1. Introduction
Synthetic fibres encompass a wide variety of polymeric materials, and their definition in the strictest sense exclude fibres that are made from regenerated cellulose and from cellulose esters. Classic examples include polyolefins, acrylics, nylons, and polyesters. As is common in other organic polymers, synthetic polymers undergo various degrees of thermal and thermo-oxidative degradations that depend on the chemical nature of the fibre and on the degree and severity of heat flux [
1]. In most cases, degrading polymers produce a variety of organic volatiles, and when these are mixed with ambient air, they most often result in flammable mixtures.
The size, shape and other morphological features of polymeric materials also have a strong effect on their overall flammability characteristics. For instance, fibres, owing to their large ratio of surface area to volume, are significantly more flammable than their moulded counterparts [
2]. In addition, the burning behaviour of derived fabrics, mainly comprised of a given fibre type, or a blend, is also influenced by a number of factors including the nature of the ignition source and the time of its impingement, the fabric orientation and point of ignition, the ambient temperature and relative humidity, the velocity of air, etc.
Successful strategies to reduce the flammability of synthetic polymers involve interrupting the complex combustion process at one or more stages with a view to reducing the rate and/or changing the mechanism of combustion at that stage. From a practical point of view, this can be achieved either via the mechanical blending of a suitable flame-retardant compound with a polymeric substrate (i.e., by introducing an additive), via a simple copolymerisation reaction, or via a chemical modification of the preformed polymer (i.e., using a reactive strategy) [
3].
Even though most synthetic polymers are usually made more flame-retardant by incorporating additives, this strategy is not very effective with fibre-forming polymers. For instance, phase separation and/or leaching out of the additive(s) are quite amenable during fibre production via wet-spinning processes in particular, or they are equally plausible during the life cycle of fibres, for example, knitted materials. For the past several years, we have been attracted to the alternative method of making synthetic fibres flame retardant, mainly polypropylene (PP) and polyacrylonitrile (PAN), via chemical modification reactions, primarily utilizing phosphorus-containing compounds. There are other significantsignificant advantages to this methodology: (a) low levels of modification may suffice, (b) modifying groups are chemically bonded and are therefore less likely to be lost during the fibre production stage and subsequent service, and (c) that the modification can more readily be molecularly dispersed throughout the polymeric matrix [
4].
In the following sections, we provide an overview of the thermal/thermo-oxidative degradation, flammability characteristics, and reactive routes specifically employed by us to make two of the most commercially important and fibre-forming polymers, PP and PAN, flame-retardant. Thus far, we have experimented with various compounds that contain penta-valent phosphorus in different chemical environments. These compounds contain phosphonates, phosphates, and phosphorilamino esters [
5]. In the case of PP, grafting reactions were either carried out in solutions under elevated temperatures, or under melt conditions using a twin-screw extruder. However, in the case of PAN, the corresponding transformations were almost exclusively affected by a copolymerisation reaction carried out via an aqueous–slurry route at moderate temperatures using a redox initiator pair. The recovered polymers were purified, dried, and subjected to a variety of spectroscopic, thermal, calorimetric, and some optional ‘hyphenated’ evaluations and techniques. In the following sections, the two polymeric systems are treated under two separate headings, i.e., PP is followed by PAN.
2. Polypropylene
Generally, polyolefin fibres include fibrous materials whose polymeric chains have a high molar mass and saturated aliphatic hydrocarbons. Among these, polypropylene (PP) is the most commercially important member, followed by polyethylene (PE); the latter has much less of a commercial significance as a fibre-forming polymer compared to PP. The other commercially important polyolefin-based fibres include poly(4-methyl-1-pentene), poly(1-butene) and poly(3-methyl-1-butene). Other examples include copolymers such as poly(ethylene-
co-propylene) and poly(ethylene-
co-octane), and blends of different polyolefins; however, these have only minor commercial significance [
6].
For α-olefin polymers, such as PP, every other carbon atom along the chain backbone is asymmetric, and therefore the constituent polymeric chains can adopt a variety of configurations. The resulting stereo-isomeric forms are known as isotactic (methyl groups are located on the same side), syndiotactic (methyl groups are alternate), and atactic (methyl groups are randomly arranged). The highly regular three-dimensional structure of isotactic PP favours the formation of fibres with superior mechanical properties. A relatively low cost of production, coupled with high tensile strengths, has made PP the most commercially important fibre-forming polyolefin polymer. Both isotactic and syndiotactic forms have fibre-forming characteristics owing to their potential to create order in the polymer matrix. Currently, isotactic PP is the main commercially available stereoisomer for use in orientated fibre films and tapes.
Generally, polyolefins are highly flammable and burn readily in air (Limiting Oxygen Index: LOI~18) causing melting and dripping and producing little or no char. LOI is only a low-level indicator of the flammability of a polymeric material, and the melt–drip behaviour of PP is considered to be a serious secondary hazard that is primarily responsible for secondary ignitions and the proliferation of fires in actual scenarios. The principal mechanism of the thermal degradation of polyolefins is homolytic chain scission, followed by inter- and intra-molecular chain transfer, resulting in the formation of volatile fragments. These long-chain fragments and the soot-like products, formed as a result of cyclization/dehydrogenation, can contribute to smoke production. Carbon dioxide and water are also formed during combustion of PP-based polymers [
7].
Oxidative chlorophosphonylation, radiative cross-linking, and the radiation grafting of vinyl phosphonate oligomers are known to be successful reactive strategies for producing flame-retardant polyethylene (PE) [
8,
9]. The flame retardance of phosphorus-modified PE is believed to arise, at least in part, from a condensed-phase mechanism. On the other hand, chemical modification with PP is less successful owing to substantial main-chain degradation encountered upon treatment with common modifying agents. Nevertheless, there are few instances where chlorinated PP has been used as a flame-retardant additive for polyolefins [
3]. In general, for polyolefins, the use of additive flame retardant materials is a more common practice, mostly driven by commercial considerations. This also includes the use of intumescent systems, largely consisting of ammonium polyphosphate and pentaerythritol. An excellent review in this field is provided by Zhang and Horrocks [
1].
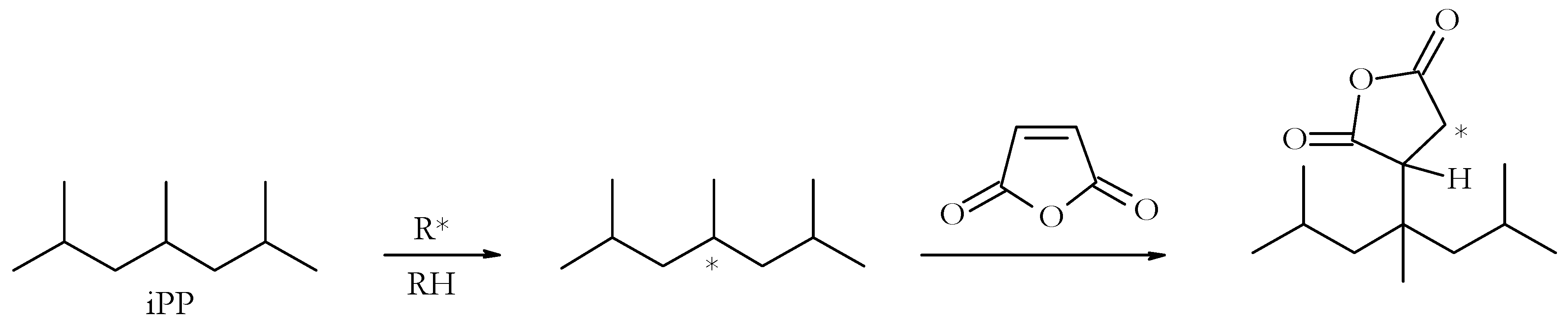
The tertiary hydrogen atoms of the PP are relatively liable and can be made to react with reactive unsaturated molecules under radical initiation, either in solution or under melt conditions, which is often referred to as a grafting reaction (
Figure 1). The latter is very commonly used, for example, with maleic anhydride on commercial scales, with the aid of a twin-screw extruder to produce malleated PP. Owing to the enhanced polarity of malleated products, these are often used as compatibilisers for PP with a relatively higher number of polar substrates [
5]. Depending on the nature and activity of the grafting reagent, and the prevailing reaction conditions, the degree and extent of the grafts can vary.
In our previous research, we compounded PP with nanoclays and melt and extruded the products into fibres; however, we also observed that, in order to improve the dispersion of the clay in polymer matrix, it is necessary to add a compatibiliser, such as maleic anhydride-grafted PP [
10]. It was observed that the optimal values of loading for the compatibiliser were between 1 and 3 wt.% for filament production. However, high loadings (i.e., 15 wt.%, or above) actually proved to be detrimental to mechanical properties at the expense of much reduced returns in terms of the exfoliation of the clay/polymer matrix. Furthermore, the cyclic anhydride structures in PP grafted with maleic anhydride are prone to slow adventitious hydrolysis, eventually leading to the intra-molecular cross-linking of the polymer chains.
