1. Introduction
Bacteriochlorophylls and their free base analogues (bacteriopheophytins) are the chief chromophores for solar light harvesting, energy transduction and charge separation in anoxygenic photosynthetic bacteria (
Chart 1) [
1]. Bacteriochlorophylls are distinguished from the other members of the tetrapyrrole family by the saturation of the chromophore and the resulting absorption in the near infrared region. Our particular interest is to employ synthetic, tailored bacteriochlorins to probe energy-transduction processes akin to those carried out by native bacteriochlorophylls in photosynthetic systems. One objective is to understand the structural origin and photophysical consequences of the vibronic properties of the macrocycles. Thus, a molecular design objective is to create a systematic collection of synthetic bacteriochlorins ranging from unsubstituted macrocycles to those bearing the full structural features of the native pigments. To probe vibronic properties, installation of isotopes at designated sites both in the core skeleton of the macrocycle as well as in substituents at the perimeter of the macrocycle is essential. This work continues a longstanding strategy in tetrapyrrole science, where the introduction of isotopic atoms—creating isotopologues and isotopomers—has been relied upon to probe physicochemical properties (as well as the biosynthesis) of tetrapyrrole macrocycles.
There are several distinct methods for the preparation of isotopically substituted tetrapyrrole macrocycles. Such methods have been applied extensively with porphyrins and chlorins [
2] but less so with bacteriochlorins. Representative strategies for access to isotopically substituted bacteriochlorins are shown in
Scheme 1 and outlined as follows.
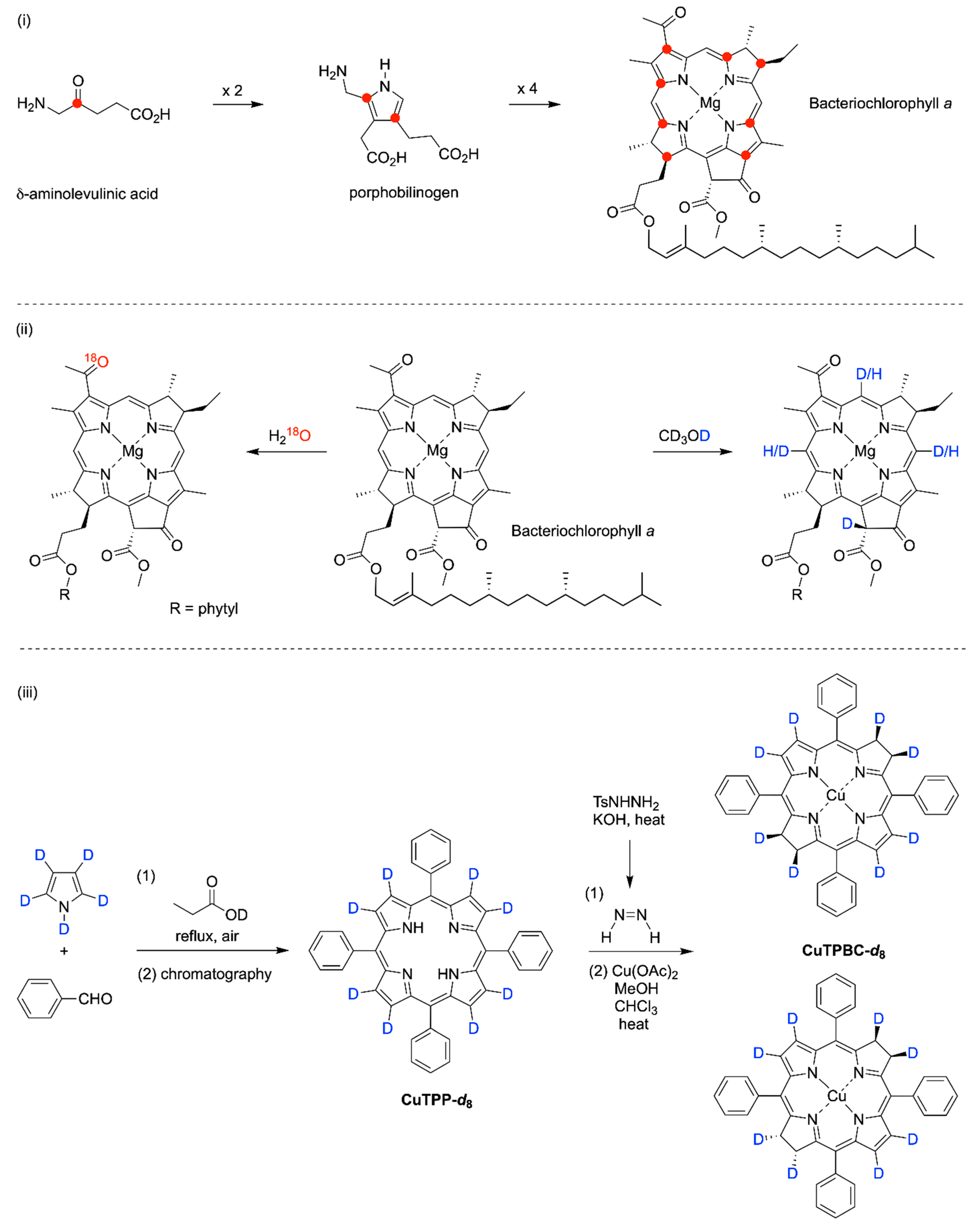
(i) Biosynthesis. Biosynthetic incorporation of isotopic atoms can be achieved by use of isotopically enriched precursors, such as
15N-ammonium,
13C-carbonate, H
218O, and/or
2H
2O, which afford global isotopic enrichment [
3,
4,
5,
6,
7,
8,
9,
10,
11]. Alternatively, use of more advanced precursors with site-specific isotopic substitution affords greater selectivity [
5,
12,
13,
14,
15,
16,
17]. Incorporation of
26Mg as the metal chelate also has been achieved via biosynthesis with use of
26MgSO
4 [
18]. Both approaches—global enrichment and site-specific incorporation—have been employed to elucidate biosynthetic pathways and for spectroscopic studies. An example of the latter makes use of δ-aminolevulinic acid [
19] and porphobilinogen [
20] with controlled isotopic incorporation at numerous sites owing to the work of Lugtenburg and coworkers. It warrants mention, however, that eight molecules of δ-aminolevulinic acid combine to form a native macrocycle, hence the presence of a single
13C or
15N-atom in δ-aminolevulinic acid typically results in four or eight such atoms in the resulting macrocycle; moreover, the pattern is irregular given the nature of the biosynthetic pathway, as shown in the example [
15];
(ii) Derivatization of intact macrocycles. The modification of intact macrocycles is generally limited to
2H/
1H exchange and
18O/
16O exchange at the macrocycle perimeter, as shown in the examples. The exchange of
2H/
1H with native bacteriochlorophylls occurs readily at the 13
2-position but more slowly at the three meso-positions [
21,
22]. The
18O/
16O exchange occurs selectively at the 3-acetyl position versus at the two esters [
23]; and
(iii) De novo synthesis. The total synthesis of chlorophylls and bacteriochlorophylls—converting small molecule precursors to the native macrocycles—has not been achieved, prompting reliance on the synthesis of model compounds to probe the effects of isotopic incorporation [
24,
25,
26,
27,
28,
29]. An example is provided of preparation of copper(II)
meso-tetraphenylbacteriochlorin containing eight
2H atoms at the β-positions (
CuTPBC-d8) [
25,
30]. The synthesis relies on deuteriation of the porphyrin, which upon reduction with diimide affords the corresponding bacteriochlorin sample as a mixture of diastereomers. Milder methods have been developed for formation of deuteriated porphyrin precursors [
31,
32]. Bacteriochlorins prepared by hydrogenation of a porphyrin are vulnerable to oxidation causing reversion to the less reduced macrocycles (chlorin or porphyrin).
