m, ArH), 6.98 (1H, dd, J = 8.2, 1.3 Hz, ArH), 6.85 (1H, td, J 7.7, 1.4 Hz, ArH) and 5.32 (2 H, s, OCH2); 13C NMR (75 MHz):155.0 (C), 134.0 (C), 133.4 (CH), 133.2 (C), 133.0 (C), 128.4 (2CH), 128.0 (CH), 127.7 (CH), 126.2 (CH), 126.1 (CH), 126.0 (CH), 124.8 (CH), 122.2 (CH), 114.0 (CH), 112.5 (C-Br) and 70.9 (CH2); HRMS (ESI+) found 335.0039. C17H1379BrNaO (M + Na) requires 335.0047.
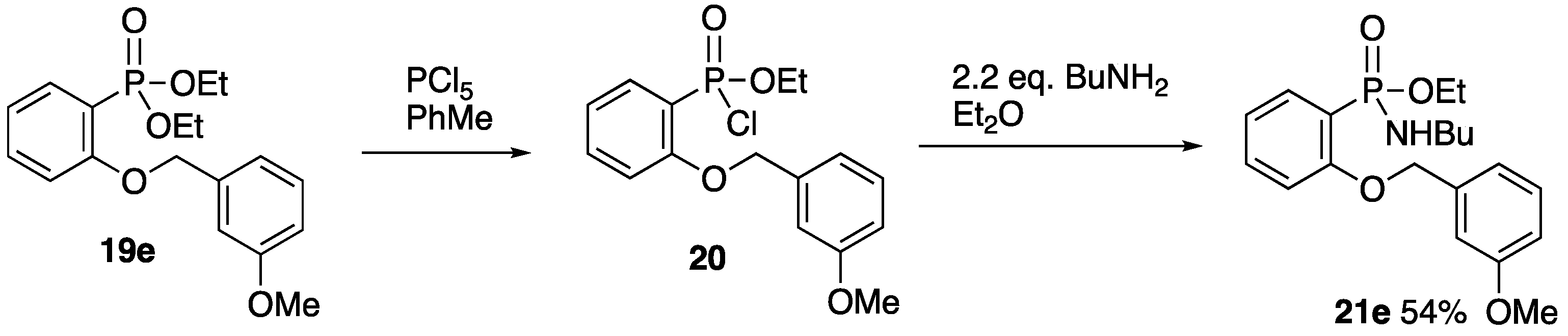
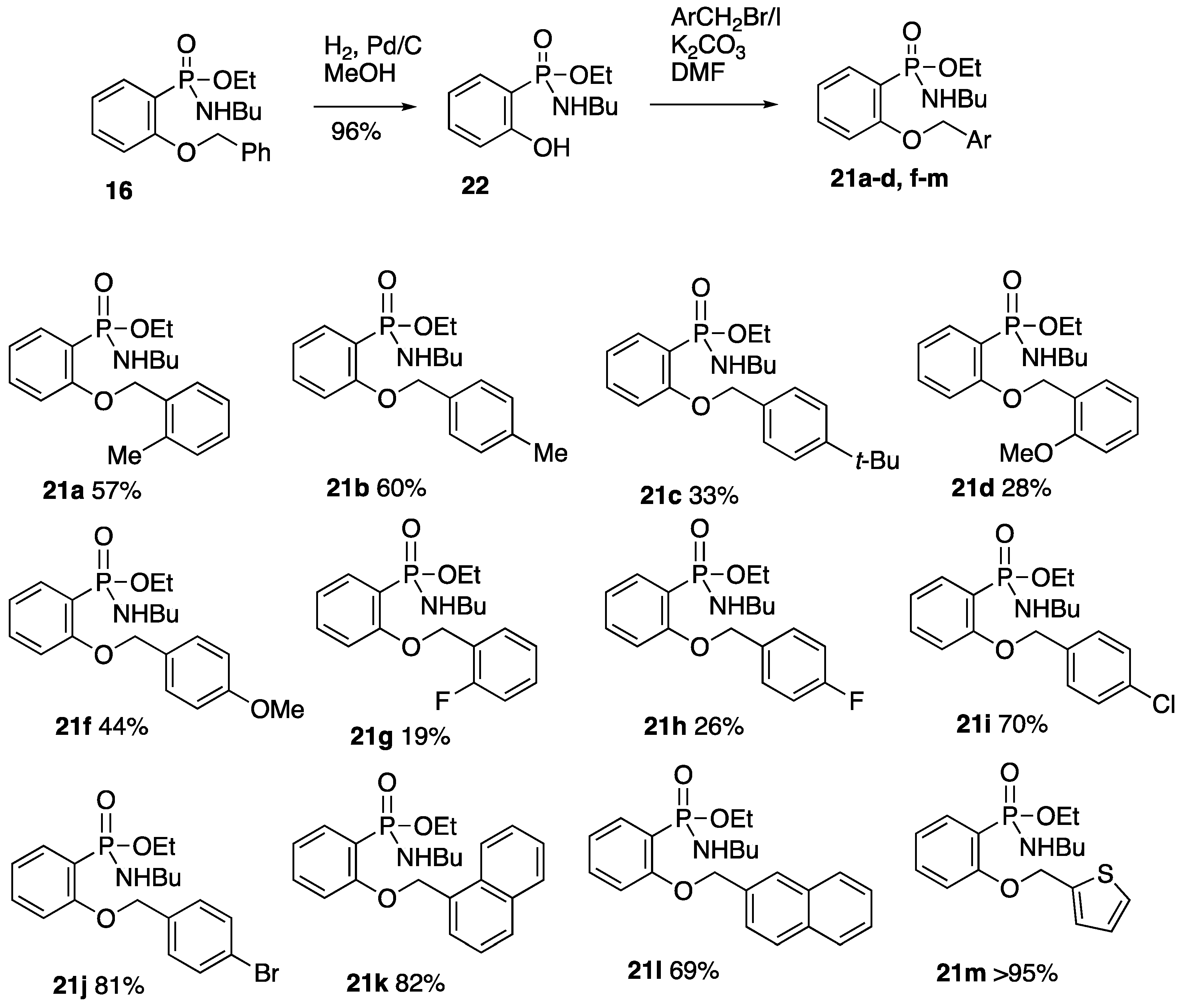
2.6. Formation and O-Benzylation of Ethyl N-Butyl-P-(2-hydroxyphenyl)phosphonamidate
2.6.1. Ethyl N-Butyl-P-(2-hydroxyphenyl)phosphonamidate 22
Using a literature procedure [
18], a solution of ethyl
P-(2-benzyloxyphenyl)-
N-butylphosphonamidate
16 (2.20 g, 6.3 mmol) in MeOH (40 mL) and 5% Pd/C (0.34 g) was stirred under a hydrogen atmosphere at rt for 2 h. The reaction mixture was filtered and concentrated to afford