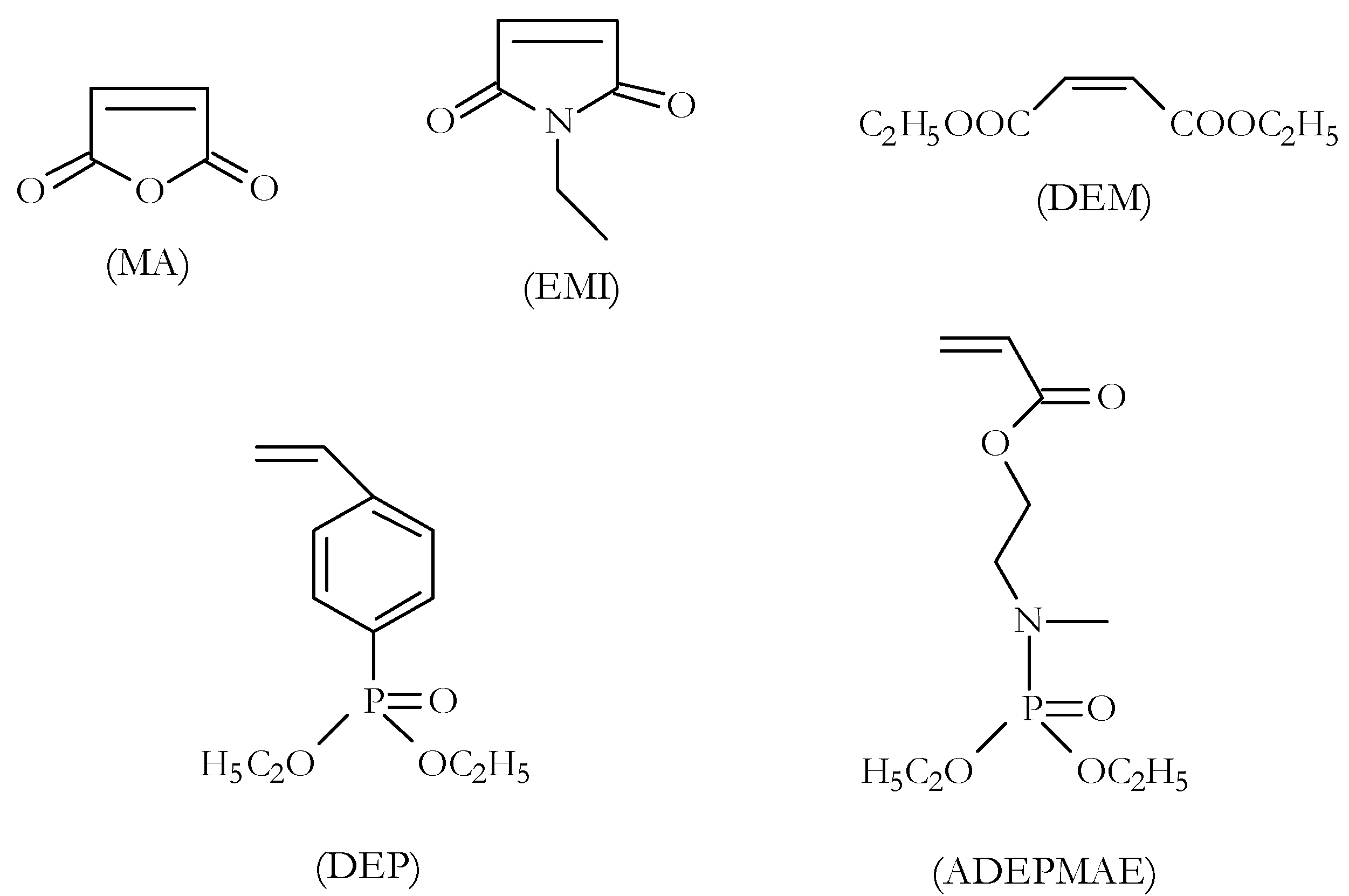
In a related study, we tried a variety of alternative grafting agents, some novel agents that are more hydrolytically stable than maleic anhydride as well as those containing flame-retardant groups [
11]. These include: ethyl maleimide (EM), diethyl maleate (DEM), diethylvinylbenzyl phosphonate (DEP), and acrylic acid-2-[(diethoxyphosphoryl)methyl amino] ethyl ester (ADEPMAE) (
Figure 2). The grafting procedure primarily involved the reaction of PP chips with a grafting agent in
ortho-dicholorobezene in the presence of the initiator, di-
tert-butylperoxide. The experimental conditions and yields for various grafting agents are also provided in our publication [
11].
Phosphorus- and phosphorus/nitrogen-containing grafting agents were synthesized by following literature precedents [
12]. The chemical structure and purity of these molecules were mainly inferred from
1H and
31P NMR spectra. The chemical structures of the grafted products were deduced via
1H NMR measurements at elevated temperatures (ca. 80–100 °C) using d
2-1,1,2,2-tetracholoroethane as the solvent. The elemental analyses of the products were also conducted using a micro-combustion method (for C, H, and N), and via inductively coupled plasma/optical emission spectroscopy (for P). The detailed characterization data for the modified polymers have been published elsewhere [
11]. Fourier-transform infrared spectroscopy (FT-IR) was carried out on films that were made via hot pressing (temperature~180 °C) at a pressure of about 10 tons (for ca. 3 min).
Isotactic PP of fibre grade and various additives were firstly hand mixed in a plastic container prior to compounding. The additives, apart from the grafting agents, included: 1 wt.% MA-grafted PP (Polybond 3200), Cloisite 20A, sodium clay Bentone HC and its modified version, and Bentone 107 (with a generic quaternary methyl, bis(dihydroxyethyl), hydrogenated tallow ammonium cation). The mixed recipe was then compounded using a Brabender mixer, with a rotor speed of 60 rpm and at a temperature profile from 180 to 190 °C. PP films (ca. 0.3 mm thickness) were prepared using compression moulding with spacer plates, between aluminium foil-covered steel plates at about 190 °C. A control sample of pure PP with a thermal history equivalent to the compounded counterparts was also produced to serve as the control.
Morphological and the associated structural features of the composites were carried out via optical microscopy, X-ray diffraction (XRD), and scanning electron microscopy (SEM). Optical microscopy was employed as a simple and fast method to gain some insights regarding nanoclay dispersion. Film samples with an approximate thickness of 0.2 mm were used, and the images were obtained using a Nikon optical microscope. In order to obtain XRD diffractograms on a thin film segment, a Siemens D500 X-ray diffractometer with Cu-Kα radiations was employed (2θ from 2 to 20° at the rate of 2°/min). SEM images were obtained using a Cambridge Stereoscan 200 SEM (at an accelerating voltage of 10 kV, CAE Company, Manchester, UK). Films were etched with chromic acid before gold sputtering to identify any underlying structural features.
Routine thermal analyses, such as differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA), were carried out on the samples using Polymer Laboratories instruments (Polymer Laboratories Company, London, UK). For DSC runs, a heating rate of 10 °C/min was employed over a range of 30 °C to 350 °C under a constant flow of nitrogen (10 cm3 min−1). TGA analyses were carried out in flowing air (10 cm3 min−1) and at a heating rate of 20 °C min−1. Approximately 6 mg of the sample was used in each case, and multiple runs were carried out to check the reproducibility of the thermograms. The concentrations of carbon monoxide and carbon dioxide from the TGA of selected samples were monitored using a non-dispersive infrared gas analyser and the concentration of oxygen was monitored using an electrochemical cell oxygen sensor.
Limiting oxygen index (LOI) values were determined using films of approximately 0.3 to 1.5 mm thickness by following a standard procedure. Cone calorimetric tests on selected samples were carried out on a Fire Testing Technology (East Grinstead, UK) cone calorimeter, and all tests were conducted according to the test methods defined in ISO 5660 using an incidental heat flux of 35 kWm−2. Samples were supported by aluminium foil and contained molten polymer during testing.
The choice of the grafting agents was primarily based on their grafting efficiency (i.e., on the reactivity of the olefinic group), polarity, hydrolytic stability (especially of EMI and DEM compared to MA), and flame-retardant properties (e.g., in the case of DEP and ADEPMAE). The recovered products were thoroughly washed with acetone (a good solvent for the unreacted grafting agents) and dried in a vacuum oven at ca. 60 °C to a constant weight. All grafting reactions were found to be quite straightforward, resulting in appreciable yields of the products.
