
Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. General Remarks
2.2. General Procedure for the Enantioselective Organocatalyzed Vinylogous Addition of TMSOF to Benzaldehyde
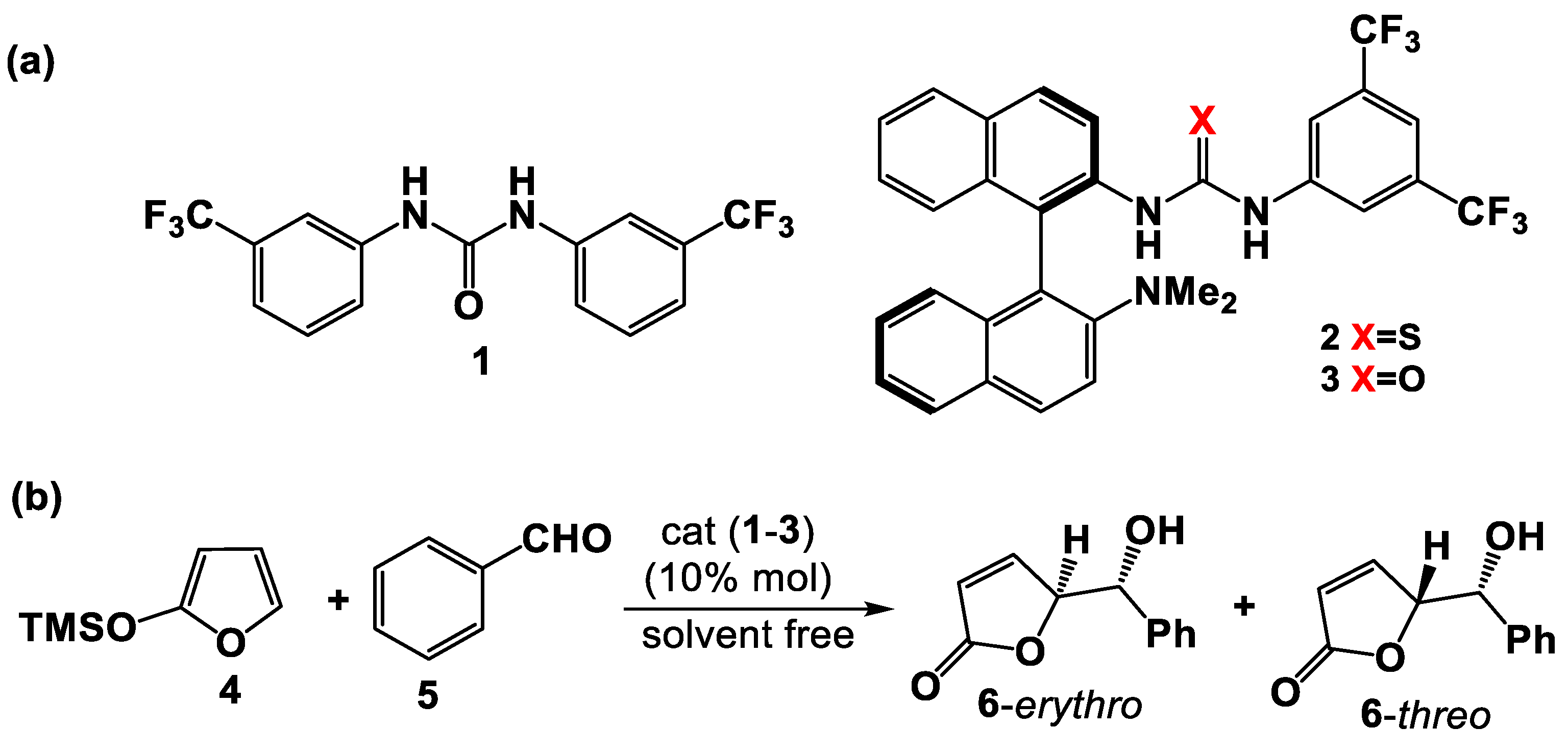
3. Results and Discussion
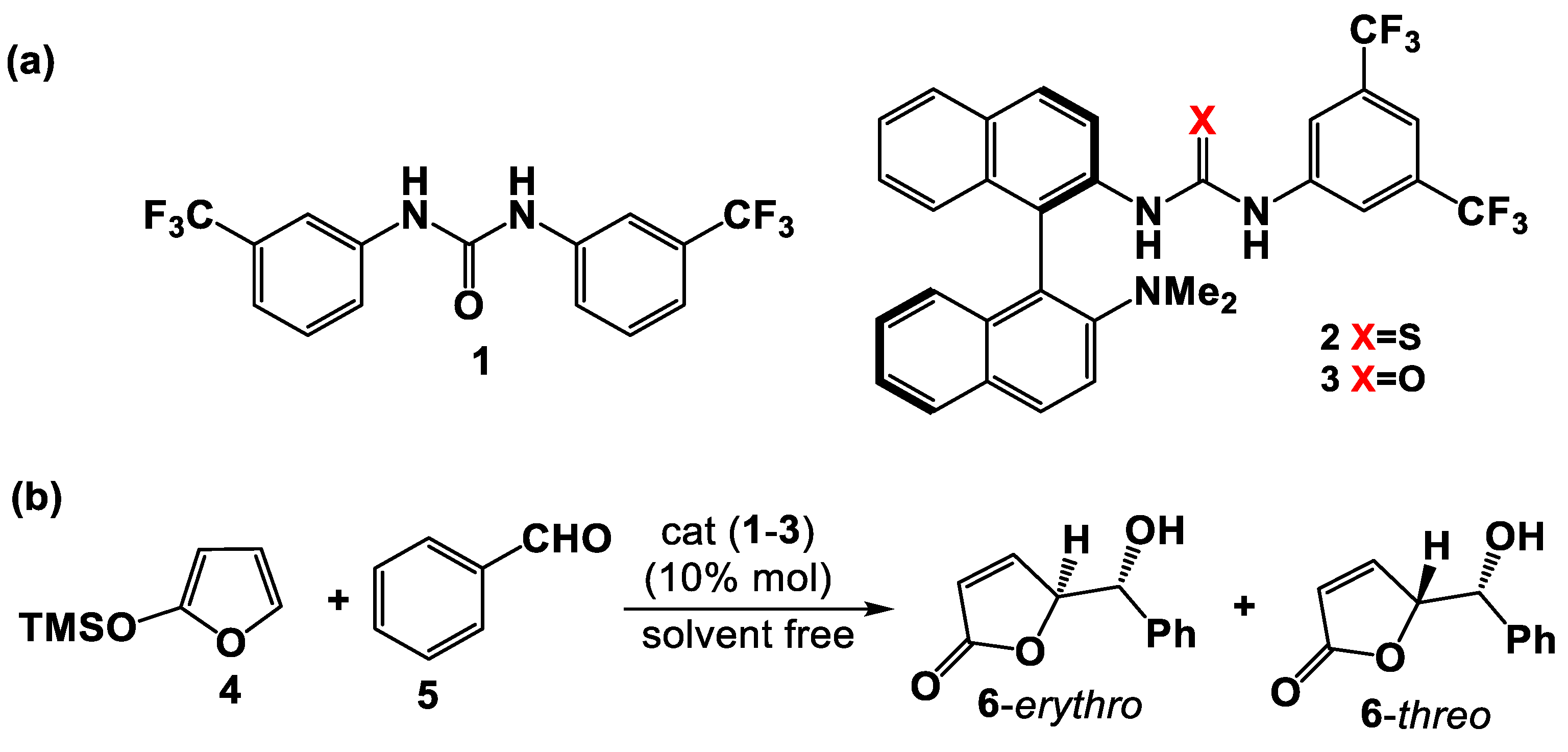
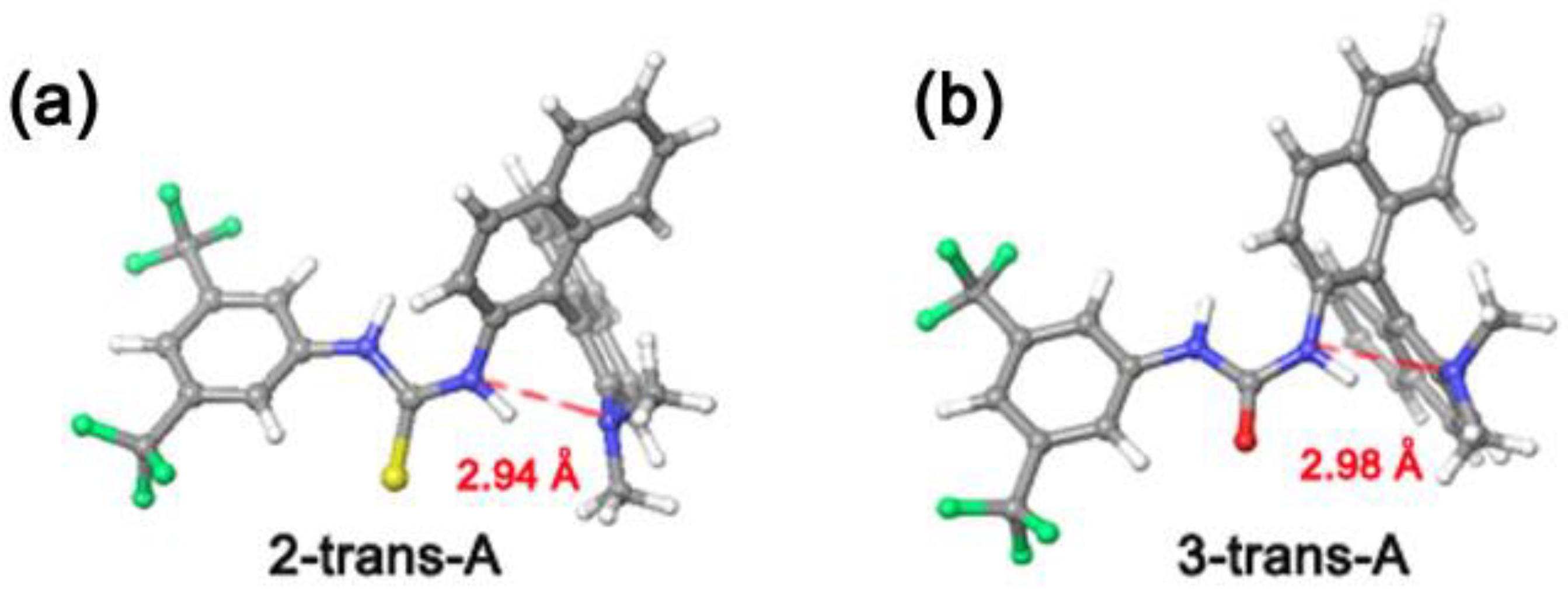
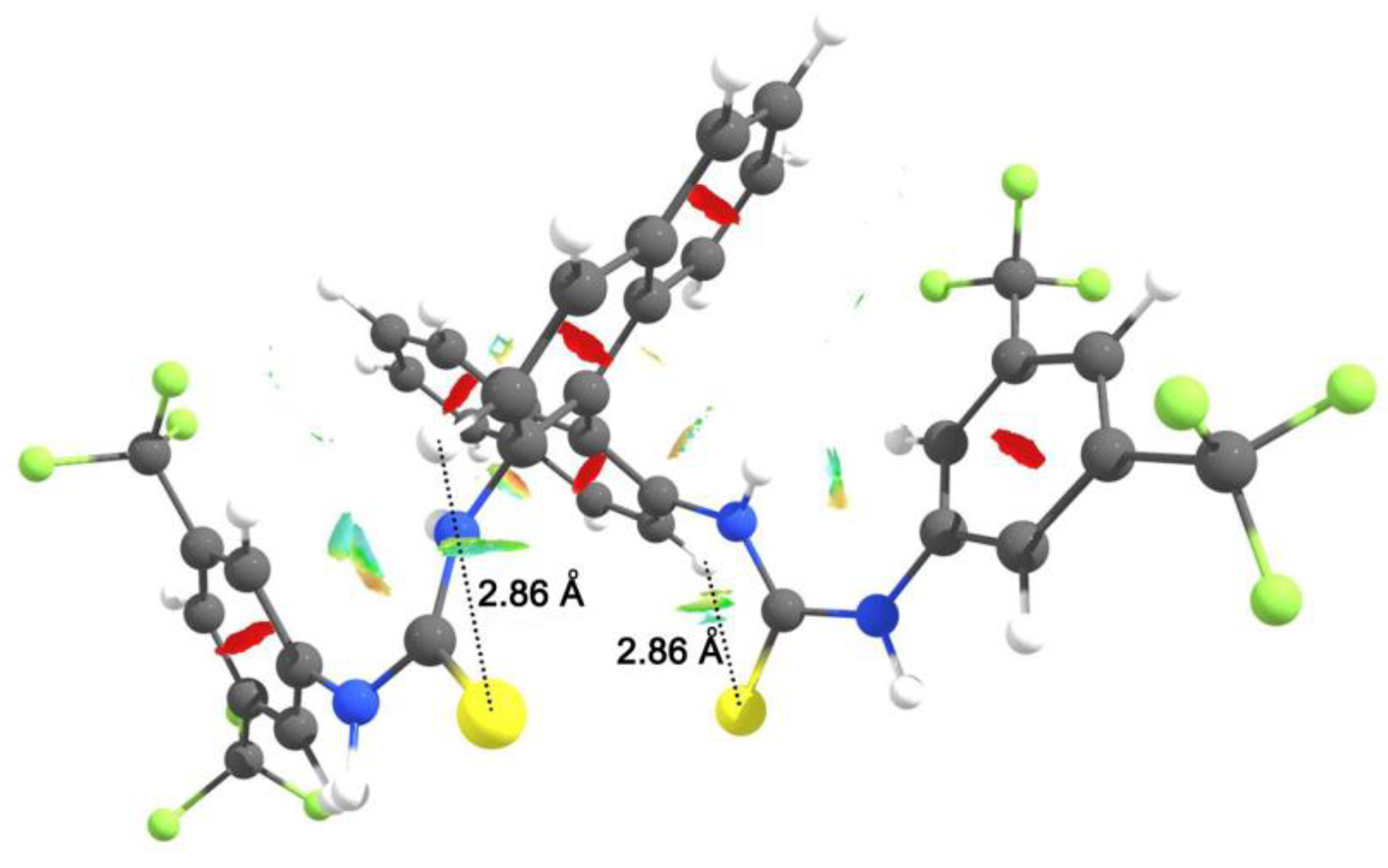
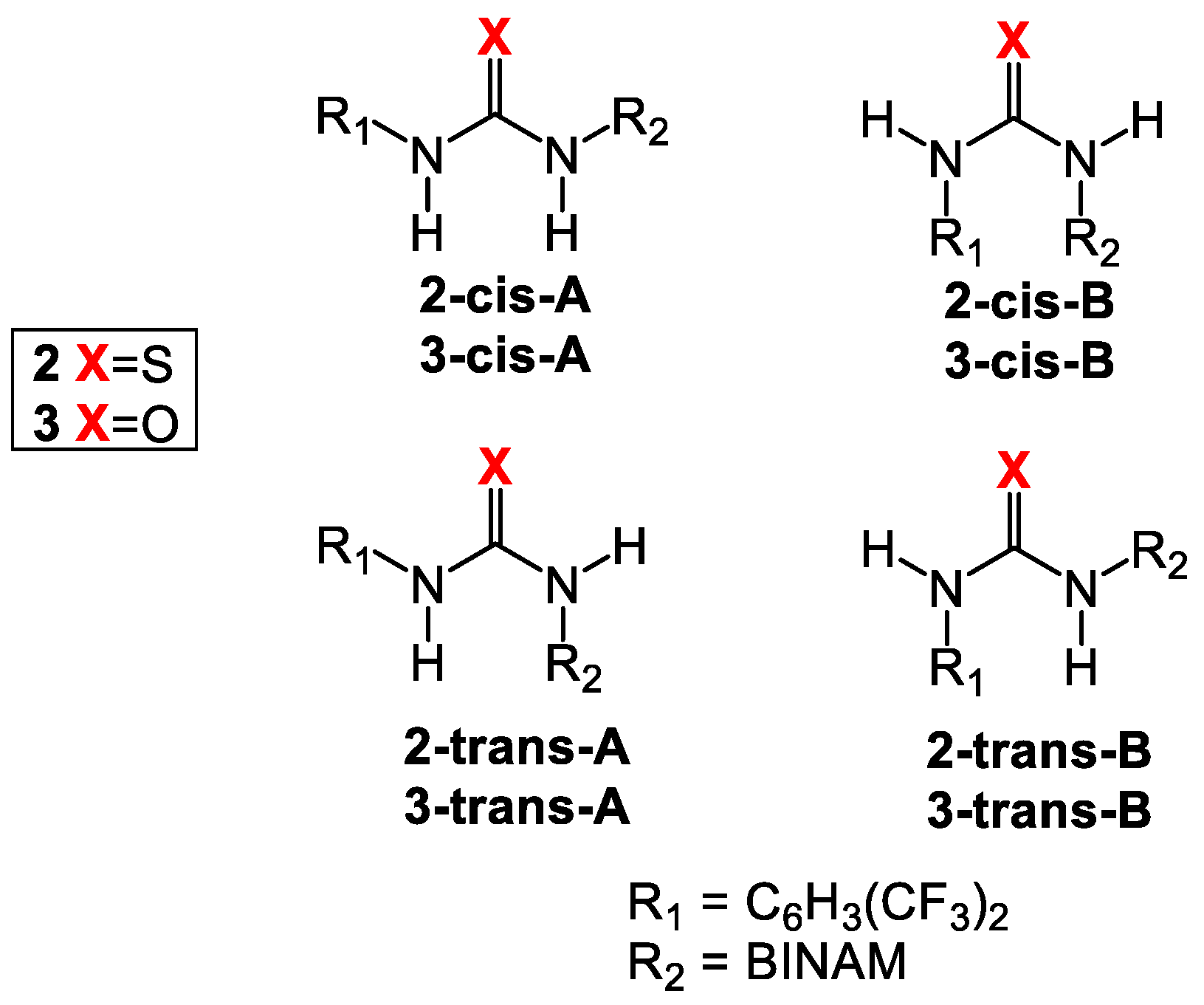
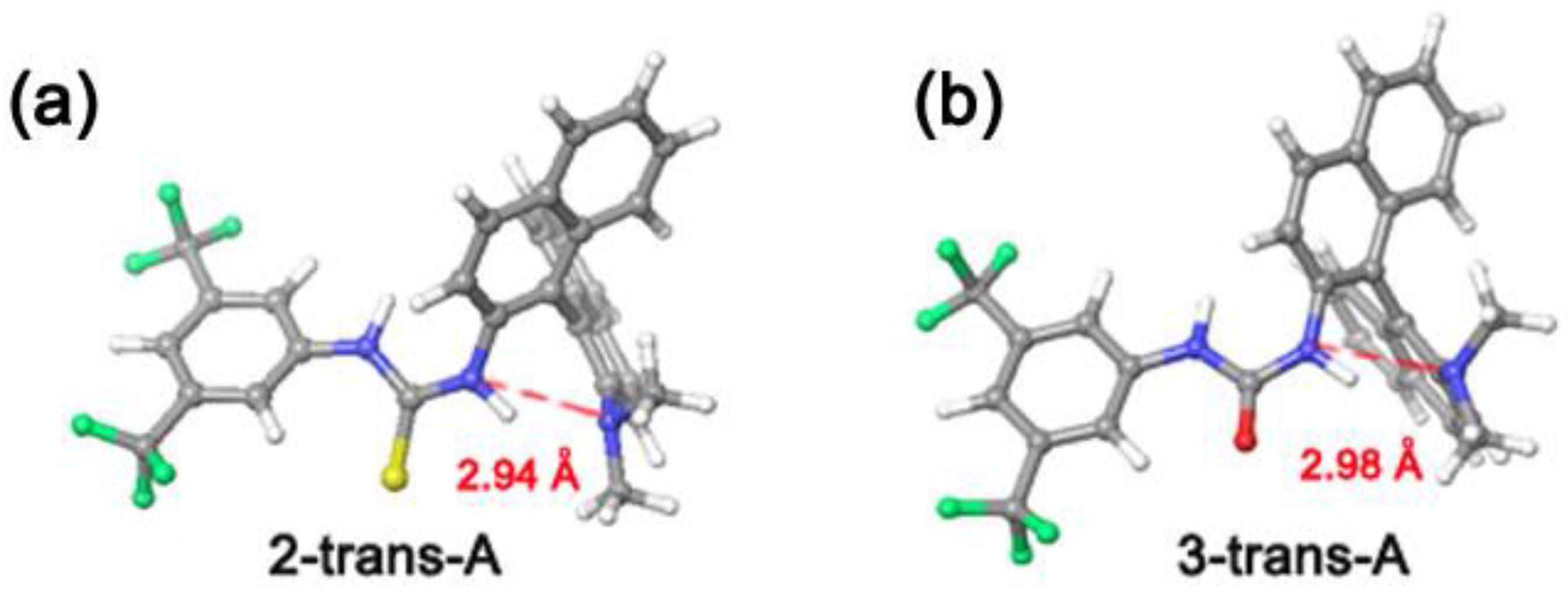
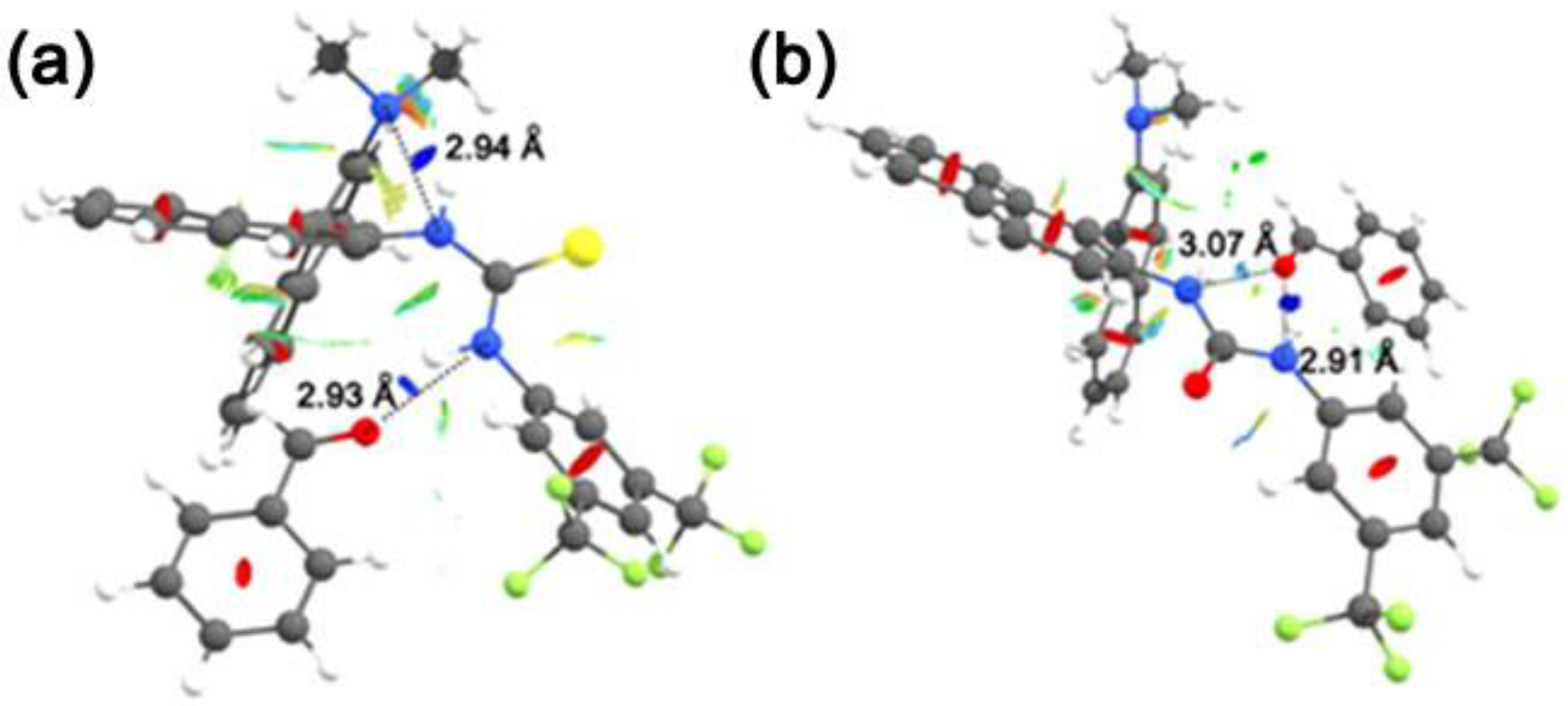
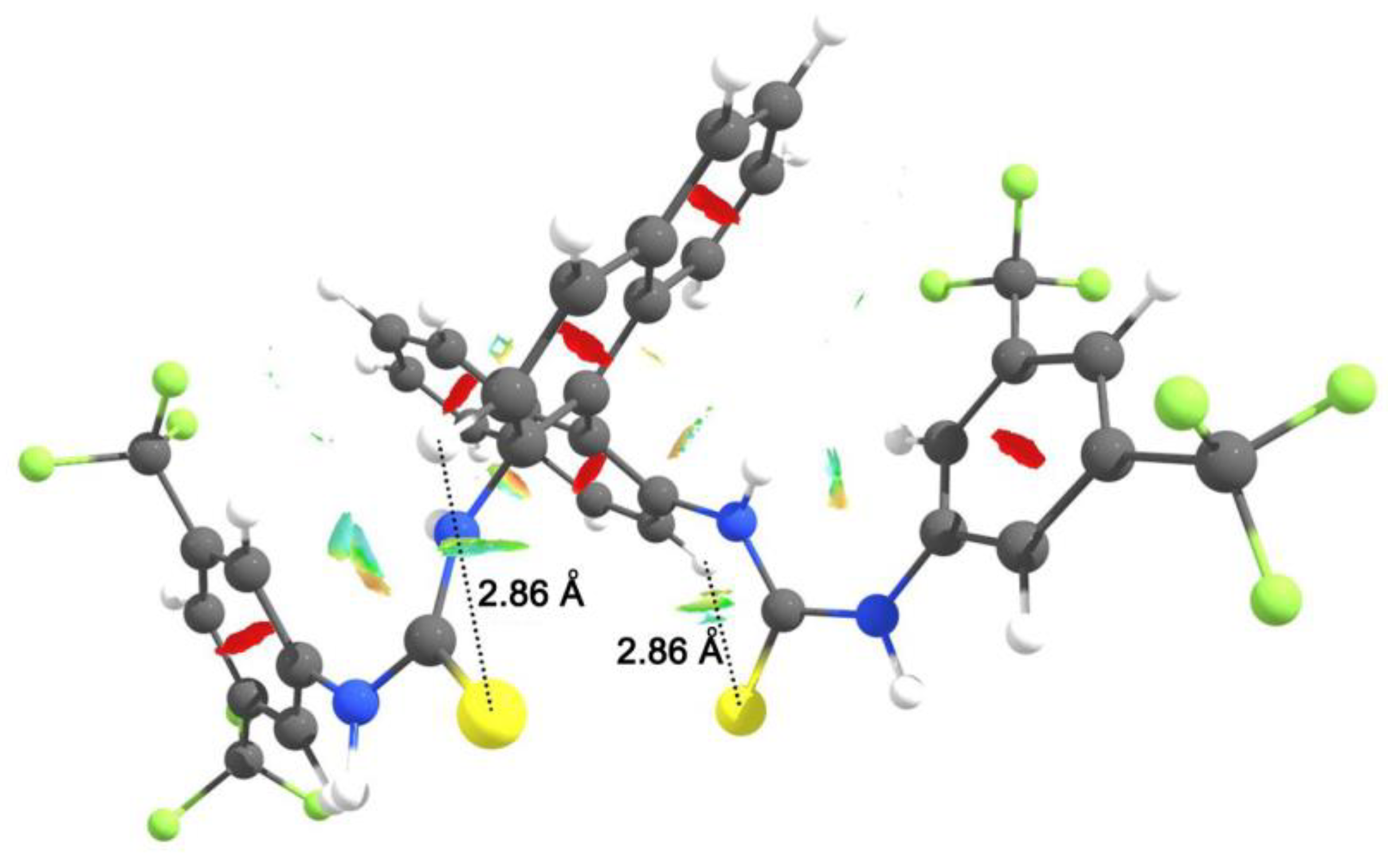
3.1. Mono(thio)urea Catalysts 2 and 3
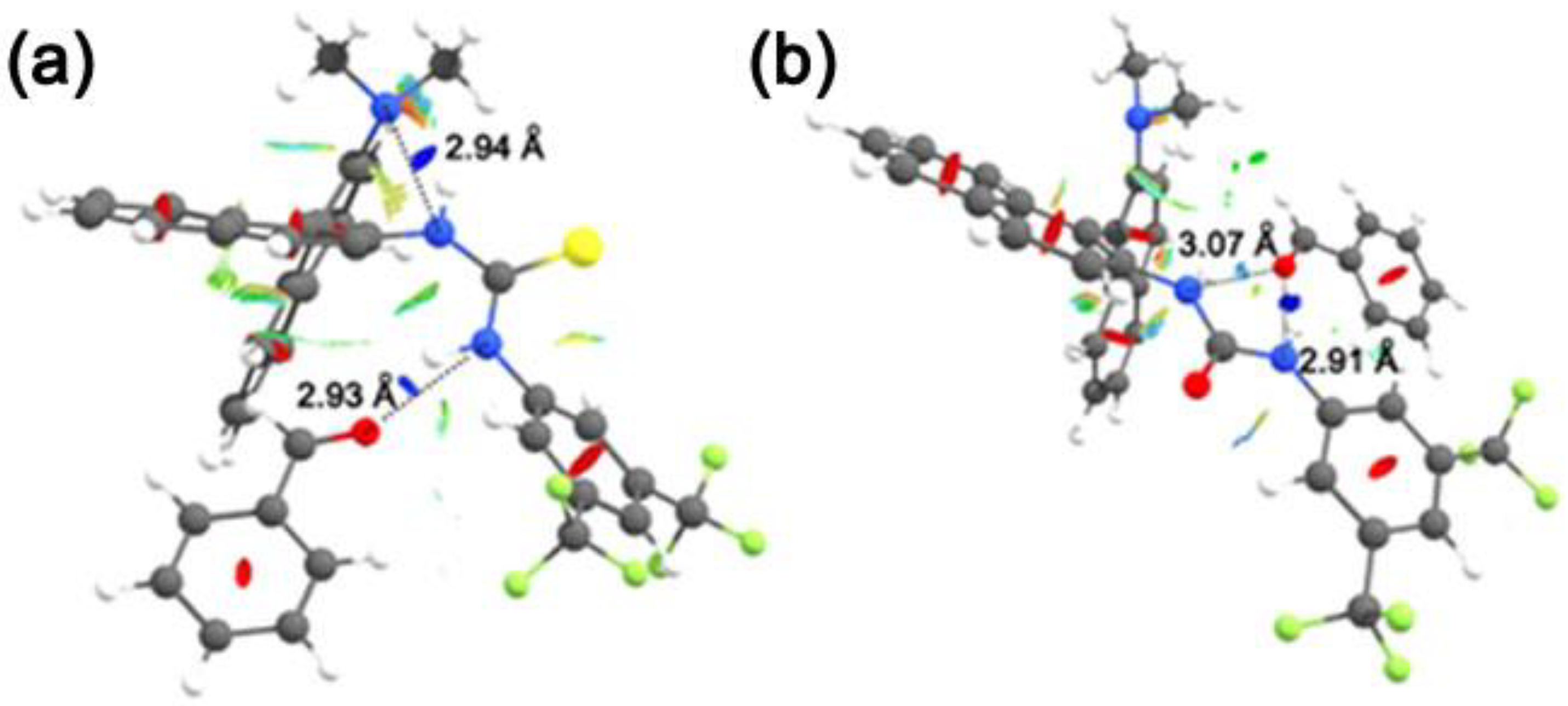
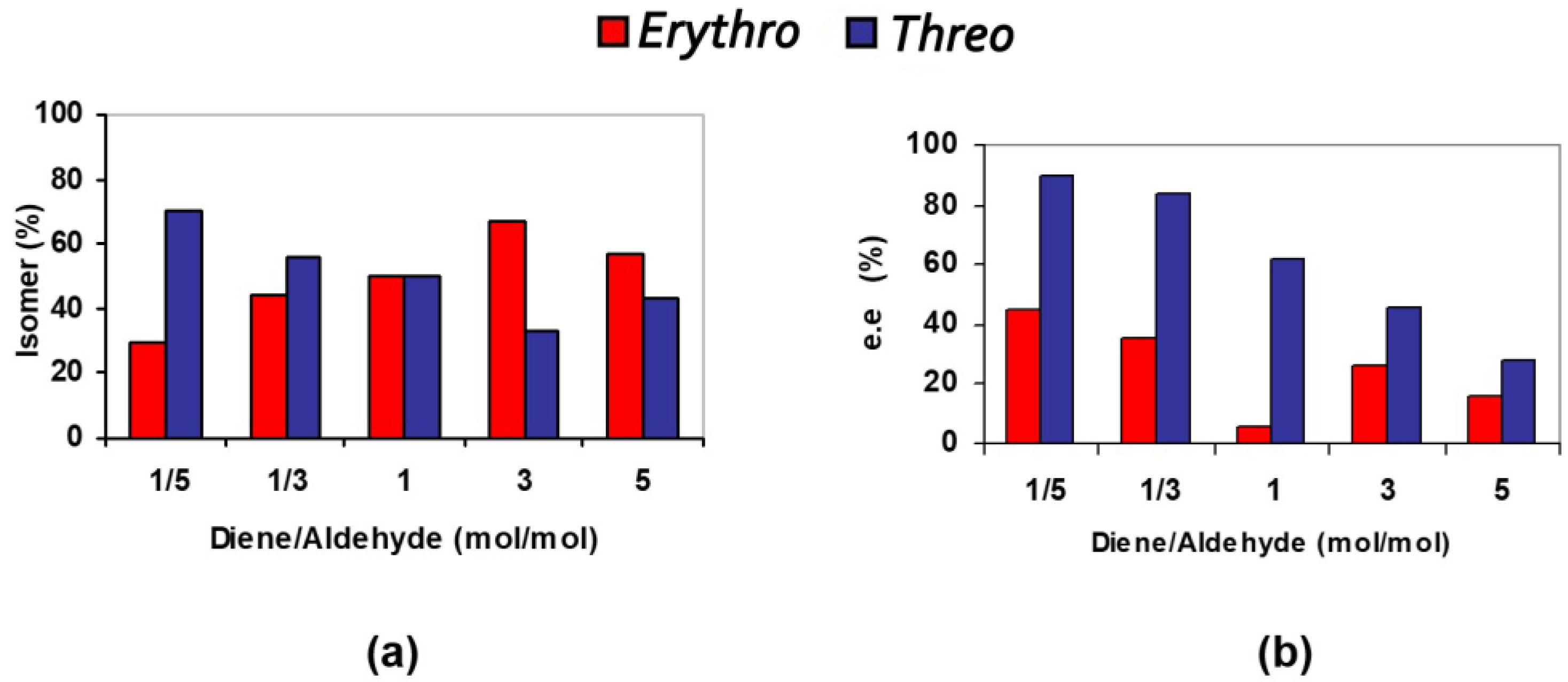
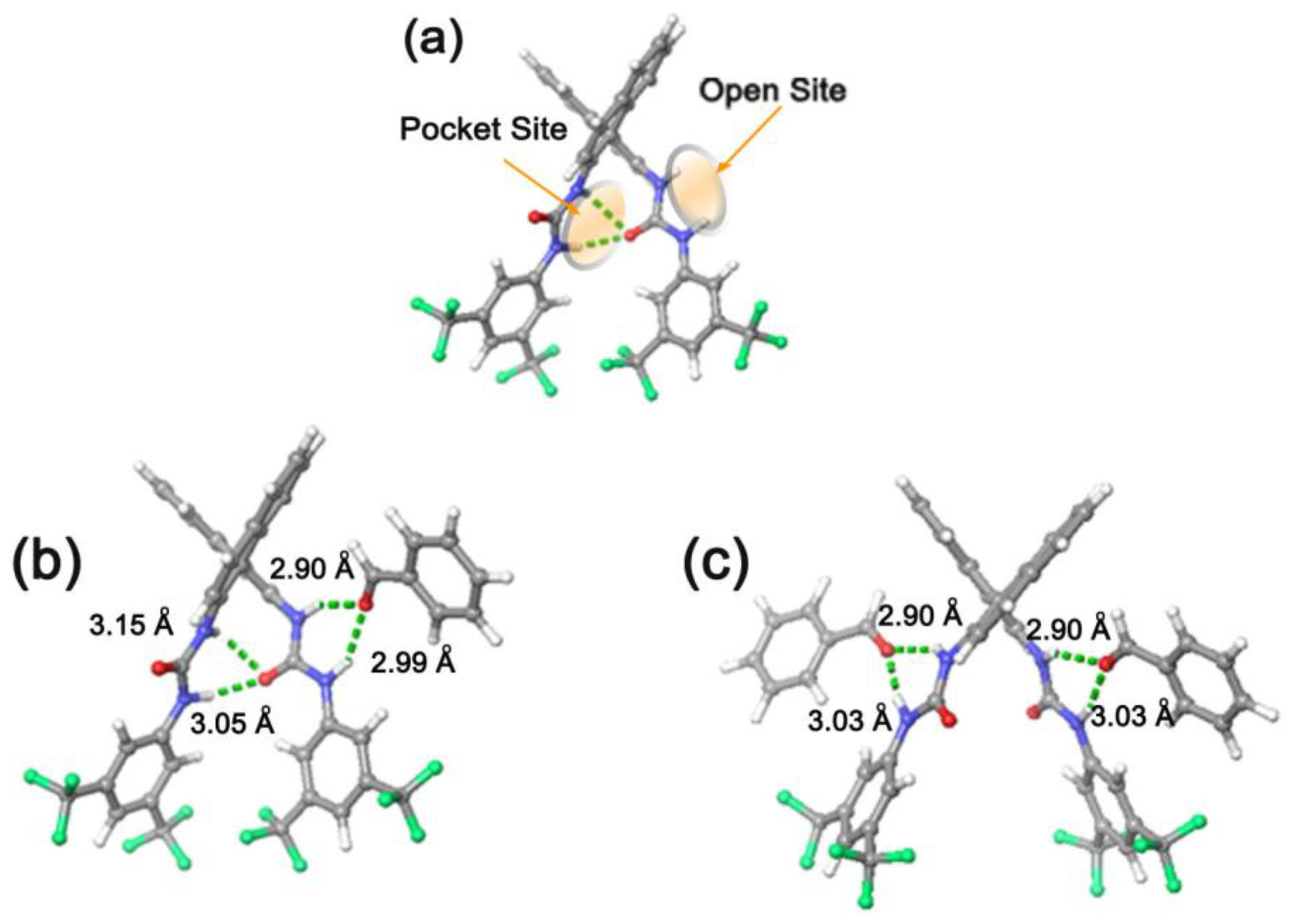
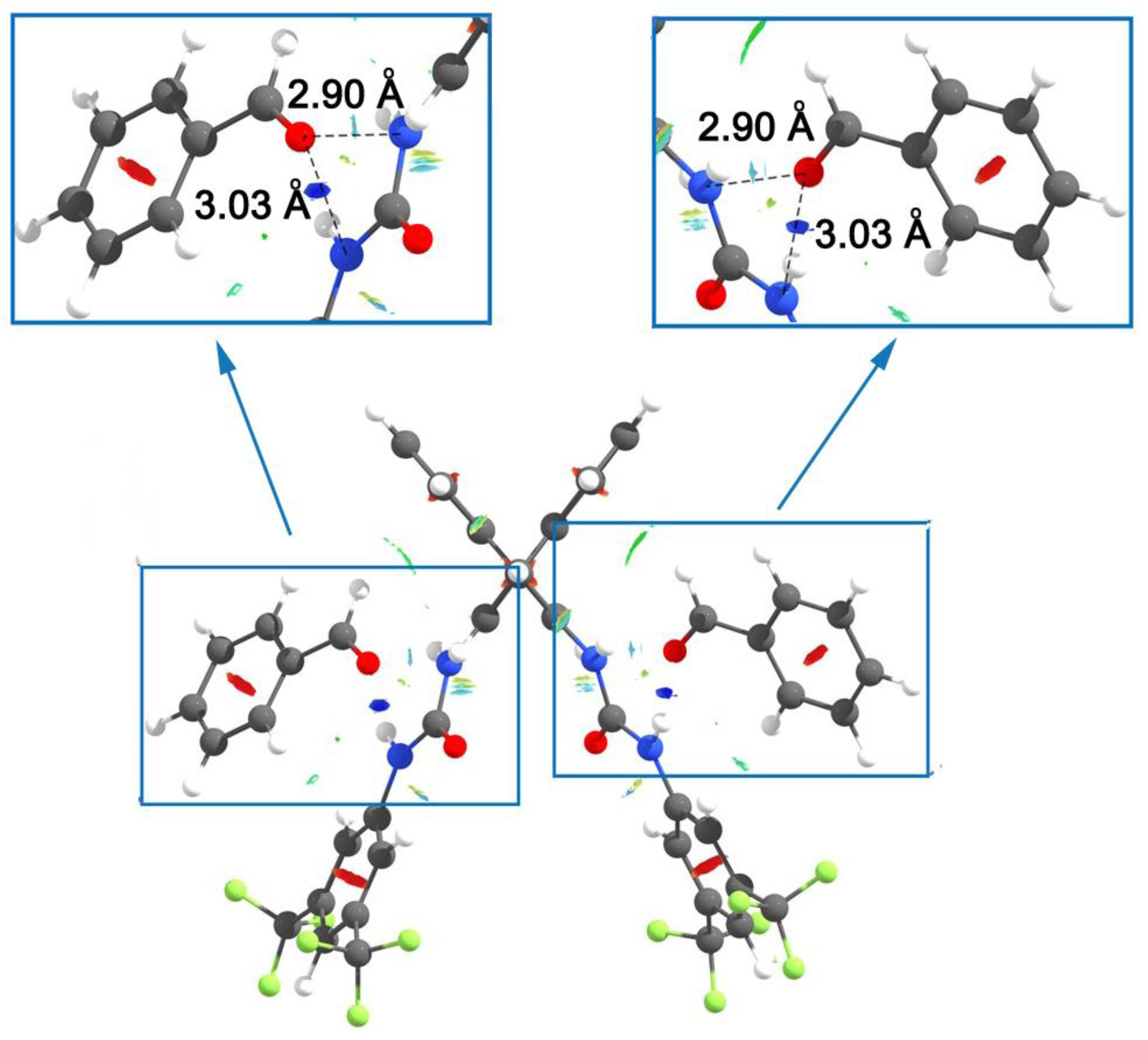
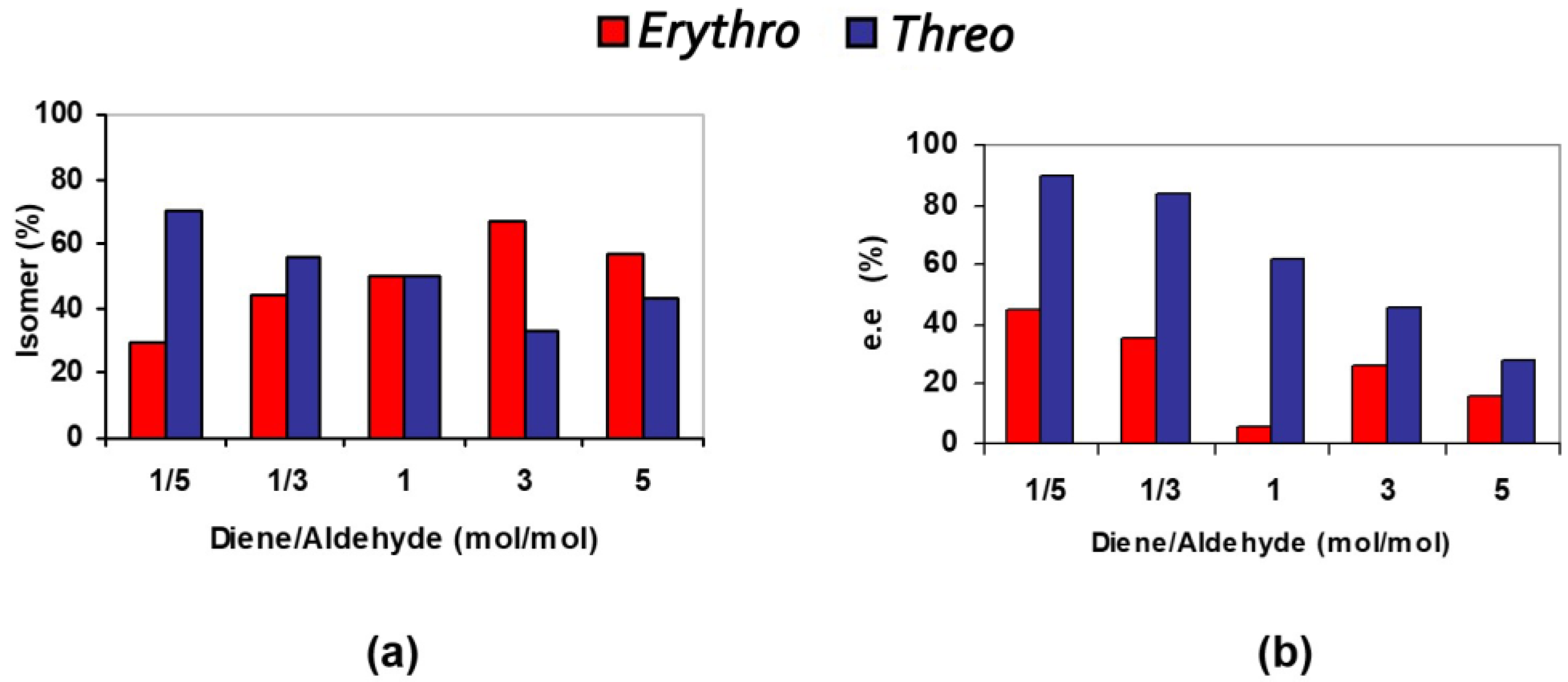
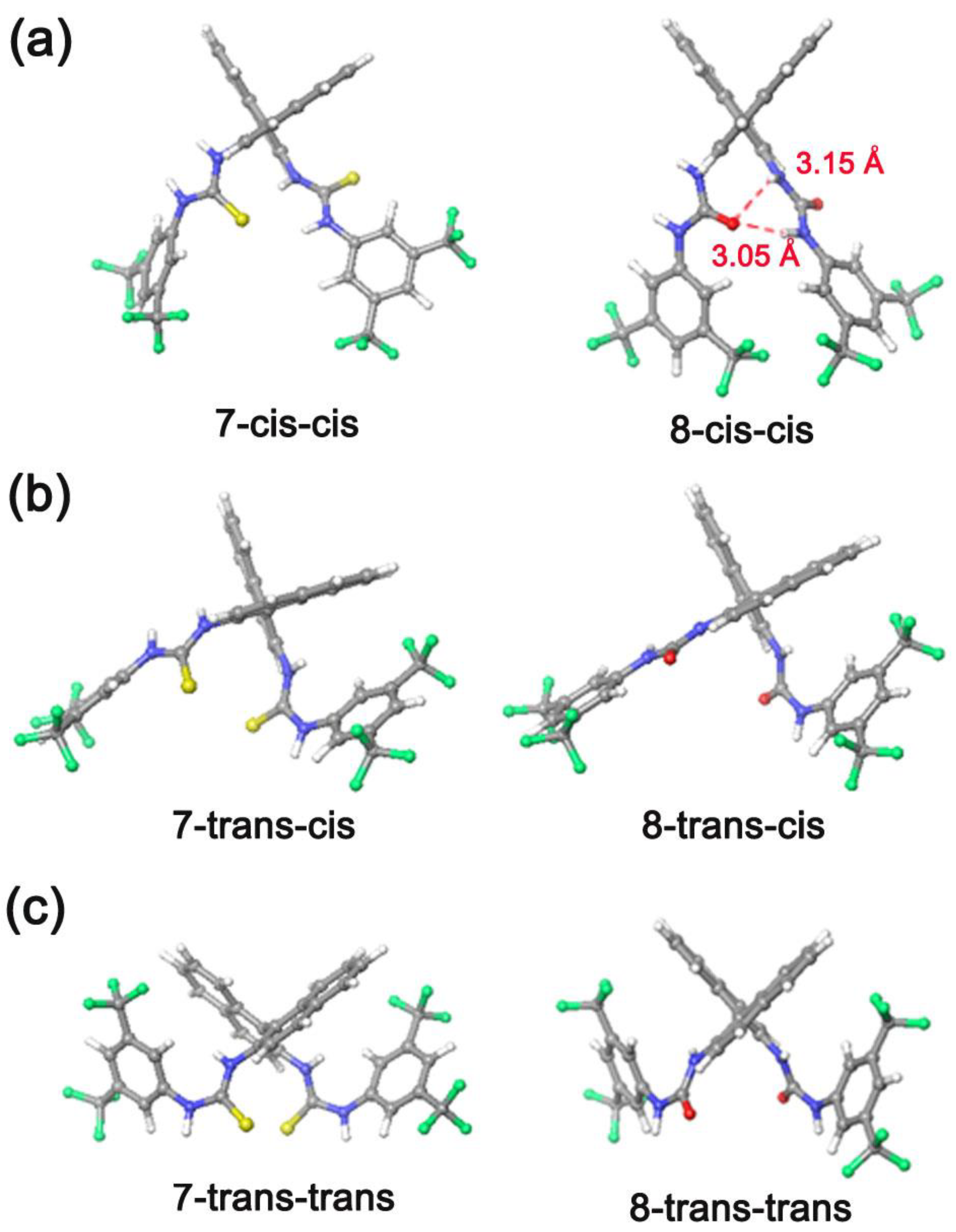
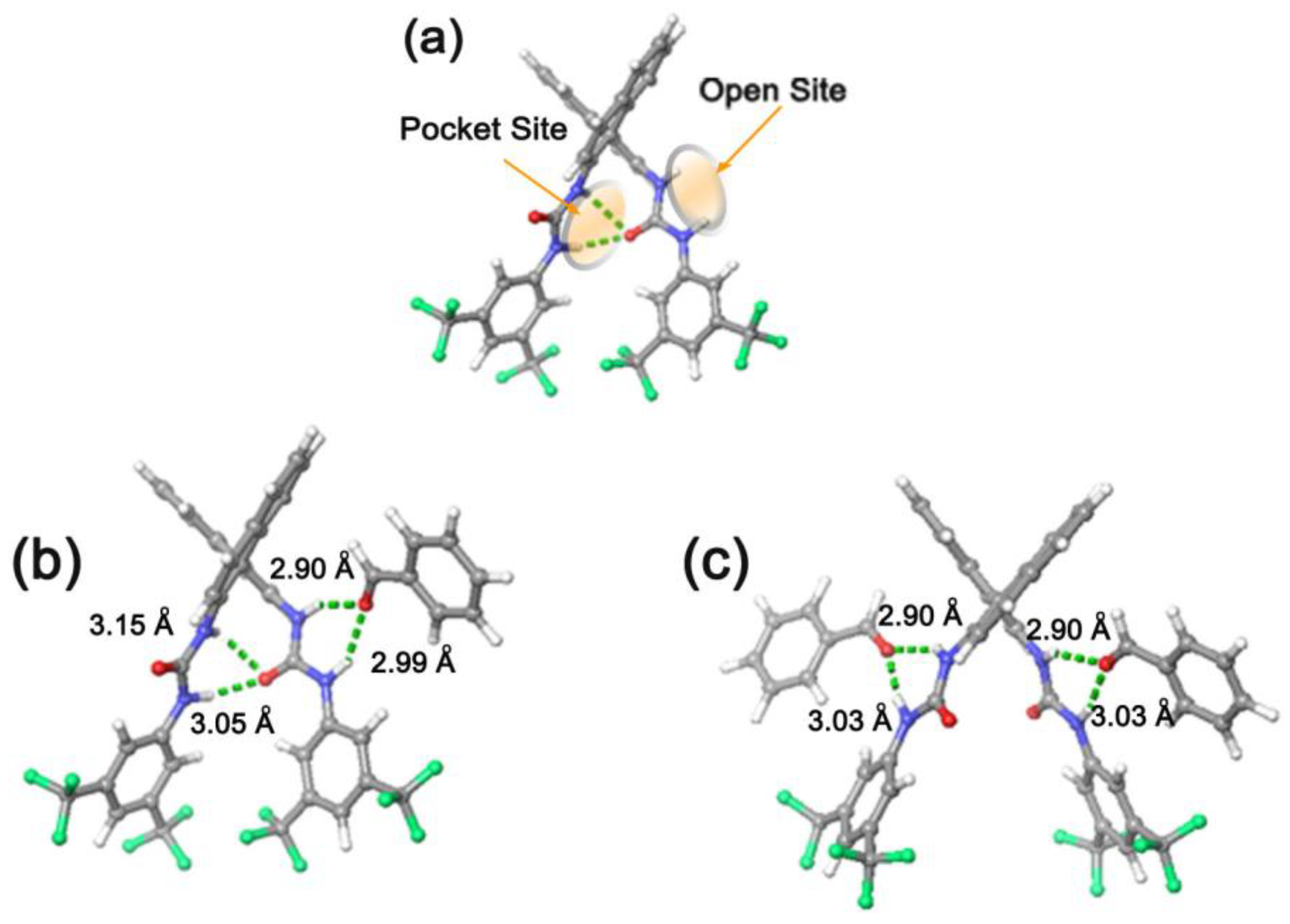
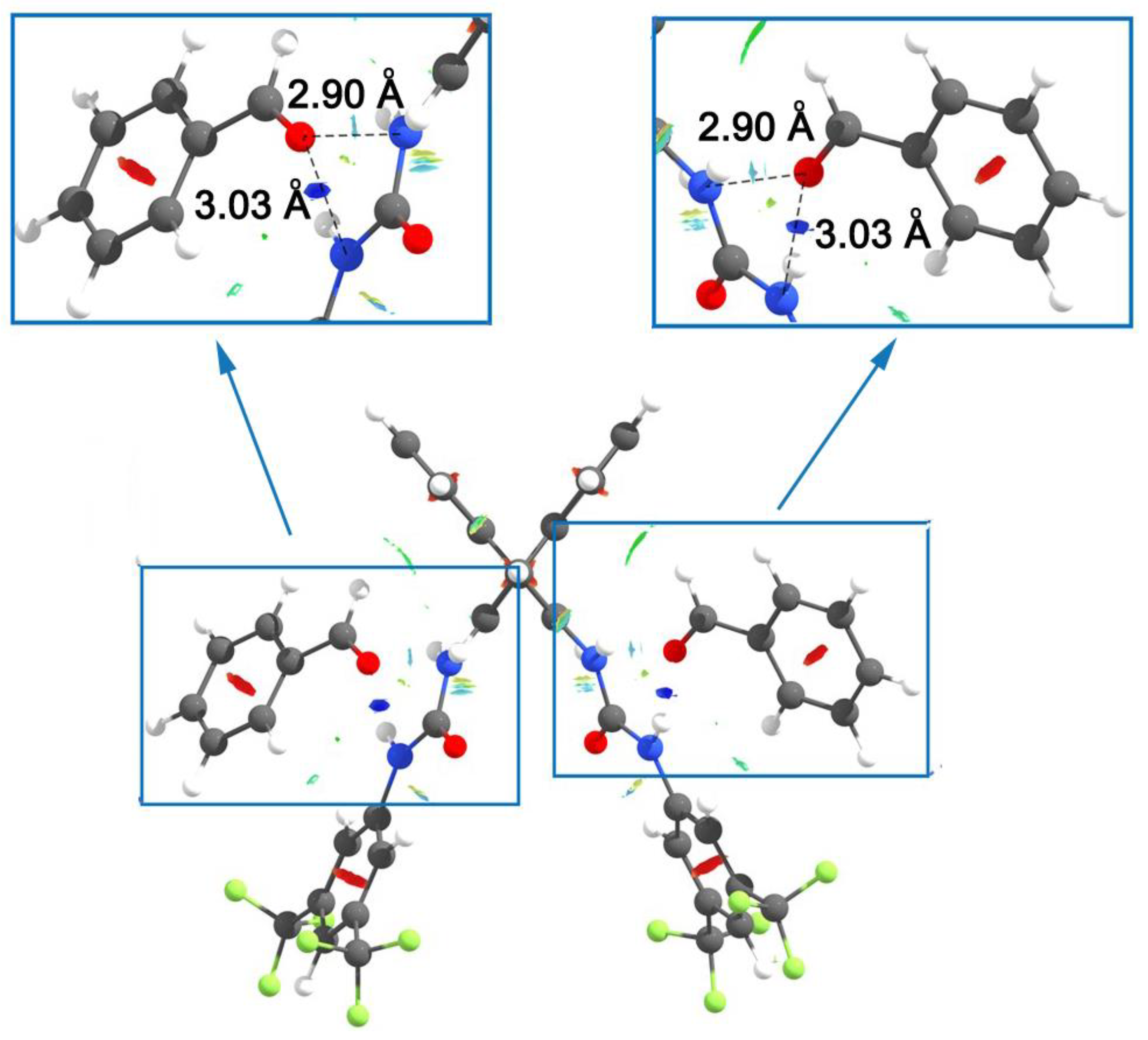
3.2. Bis(thio)urea Catalysts 7 and 8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Breslow, R. Artificial Enzymes. Science 1982, 218, 532–537. [Google Scholar] [CrossRef]
- McMillan, D.W.C. The advent and development of organocatalysis. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef]
- Purse, B.W.; Rebek, J., Jr. Functional cavitands: Chemical reactivity in structured environments. Proc. Natl. Acad. Sci. USA 2005, 102, 10777–10782. [Google Scholar] [CrossRef]
