
Inclusion and Withdrawal Criteria for Growth Hormone (GH) Therapy in Children with Idiopathic GH Deficiency—Towards Following the Evidence but Still with Unresolved Problems

Abstract
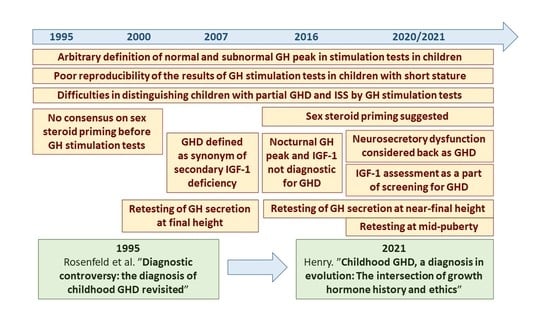
1. Introduction
2. Problems Related to Direct Assessment of GH Secretion
2.1. Arbitrarily Defined Subnormal GH Peak in Stimulation Tests
2.2. Disregarding Factors Influencing GH Secretion in Interpreting the Results of Stimulation Tests
2.3. Insufficient Diagnostic Accuracy and Poor Reproducibility of GH Stimulation Tests
2.4. Discrepancies between Spontaneous and Stimulated GH Secretion
2.5. Neurosecretory Dysfunction of GH Secretion as a Clinical Entity
3. Problems Related to Diagnosing GHD as Secondary IGF-1 Deficiency
3.1. Discrepancies between IGF-1 Levels and the Results of GH Stimulation Tests
3.2. Importance of Direct Confirmation of Secondary IGF-1 Deficiency in the Diagnosis of GHD
3.3. Re-Standardization of Assays and the Need for Validation of IGF-1 Reference Ranges
4. Problems Related to the Duration of rhGH Therapy in Children with Idiopathic GHD
4.1. Early Identification of Non-Responders
4.2. Optimal Time for rhGH Therapy Withdrawal and Retsting of GH Secretion
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Grimberg, A.; DiVall, S.A.; Polychronakos, C.; Allen, D.B.; Cohen, L.E.; Quintos, J.B.; Rossi, W.C.; Feudtner, C.; Murad, M.H. Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency. Horm. Res. Paediatr. 2016, 86, 361–397. [Google Scholar] [CrossRef] [PubMed]
- Cavarzere, P.; Gaudino, R.; Sandri, M.; Ramaroli, D.A.; Pietrobelli, A.; Zaffanello, M.; Guzzo, A.; Salvagno, G.L.; Piacentini, G.; Antoniazzi, F. Growth hormone retesting during puberty: A cohort study. Eur. J. Endocrinol. 2020, 182, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.-M.; Ranke, M.B.; Kelnar, C.J.H. The ESPE classification of paediatric endocrine diagnoses. Horm. Res. 2007, 68, 1–5. [Google Scholar]
- Savage, M.O.; Burren, C.P.; Rosenfeld, R.G. The continuum of growth hormone-IGF-I axis defects causing short stature: Diagnostic and therapeutic challenges. Clin. Endocrinol. 2010, 72, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Bidlingmaier, M.; De Bruin, C. A Proposal for the Interpretation of Serum IGF-I Concentration as Part of Laboratory Screening in Children with Growth Failure. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 130–139. [Google Scholar] [CrossRef]
- Spiliotis, B.E.; Alexandrides, T.K.; Karystianos, C.; Vassilakos, P.; Zadik, Z.; Nikolakopoulou, N.M.; Nikiforidis, G.; Beratis, N.G. The insulin-like growth factor-I (IGF-I) generation test as an indicator of growth hormone status. Hormones 2009, 8, 117–128. [Google Scholar] [CrossRef]
- Buckway, C.K.; Rosenfeld, R.G. The IGF-I Generation Test Revisited: A Marker of GH Sensitivity. J. Clin. Endocrinol. Metab. 2001, 86, 5176–5183. [Google Scholar] [CrossRef]
- Cotterill, A.M.; Camacho-Hübner, C.; Duquesnoy, P.; Savage, M.O. Changes in serum IGF-I and IGFBP-3 concentrations during the IGF-I generation test performed prospectively in children with short stature. Clin. Endocrinol. 1998, 48, 719–724. [Google Scholar] [CrossRef]