Spectroscopic analyses, primarily 1H and 13C NMR, of the purified products revealed unequivocal evidence for the presence of grafts on the PP chains. This was also supported by the examination of the corresponding FT-IR spectra in the modified polymers. Quantitative information regarding the amount of grafted units was obtained from 1H NMR and/or via elemental analyses. The results from both methods were also favourably compared. It Is a fairly well-established fact that, in the case of PP, under similar experimental conditions, grafting reactions primarily occur in the tertiary carbon atoms of the polymer backbone. Furthermore, it is highly likely that in the cases of MA, EMI, and DEM, the grafts predominantly consist of monomeric units. However, in P-containing agents (DEP and ADEPMAE), the possibility of oligomeric graft formation cannot be completely ruled out owing to their relatively higher tendency to homopolymerise.
The XRD patterns of Bentone clay before and after heating to 190 °C for 30 min revealed that there was some loss of surfactants in heated samples. As for the composites of PP with this clay, there was no shift in the clay characteristic peaks between 3° and 4° (2θ values) for PP/Bentone, indicating that there was no significantsignificant intercalation. However, for all other samples with grafted PP, there were significant peak shifts to the left, indicative of the possibility of intercalated structures. The optical microscopic images of PP control and grafted PP with nanoclays clearly showed better homogeneity in the latter case. These observations were also supported by the corresponding SEM images confirming that, generally, the clay particle dispersion in these systems improved following PP grafting.
TGA analyses demonstrated that, generally, the presence of the various additives does not change the onset of thermal decomposition of PP (232 °C). However, the presence of the grafts increased the final decomposition temperatures for all samples, slowed down the rate of decomposition, and extended the decomposition temperature range. In addition, the presence of Bentone clay seemed to have a mild char-enhancing effect on PP. The results from LOI measurements show that all the composite samples had nominal, but noticeable, increments in their LOI values compared to the PP controls (ca. from 17 to 20) as is expected from their degradation behaviours registered by the TGA runs. There were also variations in the peak heat release rates (PHRR), and for some samples, these values were not affected, but for others there were nominal reductions. The total heat released (THR) also varied among the samples, with a noticeable decrease in the case of DEP-grafted PP.
Cone calorimetric data, performed in replicates, generally showed some degree of variation, primarily arising from the considerable bubbling and flowing of the PP films. However, some general trends can be discerned. For example, there is no significant effect on time-to-ignition (TTI) with the addition of grafted PP with nanoclays as compared to virgin PP. This is in agreement with the well-known fact that nanoclays do not generally affect TTIs, and in some cases, can even lower TTIs compared to base polymers.
As the char retained after burning a polymer is also a measure of its flammability, the mass loss curves from cone calorimetric runs give some insight into the fire performance of the samples. In general, MA-grafted PP and DEP-grafted PP, which indicated better performance in TGA and LOI evaluations, were seen to be more effective in reducing the PHRR of the PP/nanoclay blends as observed in the cone calorimetric measurements. This could be attributed to the improved dispersion of clays, resulting in char enhancement in those systems. However, it should be pointed out here that the tested thermally and physically thin samples had thermal behaviours different from those of thick polymer plaques. It is believed that, in polymer clay nano-/micro-composites, a carbonaceous–silicate char builds up on the polymer surface during burning, which insulates the underlying material and slows down its mass loss/reduces the rate of decomposition, hence conferring flame-retardant properties to the polymer [
10]. However, in the case of thinner samples, the above effect could be overcome by enhanced vitalization to fuel by the surrounding polymer.
The results for smoke production, expressed as m
2/m
2, show much variation, and no real trend was observed. The evolved gas analyses (CO
2/CO ratios) also exhibited some degree of variability. The sample that contained Bentone clay alone (3 wt.%) resulted in the lowest peak CO production compared to virgin PP. The peak CO
2 production for this sample was more or less comparable with other modified samples (but much less than virgin PP), thus resulting in the highest value for peak CO
2/CO ratio. The relatively high value of peak CO
2 production caused by virgin PP points towards a higher combustion efficiency as compared to the modified samples. Generally, all modified samples had higher temperatures for peak CO
2 production [
11].
In conclusion, the degree of dispersion of nanoclays in PP can be improved via the addition of various grafted PPs. Some degree of exfoliation was believed to be achieved for the sample containing DEP-grafted PP. The grafts had a minimal effect on the melting points of the polymer but slightly enhanced the stability of the polymer below 400 °C. Samples containing grafted PP had slightly lower flammability than virgin PP, and the results for the samples with MA- and DEP-grafted PPs were particularly encouraging. Some of the most promising systems were compounded using a twin-screw extruder and melt spun into filaments with to the aim of producing flame-retardant synthetic nanocomposite fibres. The physical properties and burning behaviours of the knitted fabric samples are reported separately [
13].
3. Polyacrylonitrile
Polyacrylonitrile (PAN) is one of the most important fibre-forming polymers. On an industrial scale, large quantities of PAN are produced via radical polymerisation in an aqueous slurry medium. Homo- and co- polymers of acrylonitrile are predominantly white powders having relatively high glass transition temperatures (T
g). Owing to relatively strong intermolecular forces between polymeric chains, PAN forms fibres with superior mechanical properties. Staple acrylic fibres, being soft and resilient, are used as substitutes, or diluents for wool, and fabrics made from them show a good crease resistance and crease retention. Due to the relatively high melting point and significant carbon yields, PAN fibres are considered to be the most suitable precursors of high-performance carbon fibres [
14].
However, when subjected to heat, PAN undergoes extensive degradation involving the production of combustible volatiles (such as acrylonitrile, ammonia, organic and inorganic nitriles) and varying amounts of char, the latter strongly depending on the rate of heating [
15]. Acrylic fibres have a LOI of around 18 and burn quite readily with melting and sputtering. It was shown that the mechanism of PAN thermal degradation depends on the heating rate [
16]. At low heating rates, the intramolecular cyclization of pendant C≡N groups is the main reaction pathway of PAN degradation; however, at higher heating rates that are similar to those encountered in fires, volatile-forming chain scission prevails [
14].
Traditionally, AN-based polymers are made flame-retardant via the use of additives such as ammonium polyphosphate [
1]. Most commonly, the reactive flame retardance of PAN is achieved via the use of halogen-containing comonomers. Copolymers of AN with such comonomers (up to 15 mol.%) are referred to as ‘modacrylics’. These comonomers include vinylidene chloride, vinyl chloride, α-chloroacrylonitrile, and corresponding bromides. Although an adequate level of flame retardance has been achieved via this procedure, the perception that halogen-containing materials are ecological hazards has provided an opportunity to consider more environmentally sustainable alternatives. Owing to the potential disadvantages in using additives for fibre-forming polymers, we have turned our attention to a strategy involving the chemical attachment of flame-retardant moieties directly to polymer backbones, i.e., to a reactive strategy [
16].
Encouraged by the results from our previous studies [
16,
17,
18,
19], we have continued our efforts to make AN-based polymers flame-retardant via the chemical incorporation of P-containing comonomers into polymeric chains. It is shown that the condensed-phase mechanism of flame retardance, based on the catalysis of cyclization and char-forming processes, is enhanced in PAN bearing covalently bound phosphorus. Furthermore, there is a possibility of additional vapour phase activity, emanating from volatile degradation products that incorporate phosphorus. Additionally, the surfaces of PAN fabrics can be grafted using P-containing comonomers via plasma polymerisation to improve the flame retardance of acrylic fibres [
20,
21].
