
Total Syntheses of Chloropupukeananin and Its Related Natural Products


Abstract
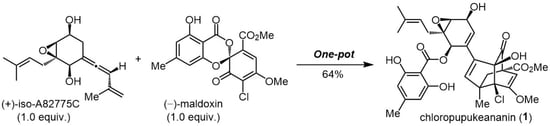
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Synthesis of the Biosynthetic Precursors of Chloropupukeananin
2.1. Pestheic Acid and Maldoxin
2.2. Iso-A82775C
3. Biomimetic Synthesis of Chloropupukeananin
3.1. Model Studies on the Intermolecular Diels–Alder Reaction
3.2. Syntheses of Chloropupukeananin and Its Related Natural Products
4. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Liu, S.; Jiang, L.; Chen, X.; Guo, L.; Che, Y. Chloropupukeananin, the first chlorinated pupukeanane derivative, and its precursors from Pestalotiopsis fici. Org. Lett. 2008, 10, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Takahashi, I.; Kawano, T.; Kimura, Y. Chloroisosulochrin, chloroisosulochrin dehydrate, and pestheic acid, plant growth regulators, produced by Pestalotiopsis theae. Z. Naturforsch. B 2001, 56, 797–803. [Google Scholar] [CrossRef]
- Ogawa, T.; Ando, K.; Aotani, Y.; Shinoda, K.; Tanaka, T.; Tsukuda, E.; Yoshida, M.; Matsuda, Y. RES-1214-1 and -2, novel non-peptidic endothelin type A receptor antagonists produced by Pestalotiopsis sp. J. Antibiot. 1995, 48, 1401–1406. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Adeboya, M.O.; Edwards, R.L.; Lassøe, T.; Maitland, D.J.; Shields, L.; Whalley, A.J.S. Metabolites of the higher fungi. Part 29. Maldoxin, maldoxone, dihydromaldoxin, isodihydromaldoxin and dechlorodihydromaldoxin. A spirocyclohexadienone, a depsidone and three diphenyl ethers: Keys in the depsidone biosynthetic pathway from a member of the fungus genus Xylaria. J. Chem. Soc. Perkin Trans. 1 1996, 1419–1425. [Google Scholar] [CrossRef]
- Burreson, B.J.; Scheuer, P.J.; Finer, J.S.; Clardy, J. 9-Isocyanopupukeanane, a marine invertebrate allomone with a new sesquiterpene skeleton. J. Am. Chem. Soc. 1975, 97, 4763–4764. [Google Scholar] [CrossRef]
- Hagadone, M.R.; Burreson, B.J.; Scheuer, P.J.; Finer, J.S.; Clardy, J. Defense allomones of the nudibranch Phyllidia varicosa Lamarck 1801. Helv. Chim. Acta. 1979, 62, 2484–2494. [Google Scholar] [CrossRef]
- Fusetani, N.; Wolstenholme, H.J.; Matsunaga, S. Co-occurrence of 9-isocyanopupukeanane and its C-9 epimer in the nudibranch Phyllidia bourguini. Tetrahedron Lett. 1990, 31, 5623–5624. [Google Scholar] [CrossRef]
- He, H.; Salva, J.; Catalos, R.F.; Faulkner, D.J. Sesquiterpene thiocyanates and isothiocyanates from Axinyssa aplysinoides. J. Org. Chem. 1992, 57, 3191–3194. [Google Scholar] [CrossRef]
- Simpson, J.S.; Garson, M.J.; Hooper, J.N.A.; Cline, E.I.; Angerhofer, C.K. Terpene metabolites from the tropical. Marine sponge Axinyssa sp. nov. Aust. J. Chem. 1997, 50, 1123–1127. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Liu, S.; Zheng, Z.; Chen, X.; Zhang, H.; Guo, L.; Che, Y. Chloropestolide A, an antitumor metabolite with an unprecedented spiroketal skeleton from Pestalotiopsis fici. Org. Lett. 2009, 11, 2836–2839. [Google Scholar] [CrossRef]