- Coutant, R.; Dorr, H.-G.; Gleeson, H.; Argente, J. Diagnosis of endocrine disease: Limitations of the IGF1 generation test in children with short stature. Eur. J. Endocrinol. 2012, 166, 351–357. [Google Scholar] [CrossRef]
- Storr, H.L.; Chatterjee, S.; Metherell, L.A.; Foley, C.; Rosenfeld, R.G.; Backeljauw, P.F.; Dauber, A.; Savage, M.O.; Hwa, V. Nonclassical GH Insensitivity: Characterization of Mild Abnormalities of GH Action. Endocr. Rev. 2019, 40, 476–505. [Google Scholar] [CrossRef]
- Collett-Solberg, P.F.; Ambler, G.; Backeljauw, P.F.; Bidlingmaier, M.; Biller, B.M.K.; Boguszewski, M.C.S.; Cheung, P.T.; Choong, C.S.Y.; Cohen, L.E.; Cohen, P.; et al. Diagnosis, Genetics, and Therapy of Short Stature in Children: A Growth Hormone Research Society International Perspective. Horm. Res. Paediatr. 2019, 92, 1–14. [Google Scholar] [CrossRef]
- Binder, G.; Reinehr, T.; Ibáñez, L.; Thiele, S.; Linglart, A.; Woelfe, J.; Saenger, P.; Bettendorf, M.; Zachurzok, A.; Goglke, B.; et al. GHD Diagnostics in Europe and the US: An Audit of National Guidelines and Practice. Horm. Res. Paediatr. 2020, 92, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.G.; Albertsson-Wikland, K.; Cassorla, F.; Frasier, S.D.; Hasegawa, Y.; Hintz, R.L.; Lafranchi, S.; Lippe, B.; Loriaux, L.; Melmed, S.; et al. Diagnostic Controversy: The Diagnosis of Childhood Growth-Hormone Deficiency Revisited. J. Clin. Endocrinol. Metab. 1995, 80, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, C.; Hawkes, C.P.; Grimberg, A. Provocative growth hormone testing in children: How did we get here and where do we go now? J. Pediatr. Endocrinol. Metab. 2021, 34, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: Summary statement of the GH Research Society. J. Clin. Endocrinol. Metab. 2000, 85, 3990–3993. [Google Scholar] [CrossRef]
- Van den Broeck, J.; Arends, N.; Hokken-Koelega, A. Growth response to recombinant human growth hormone (GH) in children with idiopathic growth retardation by level of maximum GH peak during GH stimulation tests. Horm. Res. 2000, 53, 267–273. [Google Scholar] [CrossRef]
- Smyczyńska, J.; Lewiński, A.; Hilczer, M.; Stawerska, R.; Karasek, M. Partial growth hormone deficiency (GHD) in children has more similarities to idiopathic short stature than to severe GHD. Endokrynol. Pol. 2007, 58, 182–187. [Google Scholar]
- Webb, E.A.; Dattani, M.T. Diagnosis of growth hormone deficiency. Endocr. Dev. 2010, 18, 55–66. [Google Scholar] [CrossRef]
- Wagner, I.V.; Paetzold, C.; Gausche, R.; Vogel, M.; Koerner, A.; Thiery, J.; Arsene, C.G.; Henrion, A.; Guettler, B.; Keller, E.; et al. Clinical evidence-based cutoff limits for GH stimulation tests in children with a backup of results with reference to mass spectrometry. Eur. J. Endocrinol. 2014, 171, 389–397. [Google Scholar] [CrossRef]
- Murray, P.G.; Dattani, M.T.; Clayton, P.E. Controversies in the diagnosis and management of growth hormone deficiency in childhood and adolescence. Arch. Dis. Child. 2016, 101, 96–100. [Google Scholar] [CrossRef]