In our previous publications, we reported some preliminary investigations of flame retardance in copolymers of AN and acrylic/methacrylic phosphonates [
12,
16,
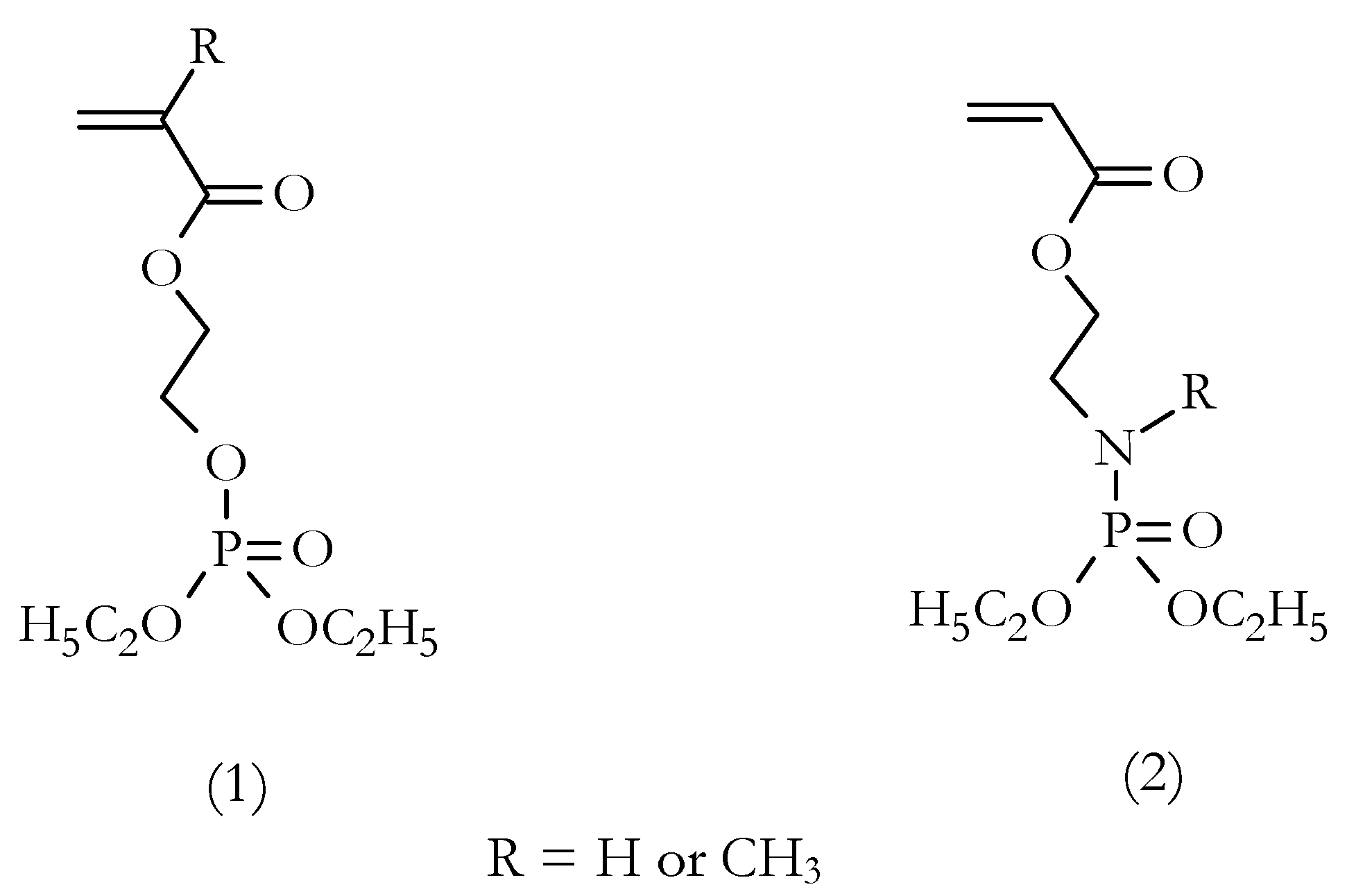
18]. The present overview also reflects our continuing research on the flame retardance of AN-based polymers via a reactive strategy involving the use of P- or P/N-containing comonomers and the incorporation of molecularly dispersed β-cyclodextrin (β-CD). In the first instance, several polymerizable unsaturated compounds (containing either P, or P and N atoms) were synthesized. These compounds included acrylic or methacrylic phosphates and phosphorylamino esters, the general structures of which are shown in
Figure 3.
All chemicals, reagents and solvents were purchased from Sigma Aldrich (Manchester, UK) with the exception of acryloyl chloride obtained from Alfa Aesar (Manchester, UK). Acrylonitrile (AN) was freed from the inhibitor, 4-methoxyphenol via passing through a column of activated basic alumina, and then it was stored over molecular sieves (4 Å type) at 0–5 °C. The solvents and other reagents were purified via standard literature procedures, as necessary [
22].
P-containing comonomers, diethyl-2-(acryloyloxy)ethyl phosphate (DEAEP) and diethyl-2-(methacryloxy)ethyl phosphate (DEMEP), were synthesized via condensation of diethylchlorophosphate with 2-hydroxyethyl acrylate and hydroxyethylmethacrylate, respectively [
23]. The detailed procedures for the preparation of P- and N-containing comonomers—i.e., phosphorylamino esters, acrylic acid-2-(diethoxyphosphorylamino)ethyl ester (ADEPAE), and acrylic acid-2-[(diethoxyphosphoryl)methyl amino] ethyl ester (ADEPMAE)—were described in our previous publication [
12]. The typical yields of the synthesized comonomers, after purification, ranged from 50 to 98%.
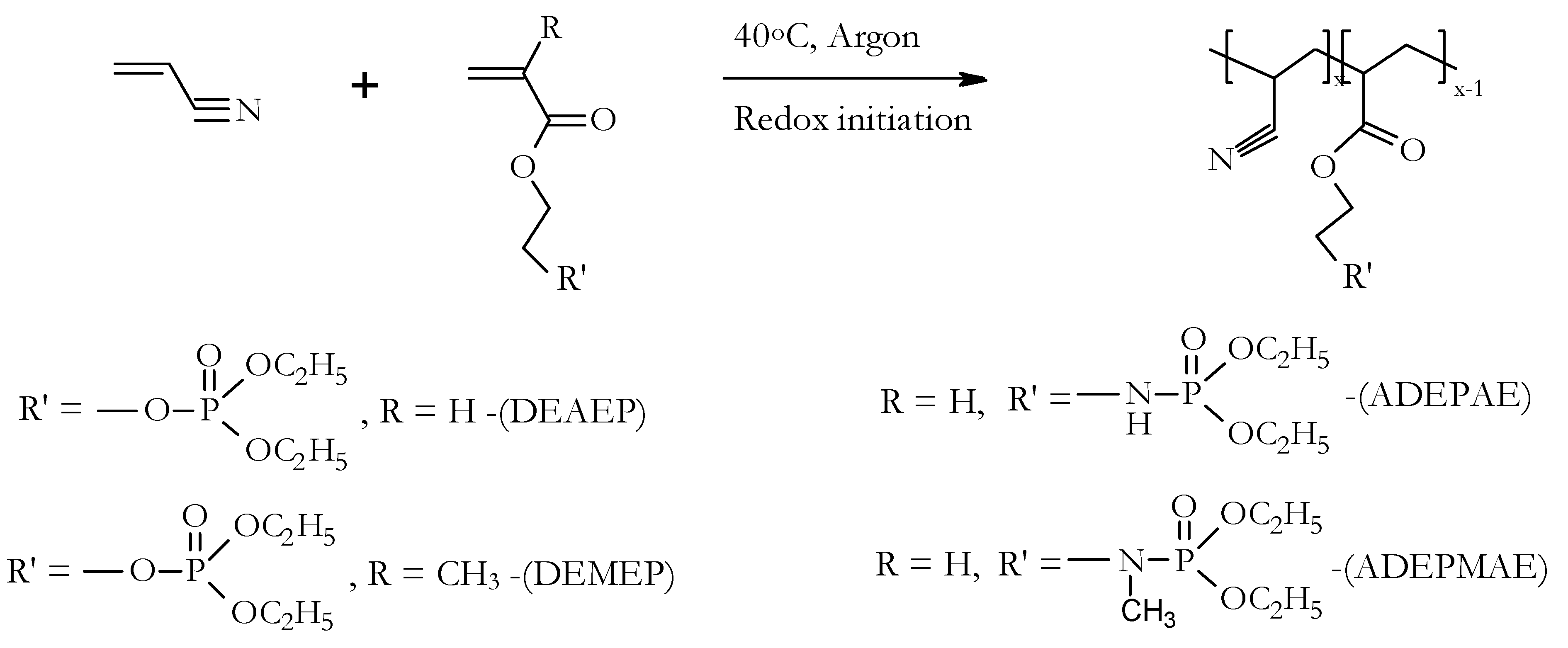
At first, the homopolymer of acrylonitrile, i.e., PAN, was synthesized in aqueous slurry with the view to be used as a control sample. In the current study, the polymerisation of AN was initiated by a redox pair consisting of ammonium persulfate and sodium metabisulfite (
Figure 4). A typical synthetic procedure for the preparation of PAN, or copolymers based on AN, by an aqueous slurry method is as follows: acrylonitrile (13 cm
3; 10.32 g), or an appropriate mixture of AN and comonomers as the case may be, was placed in a three-necked round bottomed flask, containing 250 cm
3 of deionised water, which was previously flushed with argon and maintained at 40 °C, fitted with a magnetic stirrer, a water condenser and a bubbler.
The mixture was stirred for ca. 30 min with argon bubbling through it. Sodium metabisulfite (1.0 g) dissolved in 25 cm3 of deionised water was added to the reaction mixture, followed by ammonium persulfate (0.35 g dissolved in 25 cm3 of deionised water). The argon inlet was withdrawn from the reaction mixture, and the polymerisation was allowed to proceed for 16 h under a blanket of argon. The aqueous slurry formed was filtered through a qualitative-grade filter paper, and the obtained polymer was washed with deionised water to remove traces of unreacted monomer(s). The polymer was dried in a vacuum oven at an elevated temperature (ca. 60 °C) to a constant weight before further examinations.