- Meeuwissen, J.; Reek, J.N.H. Supramolecular catalysis beyond enzyme mimics. Nat. Chem. 2010, 2, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Gambaro, S.; Talotta, C.; Della Sala, P.; Soriente, A.; De Rosa, M.; Gaeta, C.; Neri, P.J. Kinetic and Thermodynamic Modulation of Dynamic Imine Libraries Driven by the Hexameric Resorcinarene Capsule. Am. Chem. Soc. 2020, 142, 14914–14923. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, G.; Forgan, R.S. (Eds.) Reactivity in Confined Spaces. In Monographs in Supramolecular Chemistry; The Royal Society of Chemistry: London, UK, 2021. [Google Scholar]
- Iuliano, V.; Della Sala, P.; Talotta, C.; De Rosa, M.; Soriente, A.; Gaeta, C.; Neri, P. Supramolecular control on reactivity and selectivity inside the confined space of H-bonded hexameric capsules. Curr. Opin. Colloid Interface 2023, 65, 101692. [Google Scholar] [CrossRef]
- Borsato, G.; Rebek, J., Jr.; Scarso, A. From Selective Nanocatalysts and Nanoscience; Zecchina, A., Bordiga, S., Groppo, E.E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 105–168. [Google Scholar]
- Catti, L.; Zhang, Q.; Tiefenbacher, K. Advantages of Catalysis in Self-Assembled Molecular Capsules. Chem. Eur. J. 2016, 22, 9060–9066. [Google Scholar] [CrossRef]
- Hong, C.M.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Self-Assembled Tetrahedral Hosts as Supramolecular Catalysts. Acc. Chem. Res. 2018, 51, 2447–2455. [Google Scholar] [CrossRef]
- Ren, Y.; Tao, L.; Li, C.; Jayakumar, S.; Li, H.; Yang, Q. Development of Efficient Solid Chiral Catalysts with Designable Linkage for Asymmetric Transfer Hydrogenation of Quinoline Derivatives. Chin. J. Catal. 2021, 42, 1576–1585. [Google Scholar] [CrossRef]