- Suzuki, T.; Kobayashi, S. Concise approach to pupukeanane skeleton: Synthetic study of chloropupukeananin. Org. Lett. 2010, 12, 2920–2923. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Niu, S.; Lu, X.; Chen, X.; Zhang, H.; Guo, L.; Che, Y. Unique metabolites of Pestalotiopsis fici suggest a biosynthetic hypothesis involving a Diels–Alder reaction and then mechanistic diversification. Chem. Commun. 2010, 46, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Bruhn, T.; Guo, L.; Gotz, D.C.; Brun, R.; Stich, A.; Che, Y.; Bringmann, G. Chloropupukeanolides C-E: Cytotoxic pupukeanane chlorides with a spiroketal skeleton from Pestalotiopsis fici. Chem. Eur. J. 2011, 17, 2604–2613. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, L.; Cao, Y.; Guo, L.; Liu, G.; Che, Y. Spiroketals of Pestalotiopsis fici provide evidence for a biosynthetic hypothesis involving diversified Diels-Alder reaction cascades. J. Org. Chem. 2013, 78, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Han, Y.; Xiao, J.; Li, L.; Guo, L.; Jiang, X.; Kong, L.; Che, Y. Chlorotheolides A and B, spiroketals generated via diels-alder reactions in the endophytic fungus Pestalotiopsis theae. J. Nat. Prod. 2016, 79, 2616–2623. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, L.; Guan, F.; Li, E.; Jin, J.; Li, J.; Che, Y.; Liu, G. Characterization of a prenyltransferase for iso-A82775C biosynthesis and generation of new congeners of chloropestolides. ACS Chem. Biol. 2018, 13, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Klas, K.; Tsukamoto, S.; Sherman, D.H.; Williams, R.M. Natural Diels-Alderases: Elusive and irresistable. J. Org. Chem. 2015, 80, 11672–11685. [Google Scholar] [CrossRef]
- Minami, A.; Oikawa, H. Recent advances of Diels-Alderases involved in natural product biosynthesis. J. Antibiot. 2016, 69, 500–506. [Google Scholar] [CrossRef]
- Jeon, B.S.; Wang, S.A.; Ruszczycky, M.W.; Liu, H.W. Natural [4+2]-Cyclases. Chem. Rev. 2017, 117, 5367–5388. [Google Scholar] [CrossRef]
- Jamieson, C.S.; Ohashi, M.; Liu, F.; Tang, Y.; Houk, K.N. The expanding world of biosynthetic pericyclases: Cooperation of experiment and theory for discovery. Nat. Prod. Rep. 2019, 36, 698–713. [Google Scholar] [CrossRef]
- Gouverneur, V.E.; Houk, K.N.; de Pascual-Teresa, B.; Beno, B.; Janda, K.D.; Lerner, R.A. Control of the exo and endo pathways of the Diels-Alder reaction by antibody catalysis. Science 1993, 262, 204–208. [Google Scholar] [CrossRef]
- Siegel, J.B.; Zanghellini, A.; Lovick, H.M.; Kiss, G.; Lambert, A.R.; St Clair, J.L.; Gallaher, J.L.; Hilvert, D.; Gelb, M.H.; Stoddard, B.L.; et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 2010, 329, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Palma, A.; Artelsmair, M.; Wu, G.; Lu, X.; Barrow, S.J.; Uddin, N.; Rosta, E.; Masson, E.; Scherman, O.A. Cucurbit[7]uril as a supra-molecular artificial enzyme for Diels-Alder reactions. Angew. Chem. Int. Ed. 2017, 56, 15688–15692. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Su, C.; Du, X.; Wang, R.; Chen, S.; Zhou, Y.; Liu, C.; Liu, X.; Tian, R.; Zhang, L.; et al. FAD-dependent enzyme-catalysed intermolecular [4+2] cycloaddition in natural product biosynthesis. Nat. Chem. 2020, 12, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Basler, S.; Studer, S.; Zou, Y.; Mori, T.; Ota, Y.; Camus, A.; Bunzel, H.A.; Helgeson, R.C.; Houk, K.N.; Jimenez-Oses, G.; et al. Efficient Lewis acid catalysis of an abiological reaction in a de novo protein scaffold. Nat. Chem. 2021, 13, 231–235. [Google Scholar] [CrossRef]
- Beach, W.F.; Richards, J.H. The structure and biosynthesis of nidulin. J. Org. Chem. 1963, 28, 2746–2751. [Google Scholar] [CrossRef]
- Hendrickson, J.B.; Ramsay, M.V.J.; Kelly, T.R. New synthesis of depsidones. Diploicin and gangaleoidin. J. Am. Chem. Soc. 1972, 94, 6834–6843. [Google Scholar] [CrossRef]
- Yu, M.; Snider, B.B. Syntheses of chloroisosulochrin and isosulochrin and biomimetic elaboration to maldoxin, maldoxone, dihydromaldoxin, and dechlorodihydromaldoxin. Org. Lett. 2011, 13, 4224–4227. [Google Scholar] [CrossRef]