AN-based polymers incorporating β-CD were prepared using an in-situ polymerisation technique in an aqueous slurry medium (
Figure 5). In the case of polymer syntheses in the presence of β-CD, homogenisation of the additive was achieved by stirring β-CD in deionised water at ca. 40 °C for about 1 hr, prior to the addition of the monomer(s) and the initiator pair. The polymerisation procedure was similar to the homopolymerisation of AN described earlier. The recovered polymers were treated with hot water, filtered, and washed several times with hot water and methanol to remove the excess of β-CD. Physical mixtures of PAN and β-CD with various molar ratios of both components were prepared via the thorough grinding and mixing of two powdered materials (with exact weights) using a pestle and a mortar.
1H and 31P NMR spectra of starting materials, monomers, and AN-based polymers were recorded in deuterated solvents (CDCl3 or d6-DMSO) using a Bruker spectrometer, operating at 500 MHz (for protons) under ambient probe conditions. The spectra were processed using a proprietary software after being calibrated using the residual proton signals or the main carbon signals arising from the solvents. For 31P NMR spectra, 85% orthophosphoric acid was employed as an external calibrant.
Thermal gravimetric analyses (TGA) were performed on ca. 10 mg of samples using a Mettler Toledo TGA/SDTA851e at a heating rate of 10 °C min−1 in atmospheres of nitrogen and air, and optionally, oxygen, with temperatures ranging from 30 to 700 °C. Bomb calorimetric runs were carried out by using an IKA C200 bomb calorimeter (Belfast, UK). Measurements were conducted on samples, in the form of pellets, weighing about 0.5 g. The test for each sample was run in triplicate, and the calorific values displayed by the instrument were averaged over the three measurements. The ‘bomb’ cell was filled with oxygen up to 30 bars of pressure and subsequently ignited.
Pyrolysis combustion flow calorimetric measurements were carried out using a Fire Testing Technology Ltd. (FTT) (East Grinstead, UK) Micro Calorimeter, which works on the principle of oxygen consumption calorimetry [
24]. Accurately weighed (ca. 5 mg) solid polymeric samples were first heated to 900 °C at a constant heating rate of 1 K s
−1 in a stream of nitrogen flowing at a rate of 80 cm
3 min
−1. The obtained thermal degradation products were then mixed with a 20 cm
3 min
−1 stream of oxygen prior to entering a combustion chamber maintained at 900 °C. Each sample was run in triplicate and the obtained data were averaged over the three measurements. Plots of the heat release rates (HRR) against time were generated. In addition, the values of heat release capacities (kJg
−1K
−1), which could serve as reliable indicators of material flammability, were obtained.
The chemical structures and purities of used comonomers (i.e., DEAEP, DEMEP, ADEPAE and ADEPMAE), as well as their precursors, were confirmed by analysing corresponding
1H and
31P NMR spectra along with GC/MS results. Chromatograms and corresponding mass spectra were obtained using an Agilent 6890N gas chromatograph coupled with Agilent 5973N mass spectrometer. Agilent Technologies HP-Ultra 2 (25 m × 0.2 mm × 0.33 μm) column was used for GC. Chemical ionisation was carried out using methane with a mass spectrometer scanning mass range of 100–600 amu. For a number of polymeric systems, mole fractions of monomeric units present in copolymers were deduced from
1H NMR spectra following the algorithm previously described by us [
12,
16,
18].
The aqueous slurry route was found to be a relatively easy method for the preparation of polymeric products from AN, providing typical yields of (co-)polymers between 65 and 95% (
Figure 4,
Table 1). The compositions of copolymers (i.e., mole fractions of P- or P/N-containing monomeric units) were adjusted by changing the ratios of reacting comonomers and keeping the reaction time constant (approximately 16 h). However, different yields of resultant polymers were obtained in each case. The redox coupling of sodium metabisulfite and ammonium persulfate provided at least four radicals for the initiation of polymerisations [
24]. Furthermore, the ratio of metabisulfite to persulfate (which may affect both the molecular weight and the degree of oxidation of the products) remained constant for all polymerisation reactions carried out in this study (
Table 1). In appearance, the final polymeric products were white, powdery solids with no significant signs of discoloration. This indicated the absence of unwanted side reactions, such as the intramolecular cyclization of pendant nitrile groups, which could have led to extended conjugation and light absorption.
The results of TGA revealed noticeable differences between the general profiles of the thermograms (obtained in nitrogen, air and oxygen) for homopolymers and copolymers, even for those containing only minimal amounts of modifying groups (
Table 2). For example, the temperatures for the onset of thermal degradation (T
onset), the slopes of the main degradation steps and the amounts of char residue produced at 700 °C (in all three atmospheres) for a series of copolymers containing various amounts of either DEAEP or ADEPMAE monomeric units significantly differed from those of PAN.
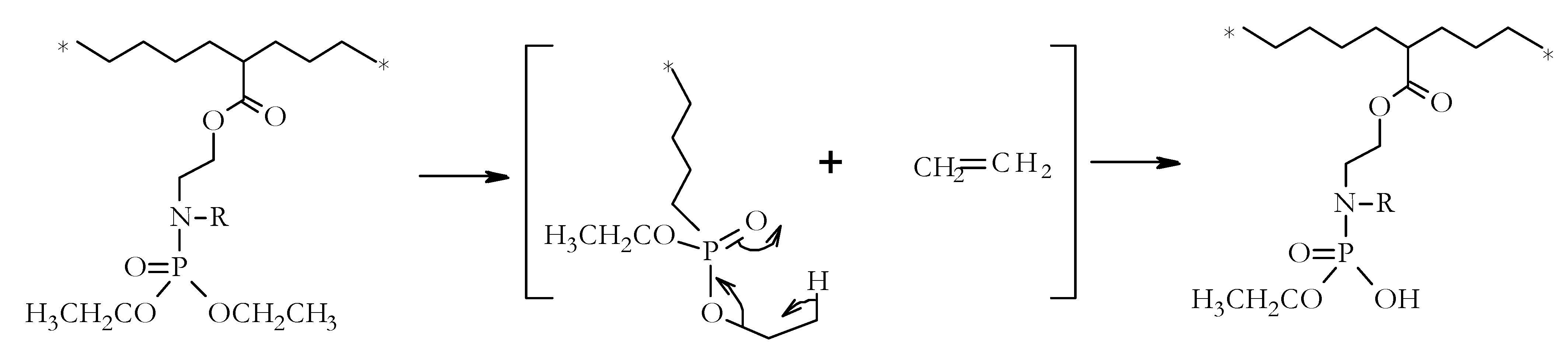
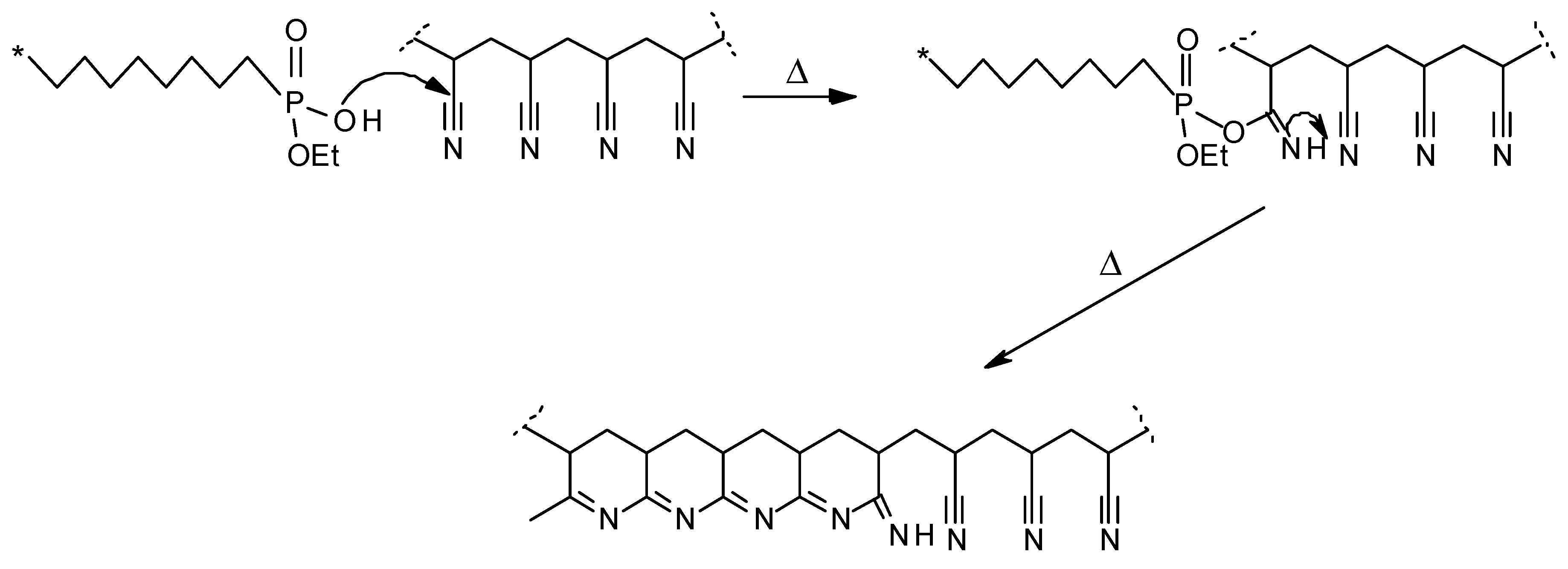
The lower values of T
onset, for example in a nitrogen atmosphere, in the case of copolymers, could be explained by an initial thermal cracking of the modifying groups (
Figure 6). The formation of the cyclic intermediate is entropically favoured owing to the elimination of ethene [
12,
14]. In addition, the calorific values (ΔH
comb) for copolymers of AN both with DEAEP and ADEPMAE were reduced compared to PAN (
Table 2). In general, the combustion (ΔH
comb) values were almost linearly reduced as the amount of covalently bound P in polymeric chains increased [
16].