- Wang, B.; Jin, C.; Shao, S.; Yue, Y.; Zhang, Y.; Wang, S.; Chang, R.; Zhang, H.; Zhao, J.; Li, X. Electron-Deficient Cu Site Catalyzed Acetylene Hydrochlorination. Green Energy Environ. 2023, 8, 1128–1140. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Wang, B.; Yue, Y.; Zhao, J. Migration: A Neglected Potential Contribution of HCl-Oxidized Au(0). Molecules 2023, 28, 1600. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 12, 5713–5743. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Li, S.; Du, J.; Zhang, B.; Liu, Y.; Mei, Q.; Meng, Q.; Dong, M.; Du, J.; Zhao, Z.; Zheng, L.; et al. Selective Hydrogenation of 5-(Hydroxymethyl)Furfural to 5-Methylfurfural by Exploiting the Synergy between Steric Hindrance and Hydrogen Spillover. Acta Phys. Chim. Sin. 2022, 38, 2206019. [Google Scholar] [CrossRef]
- Wang, K.; Wang, B.; Liu, X.; Fan, H.; Liu, Y.; Li, C. Palladium-Catalyzed Enantioselective Linear Allylic Alkylation of Vinyl Benzoxazinanones: An Inner-Sphere Mechanism. Chin. J. Catal. 2021, 42, 1227–1237. [Google Scholar] [CrossRef]
- Cafeo, G.; De Rosa, M.; Kohnke, F.H.; Neri, P.; Soriente, A.; Valenti, L. Efficient organocatalysis with a calix[4]pyrrole derivative. Tetrahedron Lett. 2008, 49, 153–155. [Google Scholar] [CrossRef]
- Cafeo, G.; De Rosa, M.; Kohnke, F.H.; Soriente, A.; Talotta, C.; Valenti, L. Calixpyrrole Derivatives: “Multi Hydrogen Bond” Catalysts for γ-Butenolide Synthesis. Molecules 2009, 14, 2594–2601. [Google Scholar] [CrossRef]
- De Rosa, M.; Citro, L.; Soriente, A. The first organocatalytic addition of 2-trimethylsilyloxyfuran to carbonyl compounds: Hydrogen-bond catalysis in γ-butenolides synthesis. Tetrahedron Lett. 2006, 47, 8507–8510. [Google Scholar] [CrossRef]