A family of gem-dimethyl-substituted bacteriochlorins bearing pairwise incorporation of
13C or
15N atoms has been prepared with the rationale to probe vibronic properties of the resulting macrocycles [
33]. The gem-dimethyl groups impart stability toward adventitious oxidation and thereby enable physicochemical studies that are less accessible with the native structures and with hydroporphyrins derived by hydrogenation of porphyrins. The self-condensation of an isotopically substituted dihydrodipyrrin causes symmetrical location (i.e., “double-stamping”) of the isotopes in the bacteriochlorin macrocycle [
33]. The general synthetic route and the resulting isotopically substituted bacteriochlorins are shown in
Chart 2. The isotopically substituted starting materials of the de novo synthesis included
13C substituted reagents (formaldehyde,
N,N-dimethylformamide, triethylorthoformate, and KCN) and
15N-substituted reagents (pyrrole and nitromethane). The collection of isotopically substituted bacteriochlorins was examined by (1)
1H,
13C, and
15N NMR spectroscopy to unambiguously assign all resonances [
34]; (2) resonance Raman spectroscopy of the copper chelates to identify vibronically active modes [
35]; and (3) electron paramagnetic spectroscopy (of the cation radicals of the zinc chelates) to identify spin-density distributions [
35].
A key theme in this work has been to employ “sparsely substituted” macrocycles so as to probe the fundamental properties of the core chromophore unadulterated by the effects of peripheral substituents. Thus, the isotopic substitutions in the macrocycles shown in
Chart 2 were entirely in the core skeleton. To extend the studies a step beyond the structures shown in
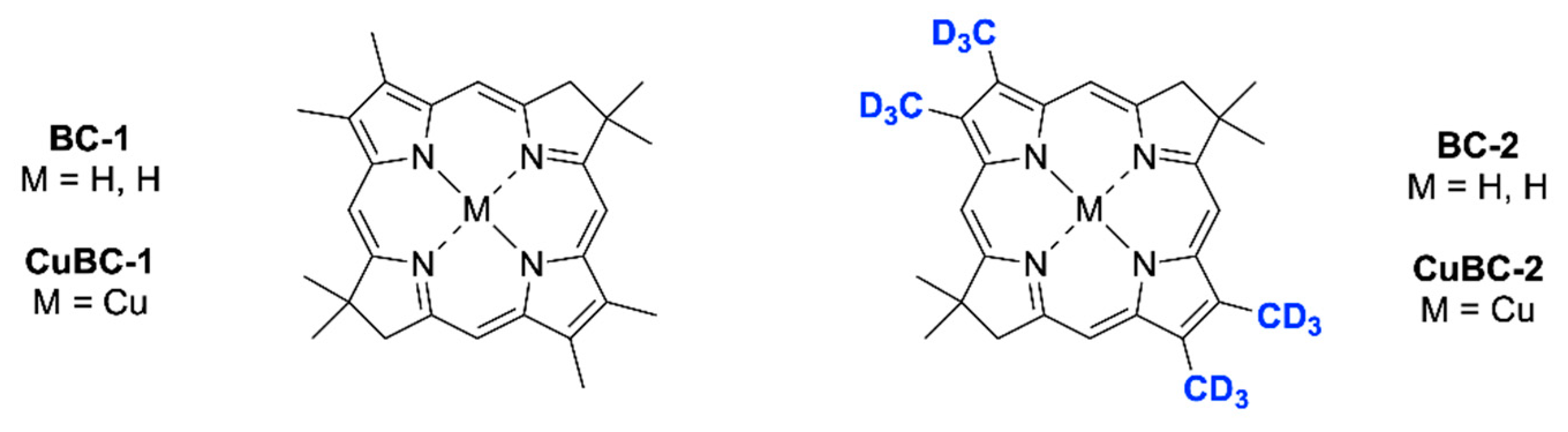
Chart 2, we sought to prepare bacteriochlorins bearing deuterium substitution of peripheral alkyl groups. Several years ago, we synthesized a bacteriochlorin bearing four methyl groups (
BC-1), one at each β-pyrrole position, and found that the resonance Raman spectrum was relatively rich in vibronic modes relative to that of the unsubstituted bacteriochlorin counterpart (
BC-0). Accordingly, we sought to prepare an analogue wherein the four methyl groups are per-deuteriated (
BC-2). The prior and target bacteriochlorins are shown in
Chart 3. Such compounds exhibit structural complexity intermediate between two extremes: the fully unsubstituted bacteriochlorins on one hand and the native bacteriochlorophylls of anoxygenic photosynthesis on the other.
The incorporation of deuterium presents challenges and opportunities that generally are not present with
13C and
15N. The distinction arises because of the possibility of
2H/
1H exchange processes. Thus, an intact tetrapyrrole macrocycle can be subjected to selective exchange at particular sites with
2H. On the other hand, a total synthesis that relies on
2H incorporation in early-stage precursors can be thwarted by inadvertent exchange processes during the course of the synthesis. Such exchange processes are generally not available (although see [
33]) for the carbon and nitrogen atoms of the tetrapyrrole macrocycle and precursors thereto.