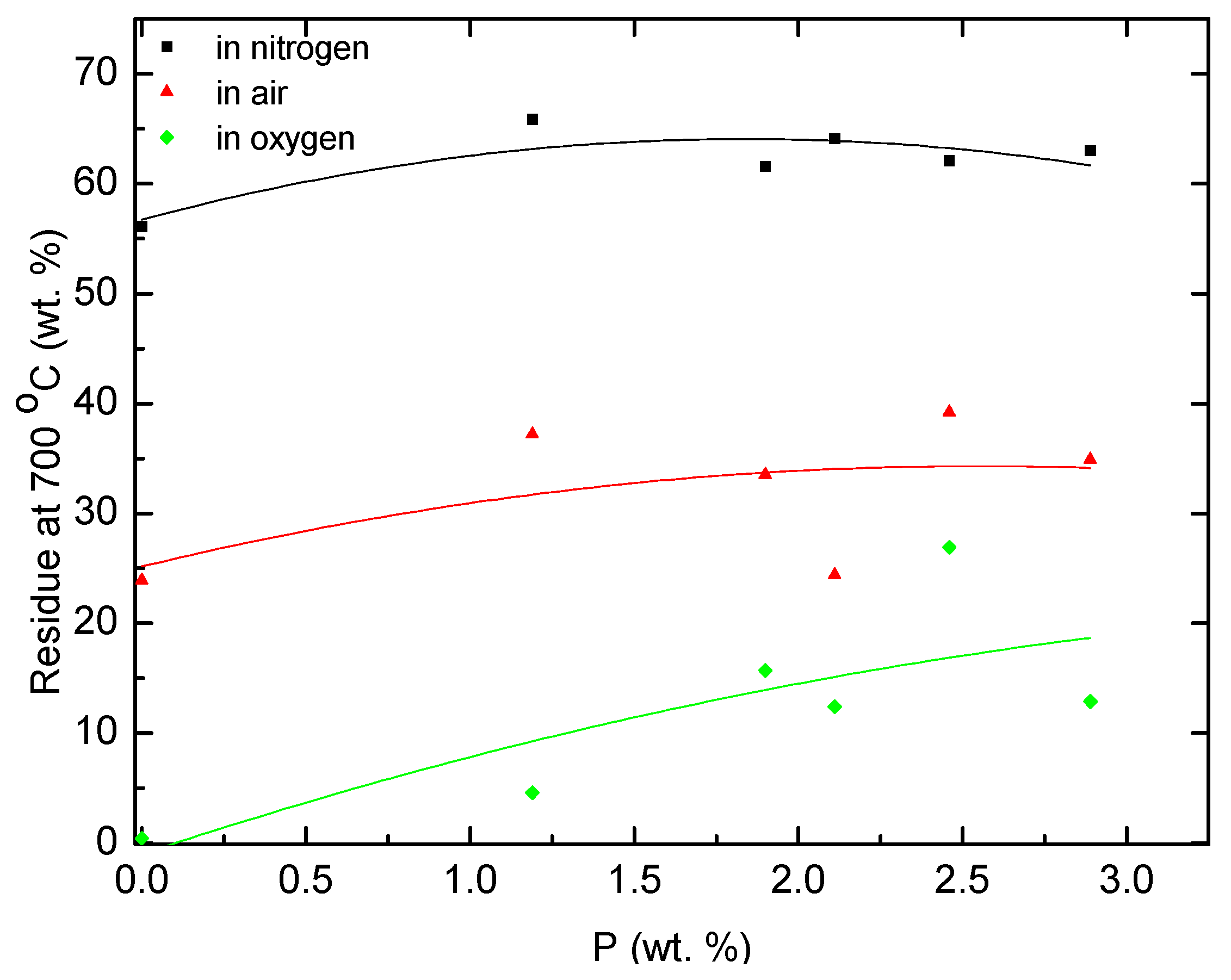
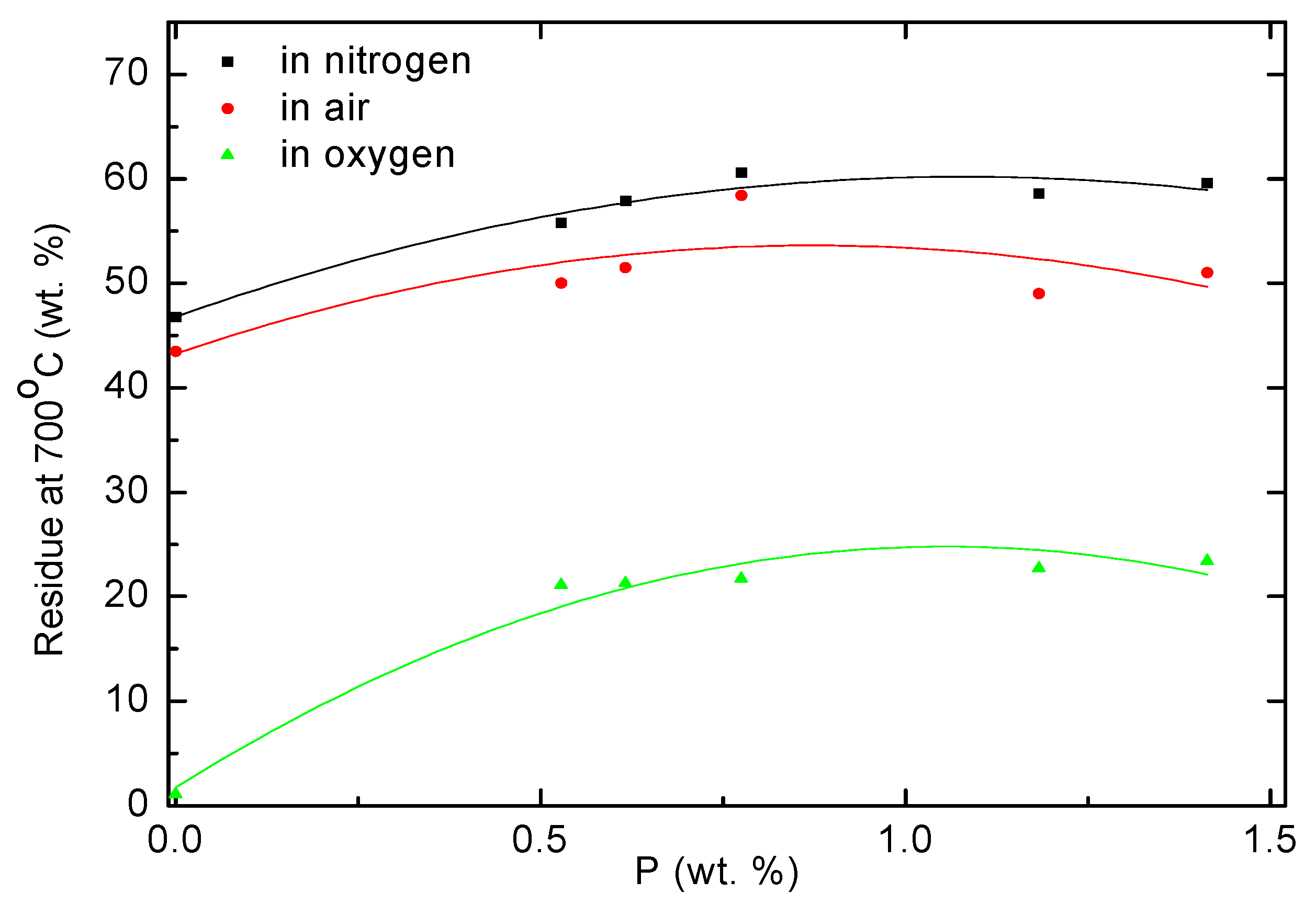
As seen from
Table 2 and plots shown in
Figure 7 and
Figure 8, the copolymers of AN generated significantly more char during TGA runs compared to PAN in all three atmospheres. This was also evident in the case of the degradation of polymeric products under an oxygen atmosphere. The char residues from the copolymers, formed under an oxygen atmosphere, seem to be somewhat resistant to further oxidative reactions at 700 °C. This might point towards the enhanced structural integrity and stability of char structures, and to a relatively more inert chemical nature that is less prone to oxidative processes. The phosphoric acid species might also present form a protective layer on the surface of the char thus preventing extended degrees of oxidative processes.