- Smith, K.; Butters, M.; Nay, B. High ortho-selectivity in the chlorination of phenols with N-chlorodialkylamines in the presence of silica. Tetrahedron Lett. 1988, 29, 1319–1322. [Google Scholar] [CrossRef]
- Gnaim, J.M.; Sheldon, R.A. Highly regioselective ortho-chlorination of phenol with sulfuryl chloride in the presence of amines. Tetrahedron Lett. 1995, 36, 3893–3896. [Google Scholar] [CrossRef]
- Gnaim, J.M.; Sheldon, R.A. Selective ortho-chlorination of phenol using sulfuryl chloride in the presence of t-butylaminomethyl polystyrene as a heterogeneous amine catalyst. Tetrahedron Lett. 2004, 45, 8471–8473. [Google Scholar] [CrossRef]
- Lambert, G.J.; Duffley, R.P.; Dalzell, H.C.; Razdan, R.K. Regioselective aromatic hydroxylation. An oxidative reaction of arylcopper(I) and lithium diarylcopper(I) ate complexes. J. Org. Chem. 1982, 47, 3350–3353. [Google Scholar] [CrossRef]
- Sala, T.; Sargent, M.V. Depsidone synthesis. Part 16. Benzophenone–grisa-3′,5′-diene-2′,3-dione–depsidone interconversion: A new theory of depsidone biosynthesis. J. Chem. Soc. Perkin Trans. 1 1981, 855–869. [Google Scholar] [CrossRef]
- Coomber, M.F.; Sargent, M.V.; Skelton, B.W.; White, A.H. Depsidone synthesis. Part 24. The synthesis of epiphorellic acid 2. A pseudodepsidone and X-ray crystal structure of a grisadienedione epoxide. J. Chem. Soc. Perkin Trans. 1 1989, 441–448. [Google Scholar] [CrossRef]
- Pulgarin, C.; Tabbachi, R. Synthèse du virensate de méthyle. Helv. Chim. Acta 1989, 72, 1061–1065. [Google Scholar] [CrossRef]
- Suzuki, T.; Watanabe, S.; Uyanik, M.; Ishihara, K.; Kobayashi, S.; Tanino, K. Asymmetric total synthesis of (−)-maldoxin, a common biosynthetic ancestor of the chloropupukeananin family. Org. Lett. 2018, 20, 3919–3922. [Google Scholar] [CrossRef]
- Churcher, I.; Hallett, D.; Magnus, P. Synthesis of the antitumor agent aglycon (±)-calicheamicinone using an o-quinone monoketal strategy. J. Am. Chem. Soc. 1998, 120, 10350–10358. [Google Scholar] [CrossRef]
- Suzuki, T.; Watanabe, S.; Ikeda, W.; Kobayashi, S.; Tanino, K. Biomimetic total syntheses of (+)-chloropupukeananin, (−)-chloropupukeanolide D, and chloropestolides. J. Org. Chem. 2021, 86, 15597–15605. [Google Scholar] [CrossRef]
- Kita, Y.; Tohma, H.; Hatanaka, K.; Takada, T.; Fujita, S.; Mitoh, S.; Sakurai, H.; Oka, S. Hypervalent iodine-induced nucleophilic substitution of para-substituted phenol ethers. Generation of cation radicals as reactive intermediates. J. Am. Chem. Soc. 1994, 116, 3684–3691. [Google Scholar] [CrossRef]
- Viswanadh, N.; Ghotekar, G.S.; Thoke, M.B.; Velayudham, R.; Shaikh, A.C.; Karthikeyan, M.; Muthukrishnan, M. Transition metal free regio-selective C–H hydroxylation of chromanones towards the synthesis of hydroxyl-chromanones using PhI(OAc)2 as the oxidant. Chem. Commun. 2018, 54, 2252–2255. [Google Scholar] [CrossRef]
- Koyama, H.; Sahoo, S.P.; Yang, G.X.-Q.; Miller, D.J. P38 Kinase Inhibiting Agents. WIPO (PCT) Patent WO2010129208A1, 11 November 2010. [Google Scholar]
- Ito, Y.; Ohmori, K.; Suzuki, K. Annulation approach to doubly linked (A-type) oligocatechins: Syntheses of (+)-procyanidin A2 and (+)-cinnamtannin B1. Angew. Chem. Int. Ed. 2014, 53, 10129–10133. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Ohmori, K.; Suzuki, K. Stereocontrolled total syntheses of (−)-rotenone and (−)-dalpanol by 1,2-rearrangement and SNAr oxycyclizations. Angew. Chem. Int. Ed. 2017, 56, 182–187. [Google Scholar] [CrossRef]