We considered the direct deuteriation of
BC-1 to prepare
BC-2. In this regard, Godziela and coworkers reported the partial exchange of the protons on the β-pyrrolic methyl groups of ClFe(III)-porphyrins upon treatment with tetrabutylammonium hydroxide in dimethylsulfoxide-
d6 [
36]. Given that (1) the exchange was incomplete and had an obligatory requirement for the presence of the ferric chelate, and (2) metalation of electron-rich bacteriochlorins is challenging [
37], we elected to pursue the de novo synthesis of
BC-2 beginning with suitable deuteriated precursors. The synthesis of pyrroles bearing deuteriated β-methyl groups is strongly precedented by work in the porphyrin arena, chiefly by Smith and coworkers [
38,
39,
40,
41,
42,
43,
44]. A key question in the present work was whether such a deuteriated pyrrole could be converted with isotopic fidelity to the corresponding bacteriochlorin. Here, we present the synthesis of
BC-2 and the copper chelate (
CuBC-2), characterization of the isotopic purity, and investigation of the resonance Raman spectrum of
CuBC-2 and analogues.
2. Results and Discussion
We followed Smith’s strategy for the preparation of pyrroles bearing deuteriated β-methyl groups [
38,
39]. Thus, acetylacetone (
1) underwent alkylation with CD
3I to give the reported 3-trideuteriomethyl-2,4-pentadione
2 [
45], which was exhaustively deuteriated via underlying keto-enol tautomerism to give 1,3-diketone
3 [
46] (
Scheme 2). Treatment of
3 and
tert-butyl acetoacetate (
4) to the Paal-Knorr reaction [
47] in hot acetic acid gave pyrrole
5 in 31% yield. The α-methyl group of
5 was found to have undergone exchange with loss of the deuterium substitution, whereas the deuterium substitution of the two β-methyl groups was found to be 91% and 97%. The selective loss of deuterium at the pyrrole α-methyl group, while fortuitous, was very convenient for the planned synthesis as deuterium was desired only at the methyl groups of the β-pyrrolic positions of the target bacteriochlorin. The α-methyl group of pyrrole
5 is destined to give the bacteriochlorin 10,20-positions, and an α-CD
3 group of pyrrole
5 would give a C–D methine at the bacteriochlorin 10,20-positions. Pyrrole
5 was treated with lead acetate to give the pyrrole-carboxaldehyde
6 in 70% yield, which upon Henry reaction gave nitrovinylpyrrole
7 in 79% yield. Conjugate reduction with NaBH
4 gave nitroethylpyrrole
8 (80% yield), which served as the nucleophile with Michael acceptor mesityl oxide in the presence of TBAF and molecular sieves [
48] to give nitrohexanone-pyrrole
9 in 52% yield. McMurry cyclization gave dihydrodipyrrin
10 (49% yield), which upon Riley oxidation gave the dihydrodipyrrin-carboxaldehyde
11 in 54% yield. Treatment of
11 with neat TFA as described for the analogous natural abundance substrate [
48] gave the tetrakis(trideuteriomethyl)bacteriochlorin
BC-2 in 14% yield, a result that is typical for self-condensations of dihydrodipyrrin-carboxaldehydes [
48] (
Scheme 2).
The isotopic purity of the compounds containing deuteriated methyl groups was examined by
1H NMR spectroscopy with comparison of peak intensities of resonances of natural abundance sites versus the target deuteriated sites. The
