Abstract
Pituitary apoplexy (PA) is a rare medical emergency. The sudden pressure increase in the sella turcica may determine compression on the surrounding structures determining the classical symptomatology associated, especially visual field impairment and/or ocular palsies and hypopituitarism; hypotonic hyponatremia may occur too, even if it is not common. Although already described in the literature, cases of isolated III cranial nerve palsies are extremely rare events. We report the case of a mid-60-year-old man with a known pituitary adenoma accessing the Emergency Department (ED) for worsening headaches unresponsive to analgesics, with a morphological picture consistent with ischemic PA, despite no dimensional increase of the pituitary lesion; upon ED access, a mild paucisymptomatic hyponatremia was also observed. Dexamethasone and mannitol were empirically introduced upon neurosurgical indication and tramadol and ketorolac were promptly administered as well, but without benefit. In the next days, a severe hypotonic hyponatremia was evidenced and a clear left III cranial nerve palsy developed, but no clear signs of cerebral bleeding or ischemia, nor a significant compression on the homolateral cavernous sinus, were observed. Upon ruling out other possible causes, a likely diagnosis of syndrome of inappropriate antidiuresis (SIAD) was made, confirmed by the quick response to fluid restriction. Overall, the sudden fall in tonicity plasma levels seemed to contribute to the exacerbation of the neurological deficit since the normalization of sodium levels was associated with a rapid and complete reversion of the III cranial nerve palsy.
1. Introduction
Pituitary apoplexy (PA) is a rare medical emergency and a potentially life-threatening condition characterized by a sudden pressure increase in the pituitary region, generally due to abrupt hemorrhaging and/or infarction within a pre-existing adenoma [1]; between them, the form due to ischemic vascular accident appears to be less common [2]. PA is characterized by a highly variable clinical presentation, ranging from thunderclap headache, hypopituitarism, and visual disturbance, to more dramatic onsets, such as neurological deficits, coma, and even death [1,3]. Involvement of the endocrine function of the pituitary gland is common, and hypotonic hyponatremia may also occur frequently due to acute central adrenal insufficiency (CAI) and just occasionally due to the increased secretion of arginine vasopressin (AVP) from the neurohypophysis with subsequent syndrome of inappropriate antidiuresis (SIAD) [4]. Neuro-ophthalmic manifestations usually refer to compression of the optic chiasm or to palsy of the III, IV, and VI cranial nerves which pass through the cavernous sinus along the sella turcica and may be compressed by the expanding mass in the pituitary region. Even if the neurological dysfunction typically affects more nerves at once, cases of isolated III cranial nerve palsies are described in the literature, though they seem to be extremely rare events [3]. Ocular palsies generally have a good prognosis, even in conservatively treated cases, but it usually takes weeks or months for a complete recovery of ocular motility [1,5].
We describe herein the excellent and extremely rapid response to conservative treatment in a case of acute isolated III cranial nerve palsy associated with severe hypotonic hyponatremia secondary to ischemic PA.
2. Case Presentation
In March 2015, a mid-60-year-old male patient was diagnosed with a pituitary macroincidentaloma with intra- and suprasellar development (diameter 19 mm × 19 mm × 13 mm) and with cystic components. From a morphological point of view (Figure 1A,B), the adenohypophysis, poorly recognizable, appeared to be displaced above the margin of the adenoma. The lesion determined the obliteration of the suprasellar cistern and was deforming the middle portion of the optic chiasm without significant visual field alterations. At the diagnosis, the patient had a preserved anterior pituitary function with just a mild asymptomatic hyperprolactinemia (Table 1). The patient’s past medical history included: non-toxic plurinodular goiter, prostatic adenoma treated with both tamsulosin and finasteride, and isolated hypercholesterolemia on a polygenic basis in treatment with phytosterols.