- Gourmelen, M.; Pham-Huu-Tsrung, M.T.; Girard, F. Transient partial hGH deficiency in prepubertal children with delay of growth. Pediatr. Res. 1979, 13, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Mauras, N.; Blizzard, R.M.; Link, K.; Johnson, M.L.; Rogol, A.D.; Veldhuis, J.D. Augmentation of growth hormone secretion during puberty: Evidence for a pulse amplitude-modulated phenomenon. J. Clin. Endocrinol. Metab. 1987, 64, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Marin, G.; Domené, H.M.; Barnes, K.M.; Blackwell, B.J.; Cassorla, F.G.; Cutler, G.B.J. The effects of estrogen priming and puberty on the growth hormone response to standardized treadmill exercise and arginine-insulin in normal girls and boys. J. Clin. Endocrinol. Metab. 1994, 79, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Gonc, E.N.; Kandemir, N.; Ozon, A.; Alikasifoglu, A. Final heights of boys with normal growth hormone responses to provocative tests following priming. J. Pediatr. Endocrinol. Metab. 2008, 21, 963–971. [Google Scholar] [CrossRef]
- Radetti, G.; Elsedfy, H.H.; Khalaf, R.; Meazza, C.; Pagani, S.; El Kholy, M.; Albertini, R.; De Stefano, A.M.; Navarra, A.; De Silvestri, A.; et al. Pegvisomant-primed growth hormone (GH) stimulation test is useful in identifying true GH deficient children. Hormones 2017, 16, 291–296. [Google Scholar] [CrossRef]
- Stanley, T.L.; Levitsky, L.L.; Grinspoon, S.K.; Misra, M. Effect of body mass index on peak growth hormone response to provocative testing in children with short stature. J. Clin. Endocrinol. Metab. 2009, 94, 4875–4881. [Google Scholar] [CrossRef]
- Barrett, J.; Maranda, L.; Nwosu, B.U. The relationship between subnormal peak-stimulated growth hormone levels and auxological characteristics in obese children. Front. Endocrinol. 2014, 5, 35. [Google Scholar] [CrossRef]
- Yang, A.; Cho, S.Y.; Kwak, M.J.; Kim, S.J.; Park, S.W.; Jin, D.K.; Lee, J.E. Impact of BMI on peak growth hormone responses to provocative tests and therapeutic outcome in children with growth hormone deficiency. Sci. Rep. 2019, 9, 16181. [Google Scholar] [CrossRef]
- Lennartsson, O.; Nilsson, O.; Lodefalk, M. Discordance Between Stimulated and Spontaneous Growth Hormone Levels in Short Children Is Dependent on Cut-Off Level and Partly Explained by Refractoriness. Front. Endocrinol. 2020, 11, 584906. [Google Scholar] [CrossRef]
- Abawi, O.; Augustijn, D.; Hoeks, S.E.; de Rijke, Y.B.; van den Akker, E.L.T. Impact of body mass index on growth hormone stimulation tests in children and adolescents: A systematic review and meta-analysis. Crit. Rev. Clin. Lab. Sci. 2021, 58, 576–595. [Google Scholar] [CrossRef]
- Kelnar, C.J.H. The evidence base for growth hormone effectiveness in children. Endocr. Dev. 2010, 18, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.J.; Hindmarsh, P.C.; Dunger, D.B. Growth hormone (GH) provocation tests and the response to GH treatment in GH deficiency. Arch. Dis. Child. 2004, 89, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Zadik, Z.; Chalew, S.A.; Gilula, Z.; Kowarski, A.A. Reproducibility of growth hormone testing procedures: A comparison between 24-hour integrated concentration and pharmacological stimulation. J. Clin. Endocrinol. Metab. 1990, 71, 1127–1130. [Google Scholar] [CrossRef]
- Hintz, R.L. Growth Hormone Neurosecretory Dysfunction as a Clinical Entity. Clin. Pediatr. Endocrinol. 1995, 4, 9–19. [Google Scholar] [CrossRef]