- Smith, C.J.; Abbanat, D.; Bernan, V.S.; Maiese, W.M.; Greenstein, M.; Jompa, J.; Tahir, A.; Ireland, C.M.J. Novel Polyketide Metabolites from a Species of Marine Fungi. Nat. Prod. 2000, 63, 142–145. [Google Scholar] [CrossRef]
- Antane, S.; Caufield, C.E.; Hu, W.; Keeney, D.; Labthavikul, P.; Morris, K.; Naughton, S.M.; Petersen, P.J.; Rasmussen, B.A.; Singh, G.; et al. Pulvinones as bacterial cell wall biosynthesis inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 176–180. [Google Scholar] [CrossRef]
- Li, R.-T.; Han, Q.-B.; Zheng, Y.-T.; Wang, R.-R.; Yang, L.-M.; Lu, Y.; Sang, S.-Q.; Zheng, Q.-T.; Zhao, Q.-S.; Sun, H.-D. Structure and anti-HIV activity of micrandilactones B and C, new nortriterpenoids possessing a unique skeleton from Schisandra micrantha. Chem. Commun. 2005, 23, 2936–2938. [Google Scholar] [CrossRef]
- Yu, C.; Ji, P.; Zhang, Y.; Meng, X.; Wang, W. Construction of Enantioenriched γ,γ-Disubstituted Butenolides Enabled by Chiral Amine and Lewis Acid Cascade Cocatalysis. Org. Lett. 2021, 23, 7656–7660. [Google Scholar] [CrossRef]
- Hug, J.J.; Kjaerulff, L.; Garcia, R.; Müller, R. New Deoxyenhygrolides from Plesiocystis pacifica Provide Insights into Butenolide Core Biosynthesis. Mar. Drugs 2022, 20, 72. [Google Scholar] [CrossRef]
- Pelter, A.; Al-Bayati, R.I.H.; Ayoub, M.T.; Lewis, W.; Pardasani, P.; Hänsel, R. Synthetic routes to the piperolides, fadyenolides, epoxypiperolides, and related compounds. J. Chem. Soc. Perkin Trans. 1987, 1, 717–742. [Google Scholar] [CrossRef]