22 (1.57 g. 96%);
1H NMR (300 MHz): 10.77 (1H, br s, OH), 7.45–7.28 (2H, m, ArH), 6.98–6.85 (2H, m, ArH), 4.10–3.85 (2H, m, OC
H2CH
3), 3.00–2.78 (3H, m, N
HC
H2), 1.50–1.20 (4 H, m, NHCH
2(C
H2)
2CH
3), 1.28 (3H, t,
J = 7.1 Hz, OCH
2C
H3) and 0.87 (3H, t,
J = 7.3 Hz, NH(CH
2)
3C
H3);
13C NMR (75 MHz): 162.2 (d,
J = 6.5 Hz, ArCOH), 134.4 (d,
J = 2.1 Hz, CH), 131.4 (d,
J = 7.1 Hz, CH), 119.2 (d,
J = 13.2 Hz, CH), 117.5 (d,
J = 10.9 Hz, CH), 111.1 (d,
J = 162.8 Hz, ArC-P), 61.6 (d,
J = 4.5 Hz, O
CH
2CH
3), 40.2 (NHCH
2), 33.8 (d,
J = 6.1 Hz, NHCH
2CH
2), 19.6 (NHCH
2CH
2CH
2), 16.2 (d,
J = 6.7 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
31P NMR (121 MHz): +28.2; HRMS (ESI
+): found 280.1073. C
12H
20NaNO
3P (M + Na) requires 280.1078.