- Uyanik, M.; Ishihara, K. Asymmetric oxidative dearomatization reaction. In Asymmetric Dearomatization Reactions; You, S.-L., Ed.; John Wiley & Sons: Weinheim, Germany, 2016; pp. 129–151. [Google Scholar]
- Wu, W.T.; Zhang, L.; You, S.-L. Asymmetric dearomatization of phenols. Chem. Soc. Rev. 2016, 45, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, G.; Hong, L.; Wang, R. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Org. Biomol. Chem. 2016, 14, 2164–2176. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Yasui, T.; Ishihara, K. Hydrogen bonding and alcohol effects in asymmetric hypervalent iodine catalysis: Enantioselective oxidative dearomatization of phenols. Angew. Chem. Int. Ed. 2013, 52, 9215–9218. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Sasakura, N.; Mizuno, M.; Ishihara, K. Enantioselective synthesis of masked benzoquinones using designer chiral hypervalent organoiodine(III) catalysis. ACS Catal. 2017, 7, 872–876. [Google Scholar] [CrossRef]
- Uyanik, M.; Yasui, T.; Ishihara, K. Chiral hypervalent organoiodine-catalyzed enantioselective oxidative spirolactonization of naphthol derivatives. J. Org. Chem. 2017, 82, 11946–11953. [Google Scholar] [CrossRef]
- Sanson, D.R.; Gracz, H.; Tempesta, M.S.; Fukuda, D.S.; Nakatsukasa, W.M.; Sands, T.H.; Baker, P.J.; Mynderse, J.S. A82775B and A82775C, novel metabolites of an unknown fungus of the order sphaeropsidales. Tetrahedron 1991, 47, 3633–3644. [Google Scholar] [CrossRef]
- Elsebai, M.F.; Kehraus, S.; Gütschow, M.; König, G.M. Spartinoxide, a new enantiomer of A82775C with inhibitory activity toward HLE from the marine-derived fungus Phaeosphaeria spartinae. Nat. Prod. Commun. 2010, 5, 1071–1076. [Google Scholar] [CrossRef]
- Liu, L.; Liu, S.; Chen, X.; Guo, L.; Che, Y. Pestalofones A–E, bioactive cyclohexanone derivatives from the plant endophytic fungus Pestalotiopsis Fici. Bioorg. Med. Chem. 2009, 17, 606–613. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, G.-D.; Zou, J.; He, R.-R.; Qin, S.-Y.; Hu, D.; Li, G.-Q.; Guo, L.-D.; Yao, X.-S.; Gao, H. Dimericbiscognienyne A: A meroterpenoid dimer from Biscogniauxia sp. with new skeleton and its activity. Org. Lett. 2017, 19, 38–41. [Google Scholar] [CrossRef]
- Suzuki, T.; Watanabe, S.; Kobayashi, S.; Tanino, K. Enantioselective total synthesis of (+)-iso-A82775C, a proposed biosynthetic precursor of chloropupukeananin. Org. Lett. 2017, 19, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Iwagawa, T.; Nakatani, M. A base catalyzed Diels-Alder reaction of 3-hydroxy-2-pyrone. Tetrahedron Lett. 1995, 36, 5939–5942. [Google Scholar] [CrossRef]
- Okamura, H.; Nakamura, Y.; Iwagawa, T.; Nakatani, M. Asymmetric base-catalyzed Diels-Alder reaction of 3-hydroxy-2-pyrone with N-methylmaleimide. Chem. Lett. 1996, 25, 193–194. [Google Scholar] [CrossRef]
- Shimizu, H.; Okamura, H.; Iwagawa, T.; Nakatani, M. Asymmetric synthesis of (−)- and (+)-eutipoxide B using a base-catalyzed Diels–Alder reaction. Tetrahedron 2001, 57, 1903–1908. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Wang, Y.-Q.; Liu, Y.; Foxman, B.M.; Deng, L. Asymmetric Diels−Alder reactions of 2-pyrones with a bifunctional organic catalyst. J. Am. Chem. Soc. 2007, 129, 6364–6365. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, F.; Lewis, J.; Heroux, K.; Cohen, S. Characterization and evaluation of pyrone and tropolone chelators for use in metalloprotein inhibitors. Inorg. Chim. Acta 2007, 360, 264–272. [Google Scholar] [CrossRef]
