
Protective Activities of Growth Hormone-Releasing Hormone Antagonists against Toxin-Induced Endothelial Injury



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Cultures
2.3. Western Blot Analysis
2.4. Fluorescein Isothiocyanate (FITC)–Dextran Assay
2.5. ROS Measurement
2.6. Densitometry and Statistical Analysis
3. Results
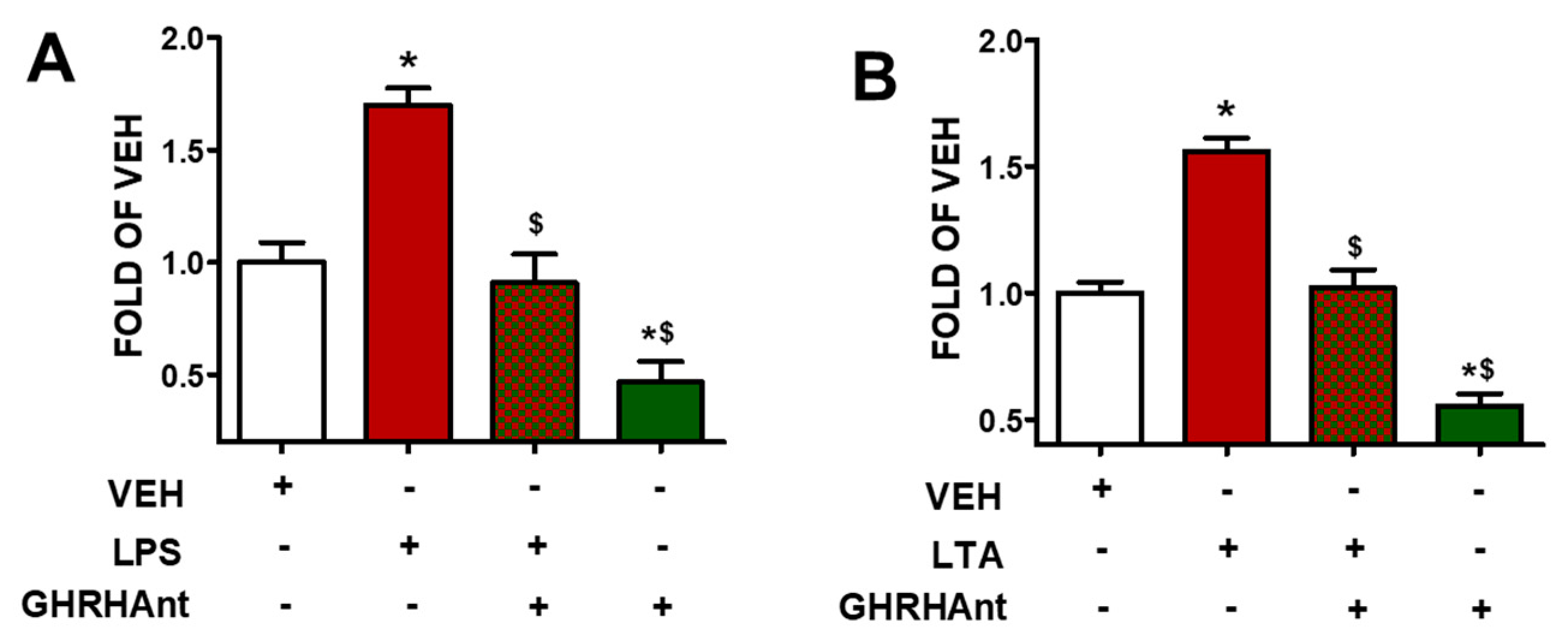
3.1. GHRHAnt Protect against Toxin-Induced Barrier Dysfunction
3.2. GHRHAnt Counteract Toxin-Induced ROS Generation
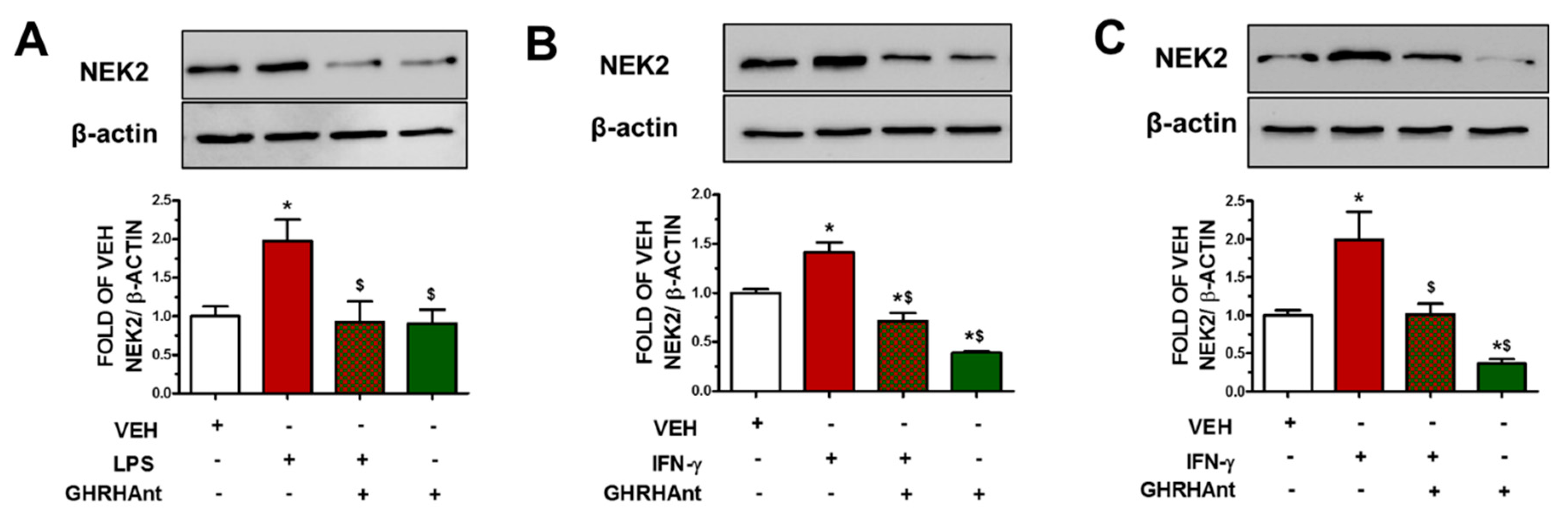
3.3. GHRHAnt Suppress Endothelial Barrier Dysfunction-Induced NEK2 Augmentation
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Balik, M.; Maly, M.; Huptych, M.; Mokotedi, M.C.; Lambert, L. Prognostic Impact of Serial Imaging in Severe Acute Respiratory Distress Syndrome on the Extracorporeal Membrane Oxygenation. J. Clin. Med. 2023, 12, 6367. [Google Scholar] [CrossRef]
- Schneider, A.; Wood, H.N.; Geden, S.; Greene, C.J.; Yates, R.M.; Masternak, M.M.; Rohde, K.H. Growth hormone-mediated reprogramming of macrophage transcriptome and effector functions. Sci. Rep. 2019, 9, 19348. [Google Scholar] [CrossRef]
- Jeay, S.; Sonenshein, G.E.; Postel-Vinay, M.C.; Kelly, P.A.; Baixeras, E. Growth hormone can act as a cytokine controlling survival and proliferation of immune cells: New insights into signaling pathways. Mol. Cell Endocrinol. 2002, 188, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Ho, B.M.; Zhou, L.; Yip, Y.W.Y.; He, J.N.; Wei, Y.; Tham, C.C.; Chan, S.O.; Schally, A.V.; Pang, C.P.; et al. Growth hormone releasing hormone signaling promotes Th17 cell differentiation and autoimmune inflammation. Nat. Commun. 2023, 14, 3298. [Google Scholar] [CrossRef]
- Zhang, C.; Cui, T.; Cai, R.; Wangpaichitr, M.; Mirsaeidi, M.; Schally, A.V.; Jackson, R.M. Growth Hormone-Releasing Hormone in Lung Physiology and Pulmonary Disease. Cells 2020, 9, 2331. [Google Scholar] [CrossRef] [PubMed]
- Halmos, G.; Szabo, Z.; Juhasz, E.; Schally, A.V. Signaling mechanism of growth hormone-releasing hormone receptor. Vitam. Horm. 2023, 123, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Zhang, X.; Cai, R.; Hare, J.M.; Granata, R.; Bartoli, M. Actions and Potential Therapeutic Applications of Growth Hormone-Releasing Hormone Agonists. Endocrinology 2019, 160, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Havt, A.; Schally, A.V.; Halmos, G.; Varga, J.L.; Toller, G.L.; Horvath, J.E.; Szepeshazi, K.; Koster, F.; Kovitz, K.; Groot, K.; et al. The expression of the pituitary growth hormone-releasing hormone receptor and its splice variants in normal and neoplastic human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 17424–17429. [Google Scholar] [CrossRef]