- Boukouvalas, J.; Maltais, F. An efficient total synthesis of the antibiotic patulin. Tetrahedron Lett. 1995, 36, 7175–7176. [Google Scholar] [CrossRef]
- Brown, S.P.; Goodwin, N.C.; MacMillan, D.W.C.J. The First Enantioselective Organocatalytic Mukaiyama−Michael Reaction: A Direct Method for the Synthesis of Enantioenriched γ-Butenolide Architecture. J. Am. Chem. Soc. 2003, 125, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Foxman, B.M.; Deng, L. Asymmetric Vinylogous Aldol Reaction of Silyloxy Furans with a Chiral Organic Salt. J. Am. Chem. Soc. 2010, 132, 9558–9560. [Google Scholar] [CrossRef] [PubMed]
- Pansare, S.V.; Paul, E.K. The Organocatalytic Vinylogous Aldol Reaction: Recent Advances. Chem. Eur. J. 2011, 17, 8770–8779. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Yu, X.; Zu, L.; Wang, W. Chiral Binaphthyl-Derived Amine-Thiourea Organocatalyst-Promoted Asymmetric Morita−Baylis−Hillman Reaction. Org. Lett. 2005, 7, 4293–4296. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Duan, W.; Zu, L.; Wang, W. Organocatalytic Asymmetric Michael Addition of 2,4-Pentandione to Nitroolefins. Org. Lett. 2005, 7, 4713–4716. [Google Scholar] [CrossRef]
- Fleming, E.M.; McCabe, T.; Connon, S.J. Novel axially chiral bis-arylthiourea-based organocatalysts for asymmetric Friedel–Crafts type reactions. Tetrahedron Lett. 2006, 47, 7037–7042. [Google Scholar] [CrossRef]
- Ollevier, T.; Bouchard, J.-E.; Desyroy, V. Diastereoselective Mukaiyama Aldol Reaction of 2-(Trimethylsilyloxy)Furan Catalyzed by Bismuth Triflate. J. Org. Chem. 2008, 73, 331–334. [Google Scholar] [CrossRef]