- Hilczer, M.; Smyczynska, J.; Lewinski, A. Limitations of clinical utility of growth hormone stimulating tests in diagnosing children with short stature. Endocr. Regul. 2006, 40, 69–75. [Google Scholar]
- Loche, S.; Bizzarri, C.; Maghnie, M.; Faedda, A.; Tzialla, C.; Autelli, M.; Casini, M.R.; Cappa, M. Results of early reevaluation of growth hormone secretion in short children with apparent growth hormone deficiency. J. Pediatr. 2002, 140, 445–449. [Google Scholar] [CrossRef]
- Hilczer, M.; Smyczyńska, J.; Stawerska, R.; Lewiński, A. Stability of IGF-I concentration despite divergent results of repeated GH stimulating tests indicates poor reproducibility of test results. Endocr. Regul. 2006, 40, 37–45. [Google Scholar]
- Wit, J.M.; Joustra, S.D.; Losekoot, M.; Van Duyvenvoorde, H.A.; De Bruin, C. Differential diagnosis of the short IGF-I-deficient child with apparently normal growth hormone secretion. Horm. Res. Paediatr. 2021, 94, 81–104. [Google Scholar] [CrossRef]
- Obara-Moszynska, M.; Kedzia, A.; Korman, E.; Niedziela, M. Usefulness of growth hormone (GH) stimulation tests and IGF-I concentration measurement in GH deficiency diagnosis. J. Pediatr. Endocrinol. Metab. 2008, 21, 569–579. [Google Scholar] [CrossRef]
- Smyczyńska, J.; Stawerska, R.; Lewiński, A.; Hilczer, M. Limited usefulness of the test of spontaneous growth hormone (GH) nocturnal secretion as a screening procedure in diagnosing GH deficiency in children with short stature. Ann. Agric. Environ. Med. 2014, 21, 893–897. [Google Scholar] [CrossRef][Green Version]
- Rose, S.R.; Ross, J.L.; Uriarte, M.; Barnes, K.M.; Cassorla, F.G.; Cutler, G.B.J. The advantage of measuring stimulated as compared with spontaneous growth hormone levels in the diagnosis of growth hormone deficiency. N. Engl. J. Med. 1988, 319, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Spiliotis, B.E.; August, G.P.; Hung, W.; Sonis, W.; Mendelson, W.; Bercu, B.B. Growth hormone neurosecretory dysfunction. JAMA 1984, 251, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Radetti, G.; Buzi, F.; Cassar, W.; Paganini, C.; Stacul, E.; Maghnie, M. Growth hormone secretory pattern and response to treatment in children with short stature followed to adult height. Clin. Endocrinol. 2003, 59, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Rogol, A.D.; Blethen, S.L.; Sy, J.P.; Veldhuis, J.D. Do growth hormone (GH) serial sampling, insulin-like growth factor-I (IGF-I) or auxological measurements have an advantage over GH stimulation testing in predicting the linear growth response to GH therapy? Clin. Endocrinol. 2003, 58, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Deeb, A.; Bin-Abbas, B.; Al Mutair, A.; Koledova, E.; Savage, M.O. Achieving optimal short- and long-term responses to paediatric growth hormone therapy. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 329–340. [Google Scholar] [CrossRef]
- Binder, G.; Hähnel, J.; Weber, K.; Schweizer, R. Adult height after treatment of neurosecretory dysfunction and comparison to idiopathic GHD. Clin. Endocrinol. 2022, 96, 184–189. [Google Scholar] [CrossRef]