2.6.2. Ethyl N-Butyl-P-(2-(2-methylbenzyloxy)phenyl)phosphonamidate 21a
To a stirred solution of ethyl N-butyl-P-(2-hydroxyphenyl)phosphonamidate 16 (0.25 g, 0.97 mmol) and K2CO3 (0.40 g, 2.92 mmol) in DMF (10 mL) at rt, 2-methylbenzyl bromide (0.13 mL, 0.18 g, 0.97 mmol) was added, and the solution was stirred for 18 h. The reaction mixture was poured into H2O (40 mL) and extracted with CH2Cl2 (20 mL) and EtOAc (3 × 20 mL). The combined organic fractions were washed with H2O (6 × 50 mL), dried over MgSO4, and concentrated to afford, after purification via column chromatography (SiO2, EtOAc/hexane 1:1), 21a (0.20 g, 57%) as a yellow oil; 1H NMR (300 MHz): 7.94 (1H, ddd, J = 14.3, 7.5, 1.8 Hz, ArH), 7.54–7.39 (2H, m, ArH), 7.33–7.20 (3H, m, ArH), 7.14–6.94 (2H, m, ArH), 5.14 and 5.08 (2H, AB pattern, J = 11.1 Hz, OCH2), 4.05–3.85 (2H, m, OCH2CH3), 3.00–2.80 (3H, m, NHCH2), 2.41 (3H, s, ArCH3), 1.28–1.05 (4H, m, NHCH2(CH2)2CH3), 1.22 (3H, t, J = 7.0 Hz, OCH2CH3) and 0.78 (3 H, t, J = 7.2 Hz, NHCH2(CH2)2CH3); 13C NMR (75 MHz): 159.2 (d, J = 2.8 Hz, ArCO), 136.4 (C), 134.5 (d, J = 6.5 Hz, CH), 134.0 (C), 133.4 (CH), 130.4 (CH), 128.5 (CH), 128.4 (CH), 126.1 (CH), 120.8 (d, J = 13.5 Hz, CH), 119.8 (d, J = 166.8 Hz, ArC-P), 111.5 (d, J = 8.6 Hz, CH), 68.7 (ArOCH2), 60.2 (d, J = 5.8 Hz, OCH2CH3), 40.2 (NHCH2), 34.0 (d, J = 6.2 Hz, NHCH2CH2), 19.6 (NHCH2CH2CH2), 18.8 (ArCH3), 16.3 (d, J = 6.9 Hz, OCH2CH3) and 13.6 (NHCH2CH2CH2CH3); 31P NMR (121 MHz): +21.3; HRMS (ESI+): found 384.1699. C20H28NaNO3P (M + Na) requires 384.1705.
2.6.3. Ethyl N-Butyl-P-(2-(4-methylbenzyloxy)phenyl)phosphonamidate 21b
To a stirred solution of NaI (0.16 g, 1.07 mmol) in acetone (1.5 mL), 4-methylbenzyl chloride (0.14 mL, 0.15 g, 1.07 mmol) was added, and the solution was stirred at rt until no further precipitation was observed. The solution was filtered and concentrated to afford 4-methylbenzyl iodide, which was used without further purification.
The 4-methylbenzyl iodide (1.07 mmol) was added to a stirred solution of ethyl N-butyl-P-(2-hydroxyphenyl)phosphonamidate 22 (0.25 g, 0.97 mmol) and K2CO3 (0.40 g, 2.92 mmol) in DMF (10 mL), and the mixture was stirred for 18 h at rt. The reaction mixture was poured into H2O (40 mL) and extracted with CH2Cl2 (20 mL) and EtOAc (3 × 20 mL). The combined organic fractions were washed with H2O (6 × 50 mL), dried over MgSO4, and concentrated to afford the product 20 (0.21 g, 60%) as an orange oil; 1H NMR (400 MHz): 7.93 (1H, ddd, J = 14.3, 7.5, 1.8 Hz, ArH), 7.50–7.42 (1H, m, ArH), 7.35 (2H, d, J = 8.0 Hz, ArH), 7.21 (2H, d, J = 8.0 Hz, ArH), 7.16–7.00 (1H, m, ArH), 7.00–6.95 (1H, m, ArH), 5.09 (2H, s, ArOCH2), 4.05–3.90 (2H, m, OCH2CH3), 3.01–2.82 (3H, m, NHCH2), 2.38 (3H, s, ArCH3), 1.34–1.21 (4H, m, NHCH2(CH2)2CH3), 1.25 (3H, t, J = 7.2 Hz, OCH2CH3) and 0.79 (3H, t, J = 7.2 Hz, NHCH2(CH2)2CH3); 13C NMR (100 MHz): 159.2 (d, J = 2.8 Hz, ArCO), 138.1 (C), 134.5 (d, J = 6.5 Hz, CH), 133.3 (CH), 133.2 (C), 129.4 (2CH), 127.5 (2CH), 120.8 (d, J = 13.1 Hz, CH), 120.0 (d, J = 170.2 Hz, ArC-P), 111.8 (d, J = 8.6 Hz, CH), 70.4 (ArOCH2), 60.3 (d, J = 5.7 Hz, OCH2CH3), 40.3 (NHCH2), 34.1 (d, J = 6.2 Hz, NHCH2CH2), 21.2 (ArCH3), 19.7 (NHCH2CH2CH2), 16.4 (d, J = 6.8 Hz, OCH2CH3) and 13.7 (NHCH2CH2CH2CH3); 31P NMR (121 MHz): +21.3.