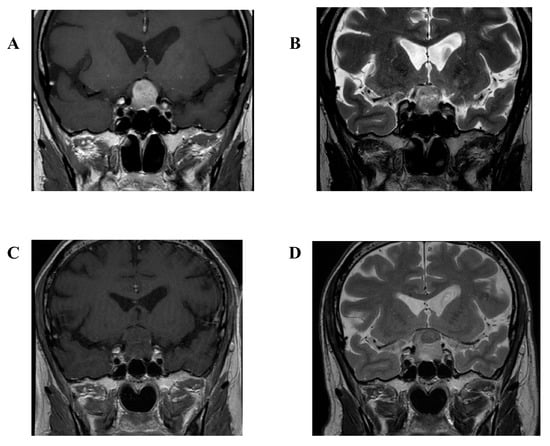
Figure 1.
T1 gadolinium enhanced (A) and T2 weighted (B) magnetic resonance imaging (MRI) of April 2019 showing the patient’s pituitary lesion at the last endocrinological control. T1 gadolinium enhanced (C) and T2 weighted (D) MRI of September 2019 showing the patient’s pituitary lesion stable in size upon Emergency Department admission.

Table 1.
Biochemical changes over time related to the patient’s electrolytes and anterior pituitary and renal function. * evaluated during normal saline infusion at +30′ and +60′. ª on two different measurements. ° after 1 µg Synacthen stimulation test. NA: not available.
The patient was repeatedly informed of the need for exeresis in order to prevent complications, including increased risk of PA and secondary panhypopituitarism [6], but he chose to postpone the surgical option. On 6 September 2019, the patient accessed the Emergency Department (ED) of our University Hospital for worsening headaches and was not responsive to either paracetamol or non-steroidal anti-inflammatory drugs (NSAIDs). In the ED, normal physical and neurological examinations, as well as vital signs, were recorded. The only remarkable biochemical alteration was a mild not-hyperglycemic hyponatremia (s-Na 131 mmol/L, serum glucose 86 mg/dL).
Neither basal brain computed tomography (CT) nor gadolinium-enhanced magnetic resonance imaging (MRI) showed a significant dimensional increase in the known pituitary adenoma (Figure 1A,B). Indeed, the suspicion of ischemic PA was placed on the basis of the radiological picture, with loss of the contrast enhancement by the pituitary lesion (Figure 1C,D). Neurosurgeon consultation indicated the immediate start of dexamethasone 8 mg bis in die and mannitol 18% 125 mL iv. Because of persistent severe headaches, tramadol 100 mg iv and ketorolac 30 mg iv were administered as well, but with no relief. Thus, the patient was admitted to the Neurological Division and therapy with dexamethasone 8 mg bis in die was continued.
The following day, a slight worsening of s-Na levels (127 mmol/L) was observed and initial signs of palsy of the III left cranial nerve appeared (Figure 2). The novel III cranial nerve deficiency was characterized by left eyelid ptosis, anisocoria with non-reactive mydriasis, and diplopia in all directions, with the exception of the ipsilateral abduction movement. An urgent second brain CT scan excluded any signs of recent bleeding, ischemia, or significant increase in the macroadenoma’s dimension. Conversely, at the biochemical analysis, a rapid decrease in s-Na levels became evident (Figure 2), confirming severe hypotonic hyponatremia in the absence of glycosuria (s-Na 110–109 mmol/L, s-K 4.4 mmol/L, p-Osm 230 mOsm/kg, spot urine osmolality 875 mOsm/kg, spot urine sodium 29 mmol/L) and the neurological deficit progressively worsened as well. At the same time, a seemingly panhypopituitarism was diagnosed (LH 0.2 mUI/L, FSH 1.6 UI/L, testosterone 0.21 ng/mL, PRL 1.1 ng/mL, IGF-I 37 ng/mL, TSH 0.21–0.09 µUI/mL, fT4 6.1–5.6 pg/mL) and levothyroxine was promptly started (Table 1); at this stage, a formal assessment of the adrenocortical axis was not possible in view of the ongoing high-dose dexamethasone therapy.