- Mayer, R. Pseudo-α-Tropolone vom typ des 3-hydroxy-α-pyrons, 3-hydroxy-γ-pyrons und der 3-hydroxy-thia-pyrone. Chem. Ber. 1957, 90, 2369–2372. [Google Scholar] [CrossRef]
- Saksena, A.K.; Mangiaracina, P. Recent studies on veratrum alkaloids: A new reaction of sodium triacetoxyborohydride [NaBH(OAc)3]. Tetrahedron Lett. 1983, 24, 273–276. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Verhoeven, T.R. Metal-catalyzed, highly selective oxygenations of olefins and acetylenes with tert-butyl hydroperoxide. Practical considerations and mechanisms. Aldrichimica Acta 1979, 12, 63–74. [Google Scholar]
- Riveiros, R.; Rodríguez, D.; Pérez Sestelo, J.; Sarandeses, L.A. Palladium-catalyzed cross-coupling reaction of triorganoindium reagents with propargylic esters. Org. Lett. 2006, 8, 1403–1406. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kim, M.J.; Chung, G.; Lee, H.-Y.; Han, S. (+)-Dimericbiscognienyne A: Total synthesis and mechanistic investigations of the key heterodimerization. Org. Lett. 2018, 20, 6886–6890. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Pan, S.C. Total synthesis of the novel antifungal agent (±)-jesterone. Org. Lett. 2004, 6, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Kumar, Y.S.; Das, M. A de novo Diels–Alder strategy toward the novel pentacyclic natural product fluostatin C: A concise synthesis of 6-deoxyfluostatin C. Tetrahedron Lett. 2011, 52, 3505–3508. [Google Scholar] [CrossRef]
- Kim, G.; Kim, T.; Han, S. Total synthesis of (+)-pestalofone A and (+)-iso-A82775C. J. Org. Chem. 2020, 85, 6815–6821. [Google Scholar] [CrossRef]
- Line, N.J.; Witherspoon, B.P.; Hancock, E.N.; Brown, M.K. Synthesis of ent-[3]-ladderanol: Development and application of intramolecular chirality transfer [2+2] cycloadditions of allenic ketones and alkenes. J. Am. Chem. Soc. 2017, 139, 14392–14395. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Snider, B.B. Diels–Alder reaction of maldoxin with an isopropenylallene. Tetrahedron 2011, 67, 9473–9478. [Google Scholar] [CrossRef]
- Suzuki, T.; Miyajima, Y.; Suzuki, K.; Iwakiri, K.; Koshimizu, M.; Hirai, G.; Sodeoka, M.; Kobayashi, S. Unexpected Diels-Alder/carbonyl-ene cascade toward the biomimetic synthesis of chloropupukeananin. Org. Lett. 2013, 15, 1748–1751. [Google Scholar] [CrossRef]
- Pinault, M.; Frangin, Y.; Genet, J.P.; Zamarlik, H. Total synthesis of siccayne. Synthesis 1990, 935–937. [Google Scholar] [CrossRef]
- Catalán, J.; Palomar, J.; Díaz, C.; de Paz, J.L.G. On solvent basicity: Analysis of the SB scale. J. Phys. Chem. A 1997, 101, 5183–5189. [Google Scholar] [CrossRef]
- Catalán, J.; Díaz, C.; López, V.; Pérez, P.; De Paz, J.-L.; Rodríguez, J.G. A Generalized solvent basicity scale: The solvatochromism of 5-nitroindoline and its homomorph 1-methyl-5-nitroindoline. Liebigs Ann. Chem. 1996, 1996, 1785–1794. [Google Scholar] [CrossRef]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, T. Total Syntheses of Chloropupukeananin and Its Related Natural Products. Organics 2022, 3, 304-319. https://doi.org/10.3390/org3030023
Suzuki T. Total Syntheses of Chloropupukeananin and Its Related Natural Products. Organics. 2022; 3(3):304-319. https://doi.org/10.3390/org3030023
Chicago/Turabian StyleSuzuki, Takahiro. 2022. "Total Syntheses of Chloropupukeananin and Its Related Natural Products" Organics 3, no. 3: 304-319. https://doi.org/10.3390/org3030023
APA StyleSuzuki, T. (2022). Total Syntheses of Chloropupukeananin and Its Related Natural Products. Organics, 3(3), 304-319. https://doi.org/10.3390/org3030023