2.6.4. Ethyl N-Butyl-P-(2-(4-tert-butylbenzyloxy)phenyl)phosphonamidate 21c
Using the method of
Section 2.6.2 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.60 g, 2.5 mmol), 4-
tert-butylbenzyl bromide [
17] (0.56 g, 2.5 mmol) and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave, after purification by column chromatography (SiO
2, hexane/EtOAc 1:1),
21c (0.33 g, 33%) as a pale-yellow oil; ν
max/cm
–1 2959, 2930, 2870, 2423, 1591, 1475, 1443, 1165, 1018, 955, 762, and 550;
1H NMR (400 MHz): 7.96–7.90 (1H, m, ArH), 7.48–7.42 (1H, m, ArH), 7.43 and 7.39 (2H, AB pattern,
J = 8.5 Hz, ArH), 7.06–6.98 (2H, m, ArH), 5.10 (2H, s, OC
H2Ar), 4.08–3.93 (2H, m, OC
H2CH
3), 2.95–2.86 (3H, m, N
HC
H2), 1.34 (9H, s, C(C
H3)
3), 1.32–1.20 (2H, m, NHCH
2C
H2), 1.25 (3H, t,
J = 7.2 Hz, OCH
2C
H3), 1.16 (2H, sextet,
J = 7.2 Hz, NHCH
2CH
2C
H2) and 0.79 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 159.2 (d,
J = 2.9 Hz, ArC-O), 151.4 (C), 134.5 (d,
J = 6.3 Hz, ArCH), 133.4 (d,
J = 1.6 Hz, ArCH), 133.2 (C), 127.3 (2CH), 125.6 (2CH), 120.8 (d,
J = 13.6 Hz, ArCH), 119.8 (d,
J = 165.5 Hz, ArC-P), 111.8 (d,
J = 8.6 Hz, ArCH), 70.3 (OCH
2Ar), 60.3 (d,
J = 5.7 Hz, O
CH
2CH
3), 40.3 (NHCH
2), 34.6 (
CMe
3), 34.1 (d,
J = 6.4 Hz, NHCH
2CH
2), 31.3 (C(
CH
3)
3), 19.6 (NHCH
2CH
2CH
2), 16.3 (d,
J = 6.6 Hz, OCH
2CH
3) and 13.7 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +21.3; HRMS (ESI
+): found 404.2336. C
23H
35NO
3P (M + H) requires 404.2355.
2.6.5. Ethyl N-Butyl-P-(2-(2-methoxybenzyloxy)phenyl)phosphonamidate 21d
Using the method of
Section 2.6.2 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.60 g, 2.5 mmol), 2-methoxybenzyl bromide (0.50 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave, after purification by column chromatography (SiO
2, hexane/EtOAc 1:1),
21d (0.24 g, 28%) as a pale-yellow oil; ν
max/cm
–1 2957, 2932, 1591, 1441, 1240, 1028, 953, 752, and 571;
1H NMR (400 MHz): 7.93 (1H, ddd,
J = 14.4, 7.6, 2.0 Hz, ArH), 7.51–7.29 (3H, m, ArH), 7.06–6.92 (4H, m, ArH), 5.21 and 5.15 (2H, AB pattern,
J = 12.0 Hz, ArOC
H2), 4.06–3.90 (2H, m, OC
H2CH
3), 3.87 (3H, s, OCH
3), 3.15–3.05 (1H, br m, NH), 3.01–2.90 (2H, m, NHC
H2), 1.36–1.29 (2H, m, NHCH
2C
H2), 1.23–1.16 (2H, m, NHCH
2CH
2C
H2), 1.21 (3H, t,
J = 7.2 Hz, OCH
2C
H3) and 0.80 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 159.2 (d,
J = 2.9 Hz, ArC-O), 156.8 (C), 134.3 (d,
J = 6.5 Hz, CH), 133.3 (d,
J = 2.1 Hz, CH), 129.3 (CH), 128.8 (CH), 124.4 (C), 120.6 (CH), 120.5 (d,
J = 11.2 Hz, CH), 119.6 (d,
J = 162.9 Hz, ArC-P), 111.6 (d,
J = 8.6 Hz, CH), 110.3 (CH), 65.5 (OCH
2Ar), 60.2 (d,
J = 5.6 Hz, O
CH
2CH
3), 55.3 (OCH
3), 40.3 (NHCH
2), 34.2 (d,
J = 6.8 Hz, NHCH
2CH
2), 19.6 (NHCH
2CH
2CH
2), 16.2 (d,
J = 6.8 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +21.5; HRMS (ESI
+): found 400.1637. C
20H
28NaNO
4P (M + Na) requires 400.1654.