- Uddin, M.A.; Akhter, M.S.; Singh, S.S.; Kubra, K.T.; Schally, A.V.; Jois, S.; Barabutis, N. GHRH antagonists support lung endothelial barrier function. Tissue Barriers 2019, 7, 1669989. [Google Scholar] [CrossRef]
- Barabutis, N.; Akhter, M.S.; Kubra, K.T.; Jackson, K. Growth Hormone-Releasing Hormone in Endothelial Inflammation. Endocrinology 2022, 164, bqac209. [Google Scholar] [CrossRef]
- Rellos, P.; Ivins, F.J.; Baxter, J.E.; Pike, A.; Nott, T.J.; Parkinson, D.M.; Das, S.; Howell, S.; Fedorov, O.; Shen, Q.Y.; et al. Structure and regulation of the human Nek2 centrosomal kinase. J. Biol. Chem. 2007, 282, 6833–6842. [Google Scholar] [CrossRef]
- Neal, C.P.; Fry, A.M.; Moreman, C.; McGregor, A.; Garcea, G.; Berry, D.P.; Manson, M.M. Overexpression of the Nek2 kinase in colorectal cancer correlates with beta-catenin relocalization and shortened cancer-specific survival. J. Surg. Oncol. 2014, 110, 828–838. [Google Scholar] [CrossRef]
- Hayward, D.G.; Fry, A.M. Nek2 kinase in chromosome instability and cancer. Cancer Lett. 2006, 237, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.A.; Akhter, M.S.; Kubra, K.T.; Barabutis, N. Induction of the NEK family of kinases in the lungs of mice subjected to cecal ligation and puncture model of sepsis. Tissue Barriers 2021, 9, 1929787. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yang, Y.; Xia, J.; Wang, H.; Salama, M.E.; Xiong, W.; Xu, H.; Shetty, S.; Chen, T.; Zeng, Z.; et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 2013, 23, 48–62. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, F.; Paronetto, M.P.; Franco, R.; Chieffi, P.; Dolci, S.; Fry, A.M.; Geremia, R.; Sette, C. Increased expression and nuclear localization of the centrosomal kinase Nek2 in human testicular seminomas. J. Pathol. 2009, 217, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, P.; Cheung, K.M.; Solanki, S.; Mas-Droux, C.; Rowan, F.; Yeoh, S.; Boxall, K.; Westlake, M.; Pickard, L.; Hardy, T.; et al. Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: Structure-activity relationship, structural biology, and cellular activity. J. Med. Chem. 2012, 55, 3228–3241. [Google Scholar] [CrossRef] [PubMed]