1H NMR spectrum of dihydrodipyrrin
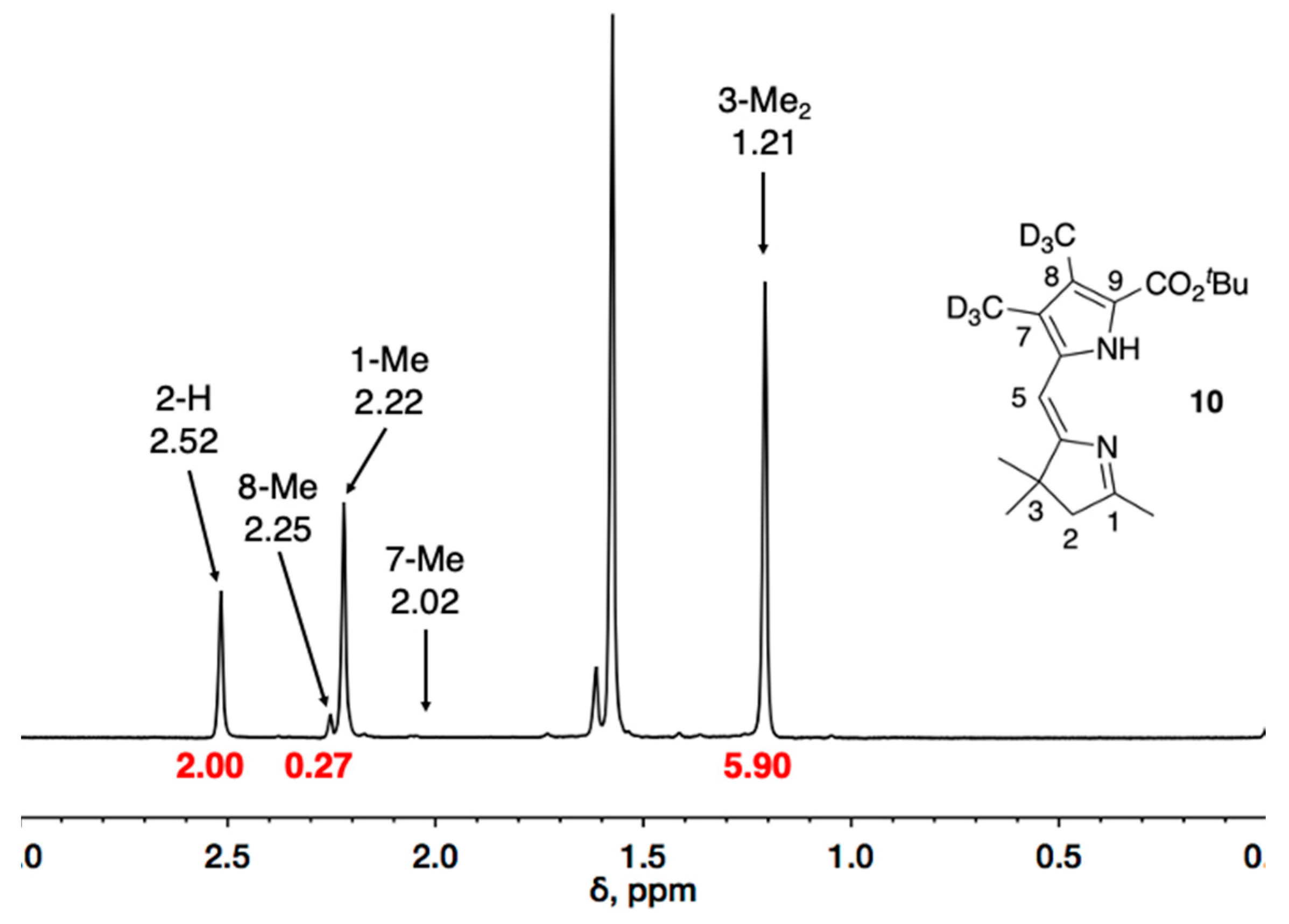
10 is shown here as an example (
Figure 1). First, integration of the peak derived from the 2-methylene group, which appears at δ = 2.52 ppm, is set to 2.00 as the standard. The resonance of the gem-dimethyl groups (δ = 1.21 ppm) affords an integration of 5.90, consistent with the expected 2:6 ratio. The 8-methyl group resonates at δ ~ 2.25 ppm, which is very slightly downfield from the resonance of the 1-methyl group (δ = 2.22 ppm). The peak due to the 8-methyl group has an estimated integrated value of 0.3, which corresponds to a residual natural abundance signal at the 10% level and hence 90% deuteriation. The deuterium incorporation of the 7-methyl in
10 is estimated to be >98% given the absence of an observable signal around 2.02 ppm.
In this manner, the isotopic incorporation for each compound was estimated. The values for a given pyrrole or dihydrodipyrrin pertain to the methyl adjacent or distal to the tert-butoxycarbonyl moiety, were, respectively: pyrroles 5 (91%, >98%), 6 (91%, 97%), 7 (91%, >98%), 8 (91%, >98%), and 9 (90%, >98%); dihydrodipyrrins 10 (90%, >98%) and 11 (91%, >98%); and tetrakis(trideuteriomethyl)bacteriochlorin BC-2 (90%). Note that the two β-methyl groups are distinct in the pyrroles and dihydrodipyrrins, but equivalent by symmetry in the bacteriochlorin. The isotopic enrichment is not expected to increase under the conditions employed for reactions downstream from that of perdeuteriated meso-methylacetylacetone (3). Accordingly, the range of values reported here (90% to >98%) likely reflects vagaries of integration of NMR signals. In short, the isotopic purity was maintained at a high level throughout the synthesis.
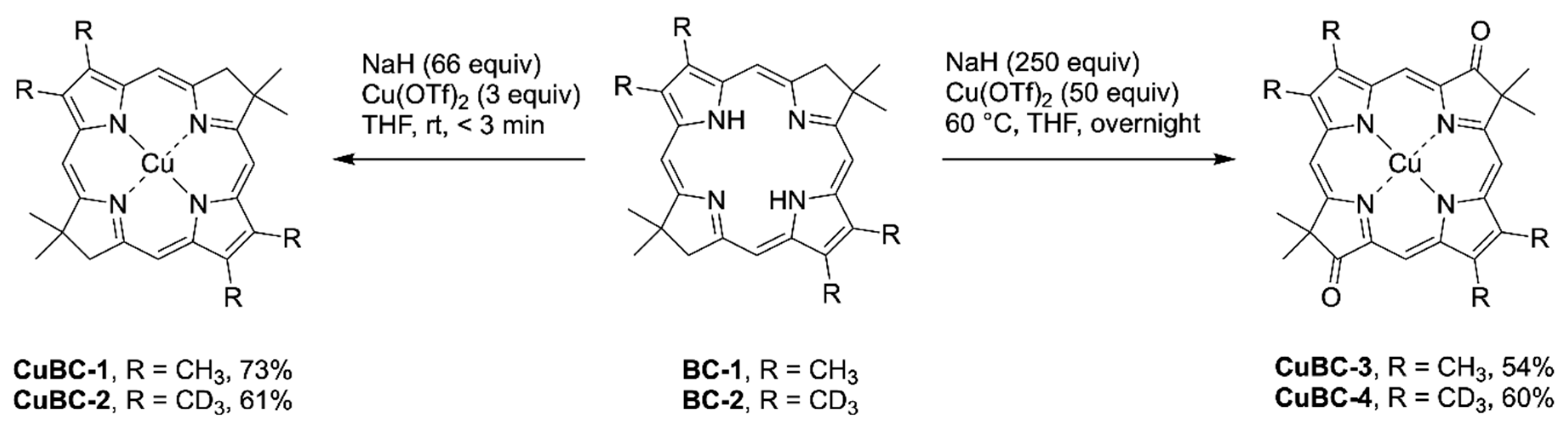
The reported procedure [
48] using copper(II) acetate of metalation for
BC-1 upon application to
BC-2 did not afford observable product. Replacement of copper(II) acetate with the more reactive copper(II) triflate in an overnight reaction under argon gave the dioxobacteriochlorin
CuBC-4 instead of the desired
CuBC-2. A similar oxidation was also found in the reaction of
BC-1 and Cu(OTf)
2 to give dioxobacteriochlorin
CuBC-3 (
Scheme 3). The conversion of
BC-2 to
CuBC-2 was achieved in a reasonable yield by decreasing the equivalents of copper(II) triflate and sodium hydride, and shortening the reaction time from 16 h to 2 min. The similar reaction of
BC-1 with the reaction time further limited to 30 s was successful in giving
CuBC-1. The appearance of dioxobacteriochlorin
CuBC-3 was observed by absorption spectroscopy when the reaction time was extended to as little as 90 s. The unexpected oxidation of the methylene group flanking the gem-dimethyl group highlights the sensitivity of this site, which has been referred to as the Achilles’ heel of the hydroporphyrins [
49]. Dioxobacteriochlorins have been prepared previously [
50] and are of fundamental interest including as the core chromophore of the naturally occurring macrocycles known as tolyporphins, which are found in a cyanobacterium [
51]. The nature of the oxidant underlying the formation of dioxobacteriochlorin in this small-scale reaction (1.0–2.0 mg) is not known.