2.6.6. Ethyl N-Butyl-P-(2-(4-methoxybenzyloxy)phenyl)phosphonamidate 21f
Using the method of
Section 2.6.2 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.60 g, 2.5 mmol), 4-methoxybenzyl bromide (0.50 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave, after purification by column chromatography (SiO
2, hexane/EtOAc 1:1),
21f (0.38 g, 44%) as a pale-yellow oil; ν
max/cm
–1 2957, 2932, 2872, 1591, 1514, 1236, 1030, 951, 820, 756, and 567;
1H NMR (400 MHz): 7.92 (1H, ddd,
J = 14.0, 7.2, 1.6 Hz, ArH), 7.48–7.43 (1H, m, ArH), 7.39 (2H, d,
J = 8.4 Hz, ArCH), 7.06–6.97 (2H, m, ArH), 6.93 (2H, d,
J = 8.4 Hz, ArCH), 5.06 (2H, s, ArOC
H2), 4.06–3.90 (2H, m, OC
H2CH
3), 3.83 (3H, s, OCH
3), 2.95–2.85 (3H, m, N
HC
H2), 1.34–1.20 (2H, m, NHCH
2C
H2), 1.25 (3H, t,
J = 7.2 Hz, OCH
2C
H3), 1.20–1.10 (2H, m, NHCH
2CH
2C
H2) and 0.79 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 159.6 (C), 159.2 (d,
J = 2.9 Hz, ArC-O), 134.4 (d,
J = 6.2 Hz, CH), 133.4 (d,
J = 2.0 Hz, CH), 129.2 (2CH), 128.2 (C), 120.8 (d,
J = 13.2 Hz, CH), 119.7 (d,
J = 166.1 Hz, ArC-P), 114.0 (2CH), 111.8 (d,
J = 8.5 Hz, CH), 70.2 (OCH
2Ar), 60.3 (d,
J = 5.7 Hz, O
CH
2CH
3), 55.3 (OCH
3), 40.3 (NHCH
2), 34.0 (d,
J = 6.4 Hz, NHCH
2CH
2), 19.6 (NHCH
2CH
2CH
2), 16.3 (d,
J = 6.8 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +21.3; HRMS (ESI
+): found 400.1631. C
20H
28NaNO
4P (M + Na) requires 400.1654.
2.6.7. Ethyl N-Butyl-P-(2-(2-fluorobenzyloxy)phenyl)phosphonamidate 21g
Using the method of
Section 2.6.3 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.64 g, 2.5 mmol), 2-fluorobenzyl iodide (0.59 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave, after purification by column chromatography (SiO
2, hexane/EtOAc 1:1),
21g (0.16 g, 19%) as a pale-yellow oil; ν
max/cm
–1 2949, 2932, 2370, 1620, 1491, 1231, 1105, 1036, 1007, 945, 833, 559 and 509;
1H NMR (400 MHz): 7.93 (1H, ddd,
J = 14.4, 7.6, 1.6 Hz, ArH), 7.57 (1H, td,
J = 5.8, 1.7 Hz, ArH), 7.50–7.45 (1H, m, ArH), 7.38–7.33 (1H, m, ArH), 7.19 (1H, td,
J = 7.6, 1.2 Hz, ArH), 7.14–7.09 (1H, m, ArH), 7.06 (1H, tdd,
J = 7.4, 2.8, 0.9 Hz, ArH), 7.04–7.01 (1H, m, ArH), 5.23 and 5.19 (2H, AB pattern,
J = 12.0 Hz, ArOC
H2), 4.06–3.90 (2H, m, OC
H2CH
3), 3.01–2.85 (3H, m, N
HC
H2), 1.36–1.29 (2H, m, NHCH
2C
H2), 1.23–1.14 (2H, m, NHCH
2CH
2C
H2), 1.23 (3H, t,
J = 7.2 Hz, OCH
2C
H3) and 0.81 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 160.5 (d,
J = 245.7 Hz, ArC-F), 158.9 (d,
J = 2.9 Hz, ArC-O), 134.6 (d,
J = 6.1 Hz, CH), 133.4 (d,
J = 2.1 Hz, CH), 130.2 (d,
J = 8.0 Hz, CH), 129.9 (d,
J = 3.8 Hz, CH), 124.4 (d,
J = 3.6 Hz, CH), 123.4 (d,
J = 14.2 Hz, C), 121.0 (d,
J = 13.2 Hz, CH), 119.9 (d,
J = 165.9 Hz, ArC-P), 115.5 (d,
J = 21.0 Hz, CH), 111.6 (d,
J = 8.5 Hz, CH), 64.2 (d,
J = 4.3 Hz, OCH
2Ar), 60.3 (d,
J = 5.6 Hz, O
CH
2CH
3), 40.4 (NHCH
2), 34.1 (d,
J = 6.1 Hz, NHCH
2CH
2), 19.7 (NHCH
2CH
2CH
2), 16.3 (d,
J = 6.7 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
19F NMR (376 MHz): –118.5;
31P NMR (162 MHz): +21.0; HRMS (ESI
+): found 388.1437. C
19H
25FNaNO
3P (M + Na) requires 388.1454.