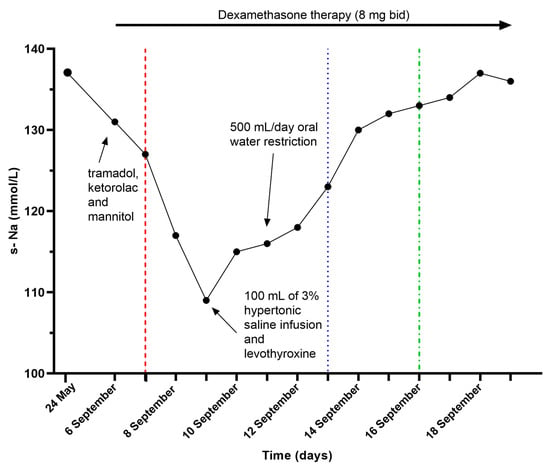
Figure 2.
Serum sodium (s-Na) trend. The red long-dashed line corresponds to neurological worsening, the blue-dotted one indicates mild clinical improvement, and the green dot-dashed one marks complete symptoms regression.
The patient was clinically euvolemic (normal blood pressure, heart rate and capillary refill time, hydrated mucous membranes and no peripheral edema, and balanced fluid intake and output) and in consideration of the compatible tests, a diagnosis of SIAD was made, possibly aggravated by the drugs administered in the ED. He was initially treated with 100 mL of 3% hypertonic saline with a slight increase in s-Na levels (114 mmol/L) and subsequently, an oral strict fluid restriction was started (500 mL/die) with a progressive increase in s-Na levels (s-Na 136–137 mmol/L) and a concomitant rapid resolution in the ocular hemiplegia and in the left eyelid ptosis (Figure 2). Thereafter, corticosteroid therapy with dexamethasone was tapered and switched to substitutive therapy with cortisone acetate.
At resolution of hyponatremia and neurological symptoms, the patient underwent an uncomplicated procedure of transsphenoidal neurosurgery. For the purposes of a pre-operative evaluation, an angio-CT scan was performed and, once again, no signs of dimensional increase of the known lesion were appreciated.
Even the surgery report excluded cavernous sinus invasion, in the presence of signs of scarce previous intralesional bleeding; histological examination revealed the presence of a pituitary adenoma FSH and LH immunoreactive with the abundance of necrotic material and fibrosclerotic tissue, Ki-67 4%, p53 negative (Figure 3). At discharge, stable eunatremia and complete neurological deficit resolution were confirmed.
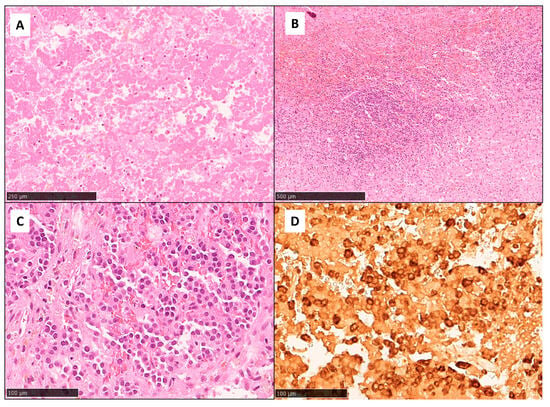
Figure 3.
Histopathological findings showed extensive coagulative necrosis ((A), HE, original magnification: 10×) and hemorrhagic changes with a patchy inflammatory infiltrate ((B), HE, original magnification: 5×). Focal viable areas consistent with a pituitary neuroendocrine tumor (PitNET)/pituitary adenoma were also present ((C), HE, original magnification: 20×) and were positive for both FSH ((D), FSH immunohistochemistry, 20×) and LH.
At the hormonal re-evaluation, performed 3 months after surgery (after a 2-week washout of levothyroxine and 24 h washout of cortisone acetate), a picture of anterior panhypopituitarism was confirmed, as shown in Table 1, with a preserved hydro-electrolytic balance (s-Na 143 mmol/L, s-K 4.7 mmol/L).
At the last evaluation, about three years after surgery, the patient remains stable over time with a normal hydro-electrolyte balance (s-Na 139 mmol/L, s-K 4.9 mmol/L) in the context of an anterior panhypopituitarism successfully treated with a complete substitution therapy and without evidence of remnant adenomatous tissue.