- Della Sala, P.; Del Regno, R.; Capobianco, A.; Iuliano, V.; Talotta, C.; Geremia, S.; Hickey, N.; Neri, P.; Gaeta, C. Confused-Prism[5]arene: A Conformationally Adaptive Host by Stereoselective Opening of the 1,4-Bridged Naphthalene Flap. Chem. Eur. J. 2023, 29, e2022030. [Google Scholar] [CrossRef]
- Zhu, J.-L.; Zhang, Y.; Liu, C.; Zheng, A.-M.; Wang, W.J. Insights into the Dual Activation Mechanism Involving Bifunctional Cinchona Alkaloid Thiourea Organocatalysts: An NMR and DFT Study. Org. Chem. 2012, 77, 9813–9825. [Google Scholar] [CrossRef] [PubMed]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, M.K.; Schreiner, P.R. (Thio)urea Organocatalyst Equilibrium Acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, 1st ed.; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Lan-Hua, W.; Yong-Bing, H.; Kuo-Xi, X.; Shun-Ying, L.; Ling-Zhi, M. Chiral Fluorescent Receptors Based on (R)-1,1′-Binaphthylene-2,2′-bisthiourea: Synthesis and Chiral Recognition. Chin. J. Chem. 2005, 23, 757–761. [Google Scholar] [CrossRef]
- Sohtome, Y.; Tanatani, A.; Hashimoto, Y.; Nagasawa, K. Development of bis-thiourea-type organocatalyst for asymmetric Baylis–Hillman reaction. Tetrahedron Lett. 2004, 45, 5589–5592. [Google Scholar] [CrossRef]
- Berkessel, A.; Roland, K.; Neudörfl, J.M. Asymmetric Morita−Baylis−Hillman Reaction Catalyzed by Isophoronediamine-Derived Bis(thio)urea Organocatalysts. Org. Lett. 2006, 8, 4195–4198. [Google Scholar] [CrossRef]
- Szlosek, M.; Figadére, B. Highly Enantioselective Aldol Reaction with 2-Trimethylsilyloxyfuran: The First Catalytic Asymmetric Autoinductive Aldol Reaction. Angew. Chem. Int. Ed. 2000, 39, 1799–1801. [Google Scholar] [CrossRef]