- Fakir, S.; Barabutis, N. Growth hormone-releasing hormone antagonists counteract interferon-gamma—Induced barrier dysfunction in bovine and human endothelial cells. Cytokine 2024, 173, 156416. [Google Scholar] [CrossRef] [PubMed]
- Franqui-Machin, R.; Hao, M.; Bai, H.; Gu, Z.; Zhan, X.; Habelhah, H.; Jethava, Y.; Qiu, L.; Frech, I.; Tricot, G.; et al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J. Clin. Investig. 2018, 128, 2877–2893. [Google Scholar] [CrossRef]
- Barabutis, N.; Akhter, M.S. Involvement of NEK2 and NEK9 in LPS—induced endothelial barrier dysfunction. Microvasc. Res. 2024, 152, 104651. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Y.; Rigor, R.R. Regulation of Endothelial Barrier Function; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar] [CrossRef]
- Mehta, D.; Ravindran, K.; Kuebler, W.M. Novel regulators of endothelial barrier function. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L924–L935. [Google Scholar] [CrossRef]
- He, P.; Talukder, M.A.H.; Gao, F. Oxidative Stress and Microvessel Barrier Dysfunction. Front. Physiol. 2020, 11, 472. [Google Scholar] [CrossRef] [PubMed]
- Takashiba, S.; Van Dyke, T.E.; Amar, S.; Murayama, Y.; Soskolne, A.W.; Shapira, L. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infect. Immun. 1999, 67, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Kraus, V.B. Does lipopolysaccharide-mediated inflammation have a role in OA? Nat. Rev. Rheumatol. 2016, 12, 123–129. [Google Scholar] [CrossRef]
- Knapp, S.; von Aulock, S.; Leendertse, M.; Haslinger, I.; Draing, C.; Golenbock, D.T.; van der Poll, T. Lipoteichoic acid-induced lung inflammation depends on TLR2 and the concerted action of TLR4 and the platelet-activating factor receptor. J. Immunol. 2008, 180, 3478–3484. [Google Scholar] [CrossRef]
- Lee, J.Y.; Sullivan, K.E. Gamma interferon and lipopolysaccharide interact at the level of transcription to induce tumor necrosis factor alpha expression. Infect. Immun. 2001, 69, 2847–2852. [Google Scholar] [CrossRef]
- Kang, S.S.; Ryu, Y.H.; Baik, J.E.; Yun, C.H.; Lee, K.; Chung, D.K.; Han, S.H. Lipoteichoic acid from Lactobacillus plantarum induces nitric oxide production in the presence of interferon-gamma in murine macrophages. Mol. Immunol. 2011, 48, 2170–2177. [Google Scholar] [CrossRef]