3. Discussion
In our case report, we describe the sudden onset of an isolated III cranial nerve palsy associated with severe hypotonic hyponatremia in a patient with a non-infiltrating, non-functioning pituitary macro-adenoma. Indeed, the contextual onset of headaches poorly responsive to analgesic therapy, a seemingly anterior hypopituitarism, and a possible mild compressive effect on the medial wall of the cavernous sinus, together with the consistent radiological finding in the ED, constitute quite a usual presentation of ischemic PA. Despite this, both the development of isolated III cranial nerve palsy and hypotonic hyponatremia are uncommon events in PA; the subsequent evidence of a rapid neurological response, together with the improvement of the hydro-electrolytic balance, makes this presentation even more peculiar.
PA is a rare clinical syndrome that usually occurs within a pre-existing adenoma, which can be complicated by an acute hemorrhagic or ischemic vascular accident. The consequent rapid increase of the sellar mass leads to the compression of the surrounding structures, thus determining the associated classical symptomatology. The upward expansion can provoke visual field impairment, while the lateral expansion can be responsible for oculomotor palsies, being the III cranial nerve affected in more than half of the cases [1]. Several reasons may explain why this nerve appears to be the more susceptible one. On the one hand, the III cranial nerve travels through the cavernous sinus in close proximity to the pituitary gland and approximately at the same horizontal level. On the other hand, the possible compression of the vasa nervorum by the expanding mass in the turcica sella can compromise the blood supply to the nerve, thus determining a motor dysfunction [7].
Endocrine alterations are frequently encountered in cases of acute PA as well, due to either the destruction and physical damage of the anterior hypophysis or to the mechanical compression on the healthy portion of the gland or the stalk [1]. CAI is the most frequent (50–80%) and potentially dangerous hormonal deficit, but other deficiencies can also be present (30–75%) even though they do not generally raise the same concerns for the patient’s health in the acute setting [1].
Eventually, in cases of PA, hyponatremia (s-Na < 135 mmol/L) can occur as well, though it does not appear to be a common finding [4]. Hyponatremia is the most frequent electrolyte disorder encountered in clinical practice, which is further distinguished on the basis of plasma osmolality into hypotonic, isotonic, and hypertonic forms. The hypotonic form, in particular, if a rapid decrease in s-Na levels occurs, can determine cerebral edema and, at least in part, can contribute to the possible appearance of neurological symptoms that can be reported at PA diagnosis. In the context of PA, hypotonic hyponatremia is generally related to the inability to eliminate free water in the kidney as both low concentrations of cortisol and the possible irritation of the hypothalamus can contribute to an excessive AVP release, determining SIAD.
In our clinical scenario, upon ruling out other possible causes, the likely underlying etiology of hypotonic hyponatremia, although uncommon in this context, was SIAD [4]. This medical condition is characterized by the simultaneous presence of euvolemia and a disruption in AVP central production or its peripheral effect. Indeed, this diagnostic hypothesis was confirmed by the quick response to water restriction, which led to the normalization of s-Na values and consequent clinical benefit. Regarding the etiology of SIAD, many factors may have contributed with a different weight in determining it. As a matter of fact, intense pain is a well-known non-osmotic stimulus for AVP secretion and its effect on the secretory AVP pathway can be enhanced by the simultaneous intake of opioids, such as tramadol, one of the most common synthetic opioids associated with SIAD [8,9]. On the other hand, NSAIDs like ketorolac lead to a decreased production of prostaglandins in the kidney, thus potentiating the renal effect of AVP [10,11]. Moreover, the administration of mannitol in the ED, prescribed empirically upon neurosurgical indication even in the absence of clear signs of cerebral edema, may have potentially contributed to hyponatremia as well, although the infused dose was modest (just 125 mL) and, thus, likely not able of determining such a reduction in sodium values. Of note, in such cases, p-Osm is typically normal or increased (isotonic/hypertonic hyponatremia) [12] and this was not our case since p-Osm was markedly reduced, confirming the finding of severe hypotonic hyponatremia. The least relevant factor in causing hyponatremia may have been hypothyroidism. This medical entity is less likely to be responsible for such a clinical presentation because, in the setting of a secondary hypothyroidism, hyponatremia is generally a manifestation of an associated secondary glucocorticoid deficiency [8]. However, this is not the case for our patient since he was on steroid treatment with dexamethasone, though it is worth noticing that it almost completely lacks the sodium-retaining property of closely related derivatives of hydrocortisone.