- Ghosh, S.; Chopra, P.; Wategaonkar, S. C–H⋯S interaction exhibits all the characteristics of conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2020, 22, 17482–17493. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
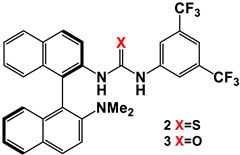 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Equivalents of Aldehyde | T/t (°C/h) | Yield (%) 2 | d.r. Erythro/Threo 3 | e.e. 4 Erythro (%) | e.e. 4 Threo (%) |
| 1 | 2 | 5 | Rt/24 | 29 | 70/30 | 9 | 21 |
| 2 | 3 | 5 | Rt/24 | 56 | 70/30 | 20 | 26 |
| Rotamers | ΔEisomer 1 (kcal/mol) | Ecoord (kcal/mol) | Yield (%) |
|---|---|---|---|
| 2-trans-A | 0.0 | - | - |
| 2-trans-B | 3.0 | - | - |
| 2-cis-A | 2.1 | - | - |
| 2-cis-B | 2.5 | - | - |
| 3-trans-A | 0.0 | - | - |
| 3-trans-B | 3.4 | - | - |
| 3-cis-A | 0.7 | - | - |
| 3-cis-B | 8.9 | - | - |
| 5@2-cis-A | 1.5 | −5.4 | 29 |
| 5@2-trans-A | 0.0 | −4.7 | - |
| 5@3-cis-A | 0.0 | −7.8 | 56 |
| 5@3-trans-A | 1.4 | −5.7 | - |
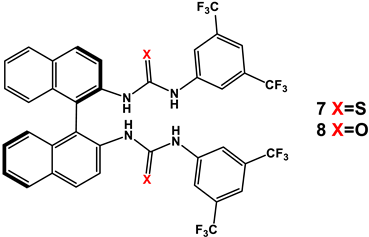 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Equivalents of Aldehyde | T/t (°C/h) | Yield (%) 2 | d.r. Erythro/Threo 3 | e.e. 4 Erythro (%) | e.e. 4 Threo (%) |
| 1 | 7 | 5 | Rt/24 | 45 | 30/70 | 32 | 93 |
| 2 | 8 | 5 | Rt/24 | 53 | 30/70 | 45 | 90 |
| 3 | 7 | 5 | Rt/48 | 45 | 30/70 | 20 | 91 |
| 4 | 8 | 5 | Rt/48 | 53 | 37/63 | 39 | 89 |
| 5 | 7 | 5 | Rt/72 | 50 | 30/70 | 15 | 91 |
| 6 | 8 | 5 | Rt/72 | 45 | 34/66 | 36 | 85 |
| Entry | TMSOF/ Aldehyde | T/t (°C/h) | Solvent | Yield (%) 2 | d.r. (Erythro/Threo) 3 | e.e. (%) Erythro 4 | e.e. (%) Threo 4 |