The phosphorus acid species produced during the early stages of the thermal degradation of copolymers (scheme shown in
Figure 6) can also act as nucleophilic centres, promoting the intramolecular cyclization of the nitrile groups in AN monomeric sequences (
Figure 9). In our previous research, a similar mechanistic pathway was shown to operate in similarly modified systems [
12,
17,
18]. Thus, the condensed-phase activity of the P- or P/N-containing modifying groups in PAN chains led to the increased production of char by enhancing the extent of intramolecular cyclization. This well-recognized cyclization scheme is the basis for making carbon fibres [
14].
The data collected from PCFC studies (
Table 3) also revealed the flame-retardant effects of modifying groups that were incorporated into PAN chains. This was particularly evident from the peak of heat release rate (PHRR), total heat released (THR) and heat release capacity (HRC) values.
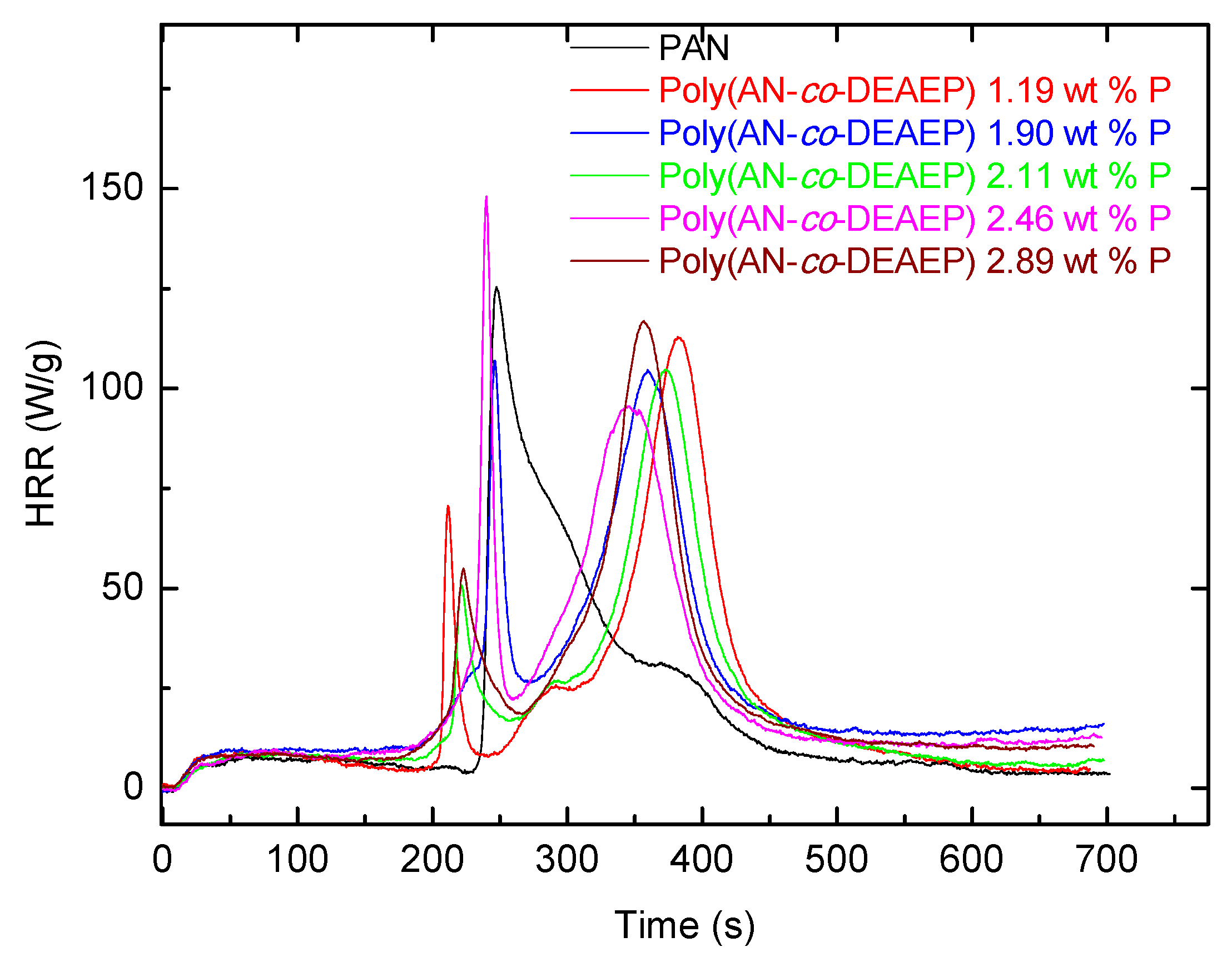
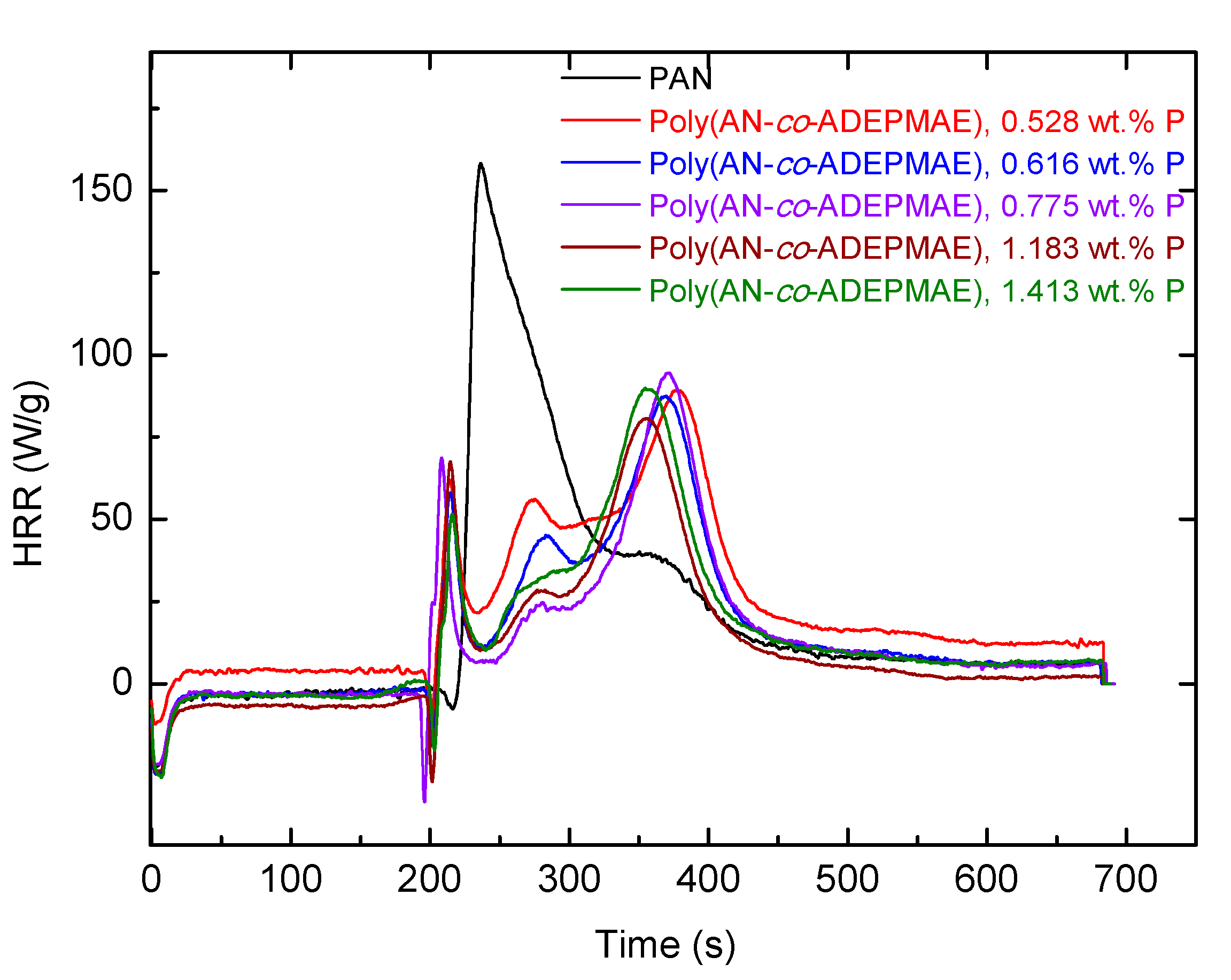
Indeed, the PHRR values, for example in AN/ADEPMAE copolymers, decreased by about 40–48% compared to the unmodified PAN. Furthermore, there were marked differences in the general profiles of the HRR vs. time curves. Evidently, the chemically modified systems showed an earlier shoulder peak, which can be attributed to the combustion of volatile fragments produced upon the thermal cracking of the P- or P/N-bearing groups. Subsequently, a shift and suppression of the main peak were also observed, which were thought to arise from the combustion-inhibitory effects exerted by the modifying groups. These effects can be also responsible for enhanced char production, as was observed in almost all cases compared to the virgin sample. For AN/DEAEP polymeric systems, the decrease in values of PHRR was less pronounced: a reduction of 10–34% was observed. An incorporation of a very nominal loading of P/N-containing monomeric units (0.009 mole fraction or 0.528 wt.% P) into polymeric chains leads to significant decreases in PHRR and HRC values by almost half (
Table 3). This can be attributed to possible phosphorus–nitrogen synergism operating in this modified system. Furthermore, the temperatures of peak heat release were substantially higher in the case of copolymers than PAN (by about 120–140 °C). The most significant improvements in flame retardance were recorded for copolymers containing a 0.059 mole fraction of DEAEP monomeric units (2.890 wt.% P) and a 0.027 mole fraction of ADEPMAE units (1.413 wt.% P). As for char production tendencies, gauged from the remaining residues (wt.%), the general trend registered from the TGA experiments correlates well with that observed from the PCFC runs. Heat releases associated with a possibly earlier induction of degradation, in the case of both types of copolymers, were also evident, as shown in the plots of
Figure 10 and
Figure 11.