As previously marked out, ischemic necrosis of a pituitary macroadenoma associated with isolated III cranial nerve palsy, although extremely rare, has already been described in the literature [3]. In our clinical context, however, no significant increase in the macroadenoma’s size nor any involvement of the cavernous sinus were reported, both at the serial morphologic imaging and at the surgery report. Since these events usually constitute the main responsible for the onset of ophthalmoplegia [13], it is likely that the worsening of plasma tonicity values reasonably aggravated the neuronal damage due to edema, unmasking an otherwise not evident neurological deficit. This theory seems supported by the temporal correlation between electrolyte alteration and motor dysfunction. In particular, while the phenomenon of apoplexy seems to be datable well before entry into ED, the acute neurological deficit was simultaneous with hypotonic hyponatremia, which was further aggravated after medical intervention in the ED. Furthermore, the clinical evolution of the picture was very rapid, with the occurrence and disappearance of the palsy in just about one week, a timing which is much lower than the data available in the literature (median time to disappearance or improvement of neurological symptoms during conservative therapy of about 90 days) [5,14]. Of note, anti-edema treatment with a potent steroid such as dexamethasone, started right upon ED admission, was not sufficient to prevent this complication. On the contrary, an adequate differential diagnosis and treatment of the hydro-electrolyte disorder favored a prompt recovery of ocular function.
Consequently, the correct interpretation of this unique clinical case was obtained by combining several factors. First, the presence of a macroadenoma in close proximity to the cavernous sinus, where the involved nerve passes through, despite the absence of a clear involvement or compression. Secondly, the radiological and clinical diagnosis of ischemic PA, confirmed by the histopathological report, might have contributed to the local inflammatory edema, despite no significant increase in the size of the adenoma being reported. Finally, the sudden drop in plasma tonicity probably aggravated the extent of neuronal edema, already affecting the III cranial nerve, and contributed to a possible brainstem edema, resulting in slowing axonal fiber conduction velocity. Of note, the neuronal edema may have facilitated, at the same time, a mild compression of the involved nerve at the entrance of the oculomotor trigone, as already proposed by other authors [15,16]. All these factors contributed to the development of the selective and acute motor dysfunction described here.
In conclusion, both hypotonic hyponatremia and isolated III cranial nerve palsy are possible, though uncommon, manifestations of acute PA. To our knowledge, however, this was the first case reporting a possible relationship between them. All this considered, in patients with pituitary macroadenoma presenting with hypotonic hyponatremia and motor dysfunction, both cavernous sinus infiltration and neurological causes should be excluded. In any case, physicians should also consider the possibility of paralysis exacerbated by plasma hypotonicity, as the normalization of the s-Na has been shown to be highly beneficial in restoring clinical symptoms. In particular, we suggest an early assessment of the hydro-electrolytic balance in every patient affected by a known pituitary-hypothalamic lesion who manifests acute neurological symptoms that cannot be otherwise explained.
Author Contributions
Writing—original draft preparation, E.V.; writing—review and editing, A.M.B. and M.M.; supervision, V.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical review and approval were/are not due for case report.