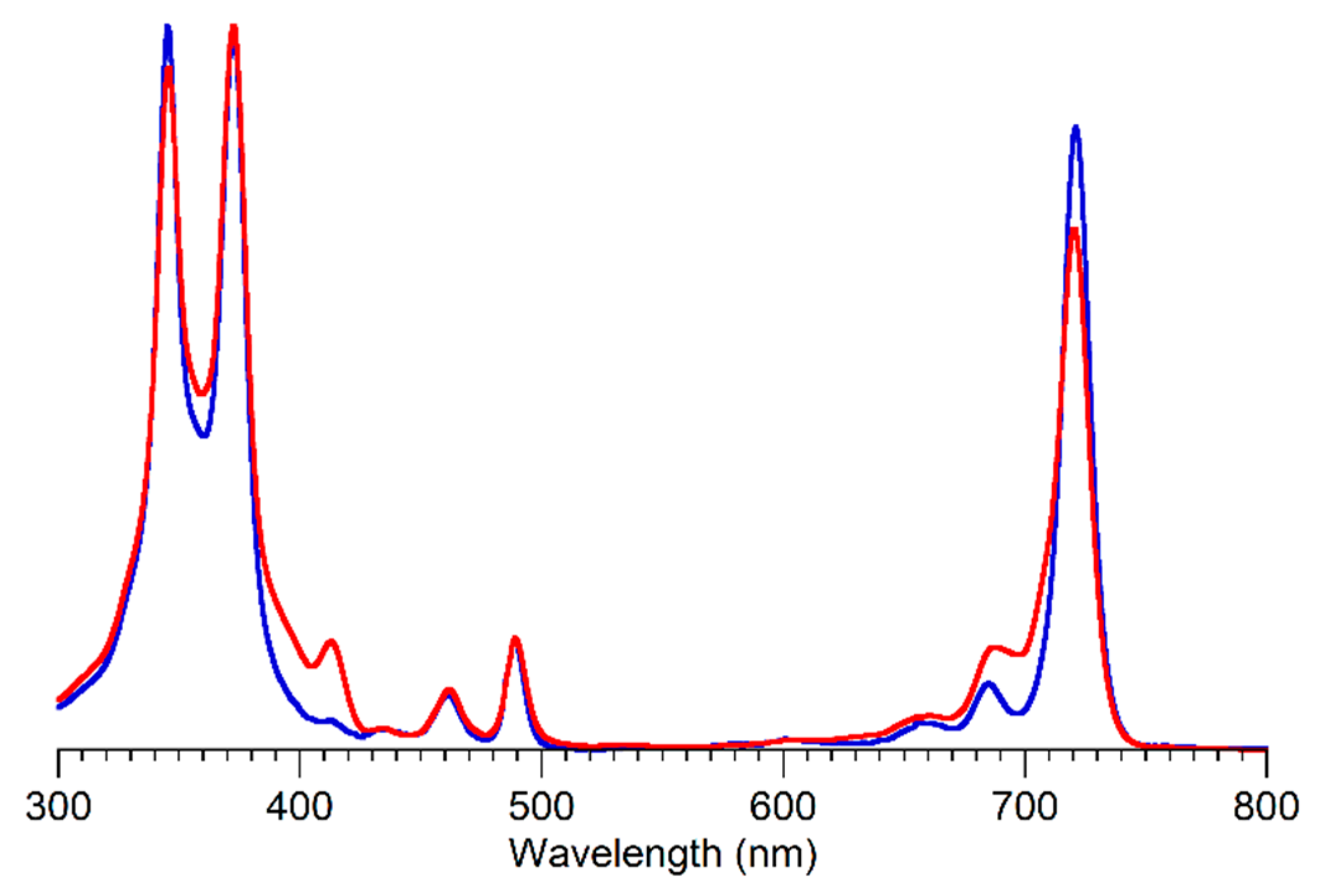
The absorption spectra of bacteriochlorins
BC-1 and
BC-2 are shown in
Figure 2. The spectra are essentially identical with the exception of a small peak (of unknown origin) at ~412 nm and a less pronounced dip between the Q
y(0,0) and Q
y(1,0) bands of the spectrum of
BC-2 versus that of
BC-1. The absorption spectra of
CuBC-1–
CuBC-4 are shown in
Figure 3. A signature hypsochromic shift (~35 nm) of the long-wavelength (Q
y) absorption band was observed for the copper dioxobacteriochlorins versus the corresponding copper bacteriochlorins.
The resonance Raman (RR) spectra of
CuBC-1 and
CuBC-2 were examined. Q
y-excitation RR studies of
CuBC-1 and
CuBC-2 are shown in
Figure 4. The RR spectra can be compared with those of previous synthetic bacteriochlorins
CuBC-0 and
CuBC-1 [
48], as well as that of
CuBChl a [
52], the copper chelate of bacteriochlorophyll
a. The diagram illustrates our initial efforts at tuning vibronic complexity. The mid frequency region (800–1700 cm
−1) of the RR spectrum of
CuBC-1 is similar to that of
CuBC-0 and exhibits only a few relatively weak bands. However, the low-frequency region (150–800 cm
−1) of the RR spectrum of
CuBC-1 exhibits additional features, particularly in the region surrounding the prominent feature at 736 cm
−1. Vibrational calculations indicate that the 736 cm
−1 band of
CuBC-1 is the analogue of the 727 cm
−1 band of
CuBC-0. The additional features are relatively weak in the spectrum of
CuBC-2.
Thus, the RR studies of CuBC-1 and CuBC-2 reveal that addition of the four beta-pyrrolic substituents increases the number of vibronically coupled modes only modestly and falls short of the vibronic complexity observed for CuBChl a. The increased vibronic activity exhibited by the natural pigments and CuBChl a must arise from the increased structural complexity of the macrocycle. In this regard, BChl a is equipped with 2,12-dimethyl substituents, a 3-acetyl group, the isocyclic ring that bridges the 13–15 positions, as well as trans-dialkyl substituents in each pyrroline ring, whereas synthetic bacteriochlorins BC-1 or BC-2 (and metal chelates) contain at the β-pyrrolic sites only the four methyl or four perdeuteriomethyl groups, respectively.