Changing the balance of flame retardance to the condensed-phase mechanism is advantageous, as this could lead to less production of volatile and other combustible components. One of the ways of achieving this would be to encourage P-bearing moieties to act even more in the condensed phase via cooperativity. With this in mind, we prepared AN-based polymers incorporating β-CD, a char promoter itself, and hoped to achieve inclusion complexes (
Figure 5) [
25]. Some results obtained for AN-based polymers containing β-CD are provided in
Table 4. For example, in comparison to PAN, the value of residue formed in a nitrogen atmosphere (at 700 °C) during TGA runs increased by 15 wt.% when β-CD (11.0 g, 0.1 mole) was used for the preparation of inclusion complex. It should be noted that the residue obtained from TGA runs in nitrogen at 700 °C for a sample of pure β-CD did not exceed 10 wt.%. The inclusion of β-CD also led to a decrease in calorific values, from 31.004 kJg
−1 for PAN to 27.915 kJg
−1. However, it appears that there are no significant indications of any cooperativity between β-CD and P- or P/N-containing monomeric units. This might be due to the fact that the modifying side groups could pose a steric hindrance for copolymer chains to ‘waive’ through the cavity of cyclodextrin cages.
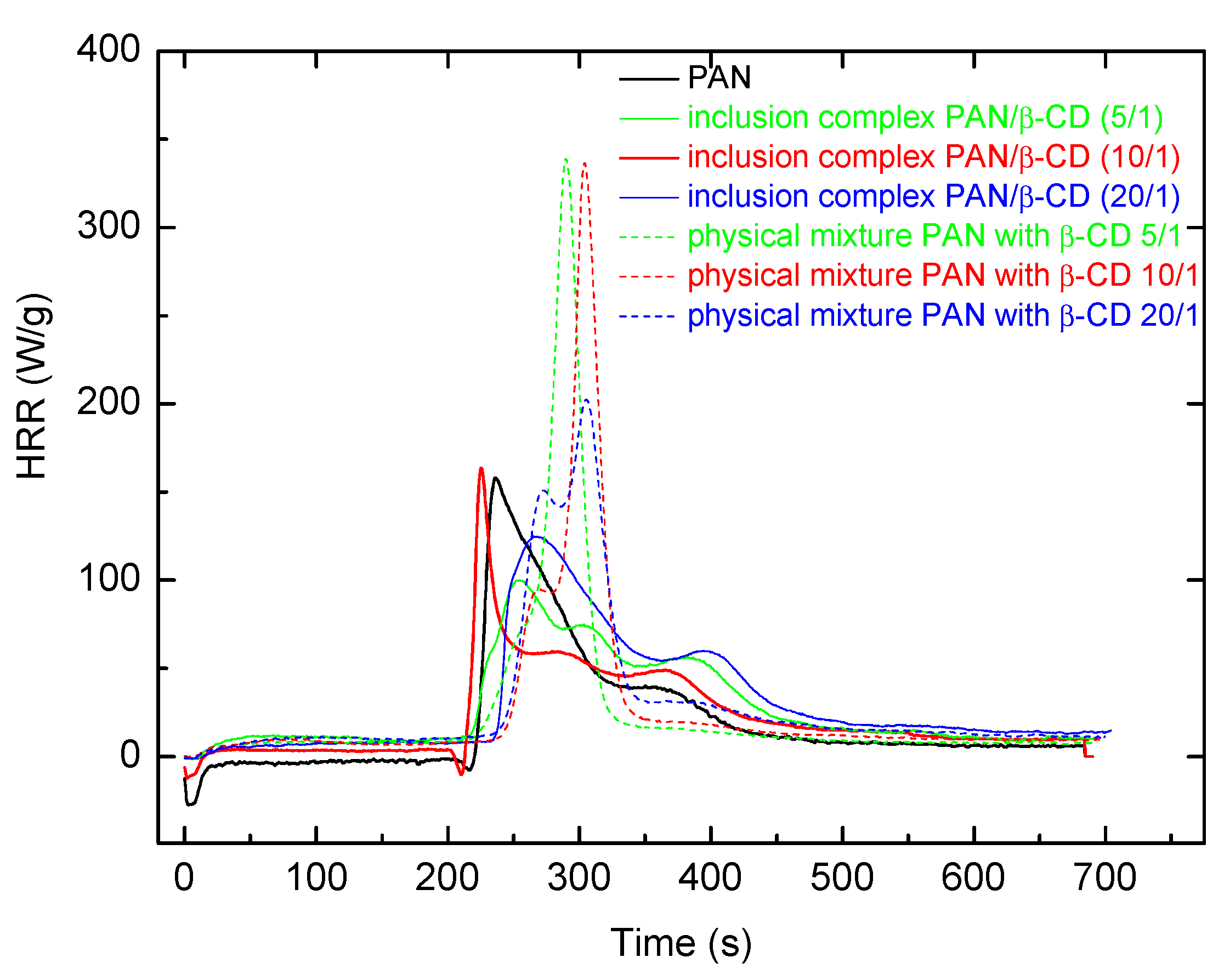
A comparison of PCFC data obtained for both inclusion complexes and physical mixtures of PAN with β-CD, where the components have similar molar ratios, also indicated that the inclusion complexes have the potential to improve flame retardance of PAN. Indeed, HR capacities were substantially reduced in the case of complexes of PAN with β-CD, whereas for physical mixtures of equimolar compositions, the trend was the opposite (
Table 5 and
Figure 12).
In summary, the chemical modification of AN-based polymers via the incorporation of DEAEP or ADEPMAE monomeric units resulted in significant improvements to their flame retardance, as gauged from increased TGA char yields and other measured combustion parameters. It is highly likely that the mechanism of flame retardance in these systems involves a significant condensed-phase activity initiated by the precursors obtained via the early thermal cracking of the P- or P/N-containing groups.
The use of phosphorus-based flame retardants in combination with other flame retardants, for example N-containing compounds, is most effective in situations where the combination proves synergistic. Although the results of our study on PAN modified with molecularly dispersed β-cyclodextrin are encouraging, the understanding of such potential synergistic effects is far from complete, and more fundamental research is required in this area. Some studies are already underway, including a detailed morphological, structural, and compositional evaluation of inclusion complexes and solid residues produced by the polymeric systems upon combustion. In addition, the exact nature of interaction of the modifying groups and the nanoscopic filler (β-CD) is yet to be identified.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}