2.6.8. Ethyl N-Butyl-P-(2-(4-fluorobenzyloxy)phenyl)phosphonamidate 21h
Using the method of
Section 2.6.3 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.64 g, 2.5 mmol), 4-fluorobenzyl iodide (0.59 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave, after purification by column chromatography (SiO
2, hexane/EtOAc 1:1),
21h (0.22 g, 26%) as a pale-yellow oil; ν
max/cm
–1 2959, 2930, 2872, 1603, 1512, 1443, 1223, 1157, 1030, 951, 756 and 563;
1H NMR (400 MHz): 7.92 (1H, ddd,
J = 14.4, 7.6, 2.0 Hz, ArH), 7.48–7.44 (3H, m, ArH), 7.10 (2H, t,
J = 8.8, Hz, ArH), 7.09–7.04 (1H, m, ArH), 6.97 (1H, dd,
J = 8.0, 6.0 Hz, ArH), 5.10 (2H, s, ArOC
H2), 4.07–3.94 (2H, m, OC
H2CH
3), 3.00–2.75 (3H, m, N
HC
H2), 1.33–1.23 (2H, m, NHCH
2C
H2), 1.26 (3H, t,
J = 7.2 Hz, OCH
2C
H3), 1.22–1.12 (2H, m, NHCH
2CH
2C
H2) and 0.80 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 162.6 (d,
J = 245.4 Hz, ArC-F), 159.0 (d,
J = 2.9 Hz, ArC-O), 134.4 (d,
J = 6.5 Hz, CH), 133.4 (d,
J = 2.1 Hz, CH), 132.0 (d,
J = 3.5 Hz, C), 129.3 (d,
J = 8.1 Hz, 2CH), 121.0 (d,
J = 13.4 Hz, CH), 119.9 (d,
J = 167.0 Hz, ArC-P), 115.6 (d,
J = 21.3 Hz, 2CH), 111.8 (d,
J = 8.4 Hz, CH), 69.7 (OCH
2Ar), 60.2 (d,
J = 5.7 Hz, O
CH
2CH
3), 40.3 (NHCH
2), 34.0 (d,
J = 5.9 Hz, NHCH
2CH
2), 19.6 (NHCH
2CH
2CH
2), 16.4 (d,
J = 6.9 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
19F NMR (376 MHz): –113.6;
31P NMR (162 MHz): +21.0; HRMS (ESI
+): found 388.1436. C
19H
25FNaNO
3P (M + Na) requires 388.1454.
2.6.9. Ethyl N-Butyl-P-(2-(4-chlorobenzyloxy)phenyl)phosphonamidate 21i
Using the method of
Section 2.6.2 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.60 g, 2.5 mmol), 4-chlorobenzyl bromide (0.40 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave, after purification by column chromatography (SiO
2, hexane/EtOAc 1:1),
21i (0.67 g, 70%) as colourless crystals, mp 75–77 °C; ν
max/cm
–1 2957, 2932, 2872, 1591, 1443, 1221, 1092, 1030, 955, 760, and 563;
1H NMR (400 MHz): 7.92 (1H, ddd,
J = 14.4, 7.6, 2.0 Hz, ArH), 7.48–7.40 (1H, m, ArH), 7.43 and 7.38 (4H, A
2B
2 pattern,
J = 8.8 Hz, ArCH), 7.06 (1H, tdd,
J = 7.6, 2.8, 0.8 Hz, ArCH), 6.98–6.94 (1H, m, ArH), 5.11 (2H, s, ArOC
H2), 4.08–3.94 (2H, m, OC
H2CH
3), 2.96–2.78 (3H, m, N
HC
H2), 1.34–1.20 (2H, m, NHCH
2C
H2), 1.28 (3H, t,
J = 7.2 Hz, OCH
2C
H3), 1.20–1.10 (2H, m, NHCH
2CH
2C
H2) and 0.80 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 158.9 (d,
J = 2.9 Hz, ArC-O), 134.7 (C), 134.5 (d,
J = 6.4 Hz, CH), 134.1 (C), 133.4 (d,
J = 2.0 Hz, CH), 128.9 (2CH), 128.7 (2CH), 121.1 (d,
J = 13.6 Hz, CH), 119.9 (d,
J = 166.6 Hz, ArC-P), 111.8 (d,
J = 8.6 Hz, CH), 69.6 (OCH
2Ar), 60.2 (d,
J = 5.6 Hz, O
CH
2CH
3), 40.4 (NHCH
2), 34.1 (d,
J = 6.2 Hz, NHCH
2CH
2), 19.6 (NHCH
2CH
2CH
2), 16.4 (d,
J = 6.9 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +20.9; HRMS (ESI
+): found 404.1138. C
19H
25ClNaNO
3P (M + Na) requires 404.1158.