4. Experimental Section
General Methods.
1H NMR and
13C NMR spectra (see
supplementary materials) were collected at room temperature in CDCl
3 unless noted otherwise. Resonances in the
13C NMR spectra due to the presence of attached deuterium are not reported. Electrospray ionization mass spectrometry (ESI-MS) data are reported for the molecular ion or protonated molecular ion. THF used was freshly distilled from Na/benzophenone unless noted otherwise. All commercially available compounds were used as received.
Non-commercial Compounds. 3-Trideuteriomethyl-2,4-pentanedione (
2) [
45] and 2,3,8,8,12,13,18,18-octamethylbacteriochlorin (
BC-1) [
48] were prepared following literature procedures.
d10-3-Methyl-2,4-pentanedione (3). Following a general procedure [
46] with modification, a mixture of
d3-3-methyl-2,4-pentanedione (
2, 14.24 g, 122.0 mmol) in deuterium oxide (70 g) was treated with potassium carbonate (2.59 g, 18.7 mmol) and refluxed for 18 h. The mixture was allowed to cool to room temperature, neutralized by the addition of deuterium chloride (20 wt% in D
2O), and extracted by CH
2Cl
2 (150 mL). The organic phase was dried (Na
2SO
4) and concentrated to give an orange liquid (14.24 g, 85%):
13C{
1H} NMR (100 MHz)
δ 13.80, 13.84, 20.54, 20.60, 60.06, 60.09, 170.9, 205.2; ESI-MS obsd 124.1316, calcd 124.1318 [(M + H)
+, M = C
6H
1D
9O
2 due to the deuterium-protium exchange in the ESI-MS instrument].
2-tert-Butoxycarbonyl-3,4-bis(trideuteriomethyl)-5-methylpyrrole (5). Following a general procedure [
47] with modification, aqueous sodium nitrite solution (5.08 g, 73.6 mmol, 15 mL) was slowly added to a solution of
tert-butyl acetoacetate (
4, 10.37 g, 65.6 mmol) in acetic acid (25 mL) while maintaining the reaction temperature < 40 °C. The mixture was stirred at room temperature for 18 h. Then
3 (8.16 g, 65.7 mmol) was added to the mixture, followed by zinc dust (8.95 g, 0.137 mol) in several portions over 10 min to maintain the reaction temperature < 60 °C. The resulting mixture was heated to 60 °C for 1 h. After allowing to cool to room temperature, the mixture was poured into water (150 mL) and extracted with ethyl acetate (450 mL). The organic phase was washed (aqueous NaHCO
3 solution), dried (Na
2SO
4) and concentrated. The residue was recrystallized in ethanol (15 mL). The mother liquor was chromatographed (silica, hexanes/ethyl acetate, (10:1)). A chromatographic fraction containing the title compound was combined with the recrystallized sample (white solid, 4.95 g, 31%): mp 138–139 °C;
1H NMR (400 MHz)
δ 1.55 (s, 9H), 2.17 (s, 2H), 8.48 (br, 1H);
13C{
1H} NMR (100 MHz)
δ 10.7, 11.0, 11.2, 11.35, 11.40, 28.6, 79.9, 116.7, 117.9, 126.4, 128.9, 161.5; ESI-MS obsd 217.1922, calcd 217.1928 [(M + H)
+, M = C
12H
12D
7NO
2]. Analysis by
1H NMR spectroscopy indicated 91% and >98% deuterium incorporation in the 3-methyl and 4-methyl groups, respectively.
2-tert-Butoxycarbonyl-5-formyl-3,4-bis(trideuteriomethyl)pyrrole (6). Following a general procedure [
47] with modification, a solution of
5 (4.90 g, 22.7 mmol) in acetic acid (100 mL) was treated with lead tetraacetate (22.5 g, 50.8 mmol). The mixture was heated to 80 °C for 3 h. Then the mixture was treated with ethylene glycol (1 mL) to inactivate any lead dioxide present. After allowing to cool to room temperature, the mixture was poured into water (500 mL) and extracted with ethyl acetate (1200 mL). The organic phase was washed (aqueous NaHCO
3 solution), dried (Na
2SO
4), concentrated and chromatographed (silica, hexanes/ethyl acetate (10:1)) to afford an off-white solid (3.63 g, 70%): mp 81–82 °C;
1H NMR (400 MHz)
δ 1.57 (s, 9H), 9.39 (br, 1H), 9.74 (s, 0.6H);
13C{
1H} NMR (100 MHz)
δ 9.7, 20.5, 28.3, 82.2, 125.9, 126.3, 129.5, 130.3, 160.3, 179.1; ESI-MS obsd 230.1653, calcd 230.1658 [(M + H)
+, M = C
12H
11D
6NO
3]. Analysis by
1H NMR spectroscopy indicated 91% and 97% deuterium incorporation in the 3-methyl and 4-methyl groups, respectively.
(E)-2-tert-Butoxycarbonyl-3,4-bis(trideuteriomethyl)-5-(2-nitrovinyl)pyrrole (7). Following a general procedure [
47] with modification, samples of
6 (3.67 g 16.0 mmol), nitromethane (3.93 g, 64.4 mmol), potassium acetate (1.74 g, 17.7 mmol) and methylamine hydrochloride (1.08 g, 16.0 mmol) were stirred in anhydrous methanol (30 mL) for 3 h. The solvent was removed under vacuum, and the residue was diluted with water (50 mL) and extracted with ethyl acetate (150 mL). The organic phase was dried (Na
2SO
4) and concentrated. The resulting solid was rinsed with hexanes to afford a yellow solid (3.46 g, 79%): mp 156–157 °C;
1H NMR (400 MHz)
δ 1.59 (s, 9H), 7.36–7.41 (m, 1H), 7.94 (d,
J = 13.6 Hz, 0.6H), 9.10 (br, 1H);
13C{
1H} NMR (100 MHz)
δ 28.3, 82.5, 123.2, 126.0, 126.8, 129.6, 132.8, 132.9, 160.9; ESI-MS obsd 273.1719, calcd 273.1716 [(M + H)
+, M = C
13H
12D
6N
2O
4]. Analysis by
1H NMR spectroscopy indicated 91% and >98% deuterium incorporation in the 3-methyl and 4-methyl groups, respectively.