|---|---|---|---|---|---|---|---|
| 1 | 1/5 | Rt/24 | - | 53 | 30/70 | 45 | 90 |
| 2 | 1/3 | Rt/24 | - | 70 | 44/56 | 36 | 84 |
| 3 | 1/1 | Rt/24 | - | 45 | 50/50 | 6 | 62 |
| 4 | 1/1 | Rt/24 | Toluene (0.2 mL) | 62 | 48/52 | 2 | 24 |
| 5 | 3/1 | Rt/24 | 43 | 67/33 | 26 | 46 | |
| 6 | 5/1 | Rt/24 | 51 | 57/43 | 16 | 28 |
| Rotamers | ΔEisomer 1 (kcal/mol) | Ecoord (kcal/mol) |
|---|---|---|
| 7-cis-cis | 1.77 | - |
| 7-trans-cis | 2.41 | - |
| 7-trans-trans | 0.00 | - |
| 8-cis-cis | 0.00 | - |
| 8-trans-cis | 5.33 | - |
| 8-trans-trans | 8.07 | - |
| 5@8-cis-cis | - | −8.9 |
| (5)2@8-cis-cis | - | −12.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iuliano, V.; Della Sala, P.; Talotta, C.; De Rosa, M.; Gaeta, C.; Neri, P.; Soriente, A. Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives. Organics 2024, 5, 32-45. https://doi.org/10.3390/org5020003
Iuliano V, Della Sala P, Talotta C, De Rosa M, Gaeta C, Neri P, Soriente A. Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives. Organics. 2024; 5(2):32-45. https://doi.org/10.3390/org5020003
Chicago/Turabian StyleIuliano, Veronica, Paolo Della Sala, Carmen Talotta, Margherita De Rosa, Carmine Gaeta, Placido Neri, and Annunziata Soriente. 2024. "Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives" Organics 5, no. 2: 32-45. https://doi.org/10.3390/org5020003
APA StyleIuliano, V., Della Sala, P., Talotta, C., De Rosa, M., Gaeta, C., Neri, P., & Soriente, A. (2024). Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives. Organics, 5(2), 32-45. https://doi.org/10.3390/org5020003