2.6.10. Ethyl P-(2-(4-Bromobenzyloxy)phenyl)-N-butylphosphonamidate 21j
Using the method of
Section 2.6.2 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.60 g, 2.5 mmol), 4-methoxybenzyl bromide (0.62 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave
21j (0.86 g, 81%) as colourless crystals, mp 80–82 °C; ν
max/cm
–1 3177, 2953, 2928, 1591,1474, 1445, 1219,1030, 943, 760, 692, and 557;
1H NMR (400 MHz): 7.92 (1H, ddd,
J = 14.0, 7.6, 1.6 Hz, ArH), 7.54 (2H, d,
J = 8.4 Hz, ArCH), 7.49–7.43 (1H, m, ArH), 7.37 (2H, d,
J = 8.4 Hz, ArCH), 7.06 (1H, tdd,
J = 7.6, 2.8, 0.8 Hz, ArCH), 6.98–6.93 (1H, m, ArH), 5.10 (2H, s, ArOC
H2), 4.10–3.95 (2H, m, OC
H2CH
3), 2.95–2.80 (3H, m, N
HC
H2), 1.34–1.28 (2H, m, NHCH
2C
H2), 1.28 (3H, t,
J = 7.2 Hz, OCH
2C
H3), 1.22–1.12 (2H, m, NHCH
2CH
2C
H2) and 0.80 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 158.9 (d,
J = 2.8 Hz, ArC-O), 135.3 (C), 134.5 (d,
J = 6.3 Hz, CH), 133.4 (d,
J = 1.8 Hz, CH), 131.9 (2CH), 129.0 (2CH), 122.2 (C-Br), 121.1 (d,
J = 13.2 Hz, CH), 120.1 (d,
J = 166.0 Hz, ArC-P), 111.8 (d,
J = 8.6 Hz, CH), 69.7 (OCH
2Ar), 60.3 (d,
J = 5.5 Hz, O
CH
2CH
3), 40.4 (NHCH
2), 34.1 (d,
J = 6.3 Hz, NHCH
2CH
2), 19.7 (NHCH
2CH
2CH
2), 16.4 (d,
J = 7.0 Hz, OCH
2CH
3) and 13.7 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +20.9; HRMS (ESI
+): found 426.0814. C
19H
2679BrNO
3P (M + H) requires 426.0834.
2.6.11. Ethyl N-Butyl-P-(2-(1-naphthylmethoxy)phenyl)phosphonamidate 21k
Using the method of
Section 2.6.2 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.60 g, 2.5 mmol), 1-bromomethylnaphthalene (0.55 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave
21k (0.81 g, 82%) as a pale-yellow oil; ν
max/cm
–1 3393, 2957, 2930, 2870, 1591, 1472, 1441, 1227, 1084, 1032, 997, 951, 758, and 542;
1H NMR (400 MHz): 8.13–8.03 (1H, m, ArH), 7.95 (1H, ddd,
J = 14.4, 7.6, 2.0 Hz, ArH), 7.94–7.78 (2H, m, ArH), 7.63–7.41 (5H, m, ArH), 7.15 (1H, dd,
J = 8.0, 6.0 Hz, ArCH), 7.08 (1H, tdd,
J = 7.6, 3.2, 0.8 Hz, ArCH), 5.57 and 5.51 (2H, AB pattern,
J = 11.0 Hz, ArOC
H2), 3.85–3.75 (2H, m, OC
H2CH
3), 2.82–2.64 (3H, m, N
HC
H2), 1.10–1.00 (2H, m, NHCH
2C
H2), 1.08 (3H, t,
J = 7.2 Hz, OCH
2C
H3), 1.00–0.90 (2H, m, NHCH
2CH
2C
H2) and 0.69 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 159.1 (d,
J = 2.9 Hz, ArC-O), 134.6 (d,
J = 6.5 Hz, CH), 133.8 (C), 133.4 (d,
J = 2.1 Hz, CH), 131.5 (C), 131.3 (C), 129.4 (CH), 128.8 (CH), 126.9 (CH), 126.7 (CH), 126.0 (CH), 125.3 (CH), 123.3 (CH), 120.9 (d,
J = 13.2 Hz, CH), 120.0 (d,
J = 168.0 Hz, ArC-P), 111.4 (d,
J = 8.3 Hz, CH), 68.8 (OCH
2Ar), 60.1 (d,
J = 5.8 Hz, O
CH
2CH
3), 40.2 (NHCH
2), 33.9 (d,
J = 5.9 Hz, NHCH
2CH
2), 19.5 (NHCH
2CH
2CH
2), 16.2 (d,
J = 6.9 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +20.9; HRMS (ESI
+): found 420.1688. C
23H
28NaNO
3P (M + Na) requires 420.1705.