2-tert-Butoxycarbonyl-3,4-bis(trideuteriomethyl)-5-(2-nitroethyl)pyrrole (8). Following a general procedure [
47] with modification, a slurry of
7 (3.46 g, 12.7 mmol) in absolute ethanol (150 mL) was treated with sodium borohydride (1.23 g, 32.5 mmol) at room temperature. After 2.5 h, the colorless mixture was concentrated. Saturated aqueous NH
4Cl solution (50 mL) was added to the residue. The mixture was treated with ethyl acetate (120 mL). The organic extract was dried (Na
2SO
4) and concentrated. The residue was washed with hexanes to give an off-white solid (2.79 g, 80%): mp 154–155 °C;
1H NMR (400 MHz)
δ 1.56 (s, 9H), 3.22–3.28 (m, 1.6H), 4.50–4.53 (m, 2H), 8.77 (br, 1H);
13C{
1H} NMR (100 MHz)
δ 24.0, 28.5, 74.2, 80.6, 118.1, 119.7, 126.0, 126.4, 161.4; ESI-MS obsd 275.1863, calcd 275.1878 [(M + H)
+, M = C
13H
14D
6N
2O
4]. Analysis by
1H NMR spectroscopy indicated 91% and >98% deuterium incorporation in the 3-methyl and 4-methyl groups, respectively.
5-tert-Butoxycarbonyl-2-(1,1-dimethoxy-4,4-dimethyl-5-nitro-2-oxohexan-6-yl)-3,4-bis(trideuteriomethyl)pyrrole (9). Following a general procedure [
48] with modification, a mixture of
8 (2.77 g, 10.1 mmol), mesityl oxide (5.03 g, 51.2 mmol), and molecular sieves 4 Å (10.0 g) in DMF (150 mL) was treated with TBAF (15.0 mL, 1.0 M in THF solution). The resulting mixture was stirred at room temperature for 16 h. The mixture was filtered through a pad of Celite
®, and the filtered material was washed with ethyl acetate. The filtrate was poured into water (500 mL) and extracted with ethyl acetate (1.2 L). The organic extract was washed with brine, dried (Na
2SO
4), concentrated and chromatographed (silica, hexanes/ethyl acetate (8:1)) to afford a light-yellow solid (1.97 g, 52%): mp 110–111 °C;
1H NMR (400 MHz)
δ 1.12 (s, 3H), 1.25 (s, 3H), 1.54 (s, 9H), 2.14 (s, 3H), 2.39, 2.62 (AB,
2J = 17.5 Hz, 2H), 2.92–3.00 (m, 0.8H), 3.22–3.30 (m, 0.8H), 5.08–5.12 (m, 1H), 8.64 (br, 1H);
13C{
1H} NMR (100 MHz)
δ 24.0, 24.3, 25.0, 28.5, 31.8, 36.7, 51.3, 80.4, 93.50, 93.56, 118.1, 119.6, 126.2, 161.1, 206.7; ESI-MS obsd 373.2597, calcd 373.2604 [(M + H)
+, M = C
19H
24D
6N
2O
5]. Analysis by
1H NMR spectroscopy indicated 90% deuterium incorporation in the 4-methyl group and >98% deuterium incorporation in the 3-methyl group of the pyrrole (note the numbering change versus prior pyrroles).
2,3-Dihydro-1,3,3-trimethyl-7,8-bis(trideuteriomethyl)-9-tert-butoxycarbonyldipyrrin (10). Following a general procedure [
48] with modification, a solution of
9 (1.63 g, 4.38 mmol) in freshly distilled THF (10 mL) under argon was treated with NaOMe (1.08 g, 20.0 mmol). The mixture was stirred at room temperature for 15 min. In a second flask, NH
4OAc (17.08 g, 221.6 mmol) in freshly distilled THF (65 mL) was bubbled with argon for 30 min. Then, TiCl
3 (45 mL, 12 wt% in 3% HCl solution) was added, and the mixture was degassed by bubbling with argon for 20 min. The contents of the first flask were then transferred via cannula to the buffered TiCl
3 solution. The resulting mixture was stirred at room temperature for 20 h under argon. The mixture was filtered through a pad of Celite
®, and the filtered material was washed with ethyl acetate. The filtrate was washed with saturated aqueous NaHCO
3 solution, water, and brine. The organic phase was dried (Na
2SO
4), concentrated and chromatographed (silica, hexanes/ethyl acetate (6:1)) to afford a yellow solid (693.8 mg, 49%): mp 128–129 °C;
1H NMR (400 MHz)
δ 1.21 (s, 6H), 1.57 (s, 9H), 2.22 (s, 3H), 2.52 (s, 2H), 5.67 (s, 1H), 11.11 (s, 1H);
13C{
1H} NMR (100 MHz)
δ 21.0, 28.7, 29.3, 41.6, 53.9, 79.8, 101.4, 118.3, 119.8, 126.2, 130.8, 161.3, 163.6, 178.4; ESI-MS obsd 323.2590, calcd 323.2606 [(M + H)
+, M = C
19H
22D
6N
2O
2]; λ
abs (CH
2Cl
2) 350 nm. Analysis by
1H NMR spectroscopy indicated 90% deuterium incorporation in the 8-methyl group and >98% deuterium incorporation in the 7-methyl group.
9-tert-Butoxycarbonyl-1-formyl-2,3-dihydro-3,3-dimethyl-7,8-bis(trideuteriomethyl)-dipyrrin (11). Following a general procedure [
48], a solution of
10 (150 mg, 0.466 mmol) in 1,4-dioxane (7.0 mL) was treated with SeO
2 (156 mg, 1.40 mmol) under argon. The reaction mixture was stirred at room temperature and monitored via absorption spectroscopy for the characteristic bathochromic shift that accompanies oxidation of the pyrroline α-methyl group to the carboxaldehyde. After 90 min, ethyl acetate was added. The mixture was washed with aqueous NaHCO
3 solution followed by brine. The organic phase was dried (Na
2SO
4), concentrated and chromatographed (silica, hexanes/CH
2Cl
2 (1:2)) to afford a dark red solid (84.0 mg, 54%): mp 153–154 °C;
1H NMR (400 MHz)
δ 1.26 (s, 6H), 1.58 (s, 9H), 2.72 (s, 2H), 6.13 (s, 1H), 9.97 (s, 1H), 10.81 (br, 1H);
13C{
1H} NMR (100 MHz)
δ 28.5, 29.1, 41.1, 46.1, 80.4, 110.8, 122.7, 129.5, 160.8, 162.2, 170.0, 190.0; ESI-MS obsd 337.2384, calcd 337.2393 [(M + H)
+, M = C
19H
20D
6N
2O
3]; λ
abs (CH
2Cl
2) 466 nm. Analysis by
1H NMR spectroscopy indicated 90% deuterium incorporation in the 8-methyl group and >98% deuterium incorporation in the 7-methyl group.