- Li, W.; Li, D.; Chen, Y.; Abudou, H.; Wang, H.; Cai, J.; Wang, Y.; Liu, Z.; Liu, Y.; Fan, H. Classic Signaling Pathways in Alveolar Injury and Repair Involved in Sepsis-Induced ALI/ARDS: New Research Progress and Prospect. Dis. Markers 2022, 2022, 6362344. [Google Scholar] [CrossRef]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef]
- Han, S.; Mallampalli, R.K. Correction: The acute respiratory distress syndrome: From mechanism to translation. J. Immunol. 2015, 194, 5569. [Google Scholar] [CrossRef]
- Bradley, K.L.; Stokes, C.A.; Marciniak, S.J.; Parker, L.C.; Condliffe, A.M. Role of unfolded proteins in lung disease. Thorax 2021, 76, 92–99. [Google Scholar] [CrossRef]
- Marciniak, S.J. Endoplasmic reticulum stress in lung disease. Eur. Respir. Rev. 2017, 26, 170018. [Google Scholar] [CrossRef]
- Tam, A.B.; Roberts, L.S.; Chandra, V.; Rivera, I.G.; Nomura, D.K.; Forbes, D.J.; Niwa, M. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev. Cell 2018, 46, 327–343.e327. [Google Scholar] [CrossRef]
- Khoonkari, M.; Liang, D.; Lima, M.T.; van der Land, T.; Liang, Y.; Sun, J.; Dolga, A.; Kamperman, M.; van Rijn, P.; Kruyt, F.A.E. The Unfolded Protein Response Sensor PERK Mediates Stiffness-Dependent Adaptation in Glioblastoma Cells. Int. J. Mol. Sci. 2022, 23, 6520. [Google Scholar] [CrossRef]
- Vance, M.L. Growth-hormone-releasing hormone. Clin. Chem. 1990, 36, 415–420. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakir, S.; Barabutis, N. Protective Activities of Growth Hormone-Releasing Hormone Antagonists against Toxin-Induced Endothelial Injury. Endocrines 2024, 5, 116-123. https://doi.org/10.3390/endocrines5010008
Fakir S, Barabutis N. Protective Activities of Growth Hormone-Releasing Hormone Antagonists against Toxin-Induced Endothelial Injury. Endocrines. 2024; 5(1):116-123. https://doi.org/10.3390/endocrines5010008
Chicago/Turabian StyleFakir, Saikat, and Nektarios Barabutis. 2024. "Protective Activities of Growth Hormone-Releasing Hormone Antagonists against Toxin-Induced Endothelial Injury" Endocrines 5, no. 1: 116-123. https://doi.org/10.3390/endocrines5010008
APA StyleFakir, S., & Barabutis, N. (2024). Protective Activities of Growth Hormone-Releasing Hormone Antagonists against Toxin-Induced Endothelial Injury. Endocrines, 5(1), 116-123. https://doi.org/10.3390/endocrines5010008