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper/case report.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank the patient for giving the consent to the publication of his medical history. The authors would like to thank Luca Bertero for kindly providing us with hystopathological slides.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Briet, C.; Salenave, S.; Bonneville, J.-F.; Laws, E.R.; Chanson, P. Pituitary Apoplexy. Endocr. Rev. 2015, 36, 622–645. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Niizuma, K.; Mugikura, S.; Tominaga, T. Ischemic Pituitary Adenoma Apoplexy-Clinical Appearance and Prognosis after Surgical Intervention. Clin. Neurol. Neurosurg. 2016, 148, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Murad-Kejbou, S.; Eggenberger, E. Pituitary Apoplexy: Evaluation, Management, and Prognosis. Curr. Opin. Ophthalmol. 2009, 20, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Capatina, C.; Inder, W.; Karavitaki, N.; Wass, J.A.H. Management of Endocrine Disease: Pituitary Tumour Apoplexy. Eur. J. Endocrinol. 2015, 172, R179–R190. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.; Ramaswamy, S.; Sucharew, H.; Vagal, A.; Anziska, Y.; Levine, S.R. Isolated Third Cranial Nerve Palsy in Pituitary Apoplexy: Case Report and Systematic Review. J. Stroke Cerebrovasc. Dis. 2021, 30, 105969. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Castro, M.; Paredes, I.; Pérez-López, C.; García Feijoo, P.; Alvarez-Escola, C.; Calatayud, M.; Lagares, A.; Soledad Librizzi, M.; Acitores Cancela, A.; Rodríguez Berrocal, V. Differences in Clinical, Hormonal, and Radiological Presentation and in Surgical Outcomes in Patients Presenting with and without Pituitary Apoplexy. A Multicenter Study of 245 Cases. Pituitary 2023, 26, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.-J.; Joo, S.-P.; Kim, T.-S.; Seo, B.-R. Pituitary Apoplexy Presenting as Isolated Third Cranial Nerve Palsy with Ptosis: Two Case Reports. J. Korean Neurosurg. Soc. 2009, 45, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G.; Goldsmith, S.R.; Greenberg, A.; Korzelius, C.; Schrier, R.W.; Sterns, R.H.; Thompson, C.J. Diagnosis, Evaluation, and Treatment of Hyponatremia: Expert Panel Recommendations. Am. J. Med. 2013, 126, S1–S42. [Google Scholar] [CrossRef] [PubMed]
- Fournier, J.-P.; Yin, H.; Nessim, S.J.; Montastruc, J.-L.; Azoulay, L. Tramadol for Noncancer Pain and the Risk of Hyponatremia. Am. J. Med. 2015, 128, 418–425.e5. [Google Scholar] [CrossRef] [PubMed]
- Varaldo, E.; Sibilla, M.; Bioletto, F.; Cuboni, D.; Prencipe, N.; Bona, C.; Ferrari, M.; Viglino, F.; Aversa, L.S.; Grottoli, S.; et al. Neuroendocrine Response to Diclofenac in Healthy Subjects: A Pilot Study. J. Endocrinol. Investig. 2023, epub ahead of print. [Google Scholar] [CrossRef]
- Liamis, G.; Milionis, H.; Elisaf, M. A Review of Drug-Induced Hyponatremia. Am. J. Kidney Dis. 2008, 52, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Kirschenbaum, M.A. Severe Mannitol-Induced Hyponatremia Complicating Transurethral Prostatic Resection. J. Urol. 1979, 121, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Hage, R.; Eshraghi, S.R.; Oyesiku, N.M.; Ioachimescu, A.G.; Newman, N.J.; Biousse, V.; Bruce, B.B. Third, Fourth, and Sixth Cranial Nerve Palsies in Pituitary Apoplexy. World Neurosurg. 2016, 94, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Prescott, H.; Ellis, E.; Soule, S. Pituitary Infarction: A Potentially Fatal Cause of Postoperative Hyponatraemia and Ocular Palsy. BMJ 2011, 342, d1221. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jiang, S.; Yang, C.; Deng, K.; Wang, R.; Bao, X. Surgical Treatment of a 72-Year-Old Patient with Headache, Hyponatremia and Oculomotor Nerve Palsy: A Case Report and Literature Review. Gland. Surg. 2021, 10, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Kawabori, M.; Terasaka, S.; Murata, J.; Houkin, K. A Possible Mechanism of Isolated Oculomotor Nerve Palsy by Apoplexy of Pituitary Adenoma without Cavernous Sinus Invasion: A Report of Two Cases. Acta Neurochir. Wien. 2011, 153, 2453–2456, discussion 2456. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).