8,8,18,18-Tetramethyl-2,3,12,13-tetrakis(trideuteriomethyl)bacteriochlorin (BC-2). Following a general procedure [
48], a sample of
11 (66.0 mg, 0.196 mmol) was dissolved in TFA (9.5 mL) under argon and stirred for 1 h at room temperature. Then, the reaction mixture was poured into saturated aqueous NaHCO
3 solution (200 mL). The resulting mixture was extracted with CH
2Cl
2 (200 mL). The organic phase was washed with brine, dried (Na
2SO
4), concentrated and chromatographed (silica, hexanes/CH
2Cl
2 (1:1)) to afford a green solid (6.2 mg, 14%):
1H NMR (400 MHz)
δ −2.47 (s, 2H), 1.93 (s, 12H), 4.43 (s, 4H), 8.54 (s, 2H), 8.67 (s, 2H);
13C{
1H} NMR (125 MHz)
δ 31.4, 46.2, 52.0, 92.7, 95.1, 127.8, 133.8, 134.6, 156.5, 168.7; ESI-MS obsd 438.3520, calcd 438.3531 [(M)
+, M = C
28H
22D
12N
4]; λ
abs (CH
2Cl
2) 343, 369, 487, 682, 718 nm. Analysis by
1H NMR spectroscopy indicated 90% deuterium incorporation in total for the four β-methyl groups.
Cu(II)-8,8,18,18-Tetramethyl-2,3,12,13-tetra(trideuteriomethyl)bacteriochlorin (CuBC-2). A sample of BC-2 (2.9 mg, 6.6 μmol) was dissolved in freshly distilled THF (1 mL) and added to a Schlenk flask. After deaeration of the sample by three freeze–pump–thaw cycles, sodium hydride (10.5 mg, 0.438 mmol) was added to the mixture under argon. The mixture was heated to 60 °C until the color turned from green to brown. Then the mixture was allowed to cool to room temperature, whereupon copper(II) triflate (7.3 mg, 20 μmol) was added under argon. After reaction at room temperature for 2 min, the mixture was quenched by the addition of water and then extracted with CH2Cl2. The organic phase was dried (Na2SO4) and concentrated, and the resulting solid was rinsed with distilled hexanes to afford a dark green solid (2.0 mg, 61%). ESI-MS obsd 499.2658, calcd 499.2671 [(M)+, M = C28H20D12N4Cu]; λabs (CH2Cl2) 337, 387, 475, 705, 737 nm.
Cu(II)-2,3,8,8,12,13,18,18-Octamethylbacteriochlorin (CuBC-1). A sample of
BC-1 (2.4 mg, 5.6 μmol) was dissolved in freshly distilled THF (1 mL) and added to a Schlenk flask. After deaeration of the sample by three freeze–pump–thaw cycles, sodium hydride (6.7 mg, 0.28 mmol) was added to the mixture under argon. The mixture was heated to 60 °C until the color turned from green to brown. Then, the mixture was allowed to cool to room temperature, whereupon copper(II) triflate (7.3 mg, 20 μmol) was added under argon. After reaction at room temperature for 30 s, the mixture was quenched by the addition of water and then extracted with CH
2Cl
2. The organic phase was dried (Na
2SO
4) and concentrated. The resulting solid was rinsed with distilled hexanes to afford a dark green solid (2.0 mg, 73%). The characterization data (absorption spectroscopy and ESI-MS) matched those obtained from an alternative route [
48].
Cu(II)-2,3,8,8,12,13,18,18-Octamethyl-7,17-dioxobacteriochlorin (CuBC-3). A sample of BC-1 (2.0 mg, 4.7 μmol) was dissolved in freshly distilled THF (1 mL) and added to a Schlenk flask. After deaeration of the sample by three freeze–pump–thaw cycles, sodium hydride (22.1 mg, 0.875 mmol) was added to the mixture under argon. The mixture was heated to 60 °C until the color turned from green to brown. Then copper(II) triflate (41.9 mg, 0.116 mmol) was added to the hot mixture under argon. The mixture was stirred overnight at 60 °C. After allowing to cool to room temperature, the mixture was diluted with CH2Cl2 and washed with water. The organic layer was dried (Na2SO4) and concentrated. The resulting solid was rinsed with distilled hexanes to afford a dark blue solid (1.3 mg, 54%): ESI-MS obsd 515.1486, calcd 515.1503 [(M)+, M = C28H28O2N4Cu]; λabs (CH2Cl2) 322, 380, 486, 670, 700 nm.
Cu(II)-8,8,18,18-tetramethyl-2,3,12,13-tetra(trideuteriomethyl)-7,17-dioxobacteriochlorin (CuBC-4). A solution of BC-2 (1.3 mg, 3.0 μmol) in freshly distilled THF (1 mL) was added to a Schlenk flask. After deaeration of the sample by three freeze–pump–thaw cycles, sodium hydride (18.1 mg, 0.754 mmol) was added to the mixture under argon. The mixture was heated to 60 °C until the solution turned from green to brown. Then, copper(II) triflate (54.3 mg, 0.150 mmol) was added to the hot mixture under argon. The resulting mixture was stirred overnight at 60 °C. After allowing to cool to room temperature, the mixture was diluted with CH2Cl2 and washed with water. The organic layer was dried (Na2SO4) and concentrated. The resulting solid was rinsed with distilled hexanes to afford a dark blue solid (1.0 mg, 60%): ESI-MS obsd 527.2265, calcd 527.2256 [(M)+, M = C28H16D12O2N4Cu]; λabs (CH2Cl2) 321, 380, 521, 669, 700 nm.
Resonance Raman Spectroscopy. The resonance Raman spectra were acquired using instrumentation and methods previously described [
35].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