2.6.12. Ethyl N-Butyl-P-(2-(2-naphthylmethoxy)phenyl)phosphonamidate 21l
Using the method of
Section 2.6.2 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.60 g, 2.5 mmol), 2-bromomethylnaphthalene (0.55 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave
21l (0.81 g, 82%) as a pale-yellow oil; ν
max/cm
–1 2957, 2930, 2870, 1591, 1441, 1227, 1086, 1030, 951, 813, 756, 565, and 475;
1H NMR (400 MHz): 7.96 (1H, ddd,
J = 15.2, 7.2, 1.6 Hz, ArH), 7.93–7.72 (4H, m, ArH), 7.60–7.44 (4H, m, ArH), 7.08–7.01 (2H, m, ArCH), 5.31 and 5.29 (2H, AB pattern,
J = 11.4 Hz, ArOC
H2), 4.10–3.93 (2H, m, OC
H2CH
3), 3.00–2.82 (3H, m, N
HC
H2), 1.30–1.20 (2H, m, NHCH
2C
H2), 1.28 (3H, t,
J = 7.2 Hz, OCH
2C
H3), 1.12–1.02 (2H, m, NHCH
2CH
2C
H2) and 0.71 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 159.1 (d,
J = 2.9 Hz, ArC-O), 134.6 (d,
J = 6.5 Hz, CH), 133.8 (C), 133.4 (d,
J = 2.1 Hz, CH), 131.5 (C), 131.3 (C), 129.4 (CH), 128.9 (CH), 126.9 (CH), 126.7 (CH), 126.0 (CH), 125.3 (CH), 123.3 (CH), 120.9 (d,
J = 13.3 Hz, CH), 120.0 (d,
J = 165.7 Hz, ArC-P), 111.4 (d,
J = 8.5 Hz, CH), 68.8 (OCH
2Ar), 60.1 (d,
J = 5.8 Hz, O
CH
2CH
3), 40.2 (NHCH
2), 33.9 (d,
J = 5.9 Hz, NHCH
2CH
2), 19.5 (NHCH
2CH
2CH
2), 16.2 (d,
J = 6.9 Hz, OCH
2CH
3) and 13.6 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +21.2; HRMS (ESI
+): found 420.1686. C
23H
28NaNO
3P (M + Na) requires 420.1705.
2.6.13. Ethyl N-Butyl-P-(2-(2-thienylmethoxy)phenyl)phosphonamidate 21m
Using the method of
Section 2.6.2 with ethyl
N-butyl-
P-(2-hydroxyphenyl)phosphonamidate
22 (0.60 g, 2.5 mmol), 2-bromomethylthiophene (0.44 g, 2.5 mmol), and K
2CO
3 (1.04 g, 7.5 mmol) in DMF (4 mL) gave
21m (0.88 g, >95%) as a dark-orange oil; ν
max/cm
–1 3401, 2957, 2930, 2872, 1591, 1441, 1232, 1032, 953, 700, 577, and 540;
1H NMR (400 MHz): 7.94 (1H, ddd,
J = 14.0, 7.2, 1.6 Hz, ArH), 7.50–7.45 (1H, m, ArH), 7.36 (1H, dd,
J = 4.8, 1.2 Hz, ArCH), 7.14 (1H, dd,
J = 3.6, 1.2 Hz, ArCH), 7.07 (1H, tdd,
J = 7.6, 3.2, 0.6, ArH), 7.04–6.99 (2H, m, ArCH), 5.31 and 5.28 (2H, AB pattern,
J = 11.8 Hz, ArOC
H2), 4.07–3.90 (2H, m, OC
H2CH
3), 3.00–2.85 (3H, m, N
HC
H2), 1.35–1.25 (2H, m, NHCH
2C
H2), 1.24 (3H, t,
J = 7.2 Hz, OCH
2C
H3), 1.25–1.15 (2H, m, NHCH
2CH
2C
H2) and 0.81 (3H, t,
J = 7.2 Hz, NHCH
2CH
2CH
2C
H3);
13C NMR (100 MHz): 158.6 (d,
J = 2.9 Hz, ArC-O), 138.2 (C), 134.6 (d,
J = 6.4 Hz, CH), 133.3 (d,
J = 2.1 Hz, CH), 127.1 (CH), 126.9 (CH), 126.5 (CH), 121.2 (d,
J = 13.2 Hz, CH), 120.2 (d,
J = 165.4 Hz, ArC-P), 111.7 (d,
J = 8.6 Hz, CH), 65.3 (OCH
2Ar), 60.3 (d,
J = 5.8 Hz, O
CH
2CH
3), 40.4 (NHCH
2), 34.1 (d,
J = 6.0 Hz, NHCH
2CH
2), 19.7 (NHCH
2CH
2CH
2), 16.3 (d,
J = 7.0 Hz, OCH
2CH
3) and 13.7 (NHCH
2CH
2CH
2CH
3);
31P NMR (162 MHz): +20.8; HRMS (ESI
+): found 376.1097. C
17H
24NaNO
3PS (M + Na) requires 376.1112.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}