- Inoue-Lima, T.H.; Vasques, G.A.; Scalco, R.C.; Nakaguma, M.; Mendonca, B.B.; Arnhold, I.J.P.; Jorge, A.A.L. IGF-1 assessed by pubertal status has the best positive predictive power for GH deficiency diagnosis in peripubertal children. J. Pediatr. Endocrinol. Metab. 2019, 32, 173–179. [Google Scholar] [CrossRef]
- Blum, W.F.; Albertsson-Wikland, K.; Rosberg, S.; Ranke, M.B. Serum levels of insulin-like growth factor I (IGF-I) and IGF binding protein 3 reflect spontaneous growth hormone secretion. J. Clin. Endocrinol. Metab. 1993, 76, 1610–1616. [Google Scholar] [CrossRef]
- Ho, K.K.Y. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: A statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur. J. Endocrinol. 2007, 157, 695–700. [Google Scholar] [CrossRef]
- Ibba, A.; Corrias, F.; Guzzetti, C.; Casula, L.; Salerno, M.; Di Iorgi, N.; Tornese, G.; Patti, G.; Radetti, G.; Maghnie, M.; et al. Igf1 for the diagnosis of growth hormone deficiency in children and adolescents: A reappraisal. Endocr. Connect. 2020, 9, 1095–1102. [Google Scholar] [CrossRef]
- Iwayama, H.; Kitagawa, S.; Sada, J.; Miyamoto, R.; Hayakawa, T.; Kuroyanagi, Y.; Muto, T.; Kurahashi, H.; Ohashi, W.; Takagi, J.; et al. Insulin-like growth factor-1 level is a poor diagnostic indicator of growth hormone deficiency. Sci. Rep. 2021, 11, 16159. [Google Scholar] [CrossRef] [PubMed]
- Smyczyńska, J.; Lewiński, A.; Stawerska, R.; Hilczer, M.; Karasek, M. Assessment of insulin-like growth factor-I serum concentration as a screening procedure in diagnosing children with short stature. Neuroendocrinol. Lett. 2007, 28, 274–278. [Google Scholar] [PubMed]
- Meazza, C.; Elsedfy, H.H.; Khalaf, R.I.; Lupi, F.; Pagani, S.; El Kholy, M.; Tinelli, C.; Radetti, G.; Bozzola, M. Serum α-klotho levels are not informative for the evaluation of growth hormone secretion in short children. J. Pediatr. Endocrinol. Metab. 2017, 30, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.O.; Blum, W.F.; Ranke, M.B.; Postel-Vinay, M.C.; Cotterill, A.M.; Hall, K.; Chatelain, P.G.; Preece, M.A.; Rosenfeld, R.G. Clinical features and endocrine status in patients with growth hormone insensitivity (Laron syndrome). J. Clin. Endocrinol. Metab. 1993, 77, 1465–1471. [Google Scholar] [CrossRef]
- Rudman, D.; Kutner, M.H.; Blackston, R.D.; Cushman, R.A.; Bain, R.P.; Patterson, J.H. Children with normal-variant short stature: Treatment with human growth hormone for six months. N. Engl. J. Med. 1981, 305, 123–131. [Google Scholar] [CrossRef]
- Jorge, A.A.; Souza, S.C.; Arnhold, I.J.; Mendonca, B.B. Poor reproducibility of IGF-I and IGF binding protein-3 generation test in children with short stature and normal coding region of the GH receptor gene. J. Clin. Endocrinol. Metab. 2002, 87, 469–472. [Google Scholar] [CrossRef][Green Version]
- Smyczyńska, J.; Smyczyńska, U.; Hilczer, M.; Stawerska, R.; Lewiński, A. Significance of direct confirmation of growth hormone insensitivity for the diagnosis of primary IGF-I deficiency. J. Clin. Med. 2020, 9, 240. [Google Scholar] [CrossRef]
- Smyczyńska, J.; Hilczer, M.; Stawerska, R.; Lewiński, A. Significant increase of IGF-I concentration and of IGF-I/IGFBP-3 molar ratio in generation test predicts the good response to growth hormone (GH) therapy in children with short stature and normal results of GH stimulating tests. Neuroendocrinol. Lett. 2013, 34, 222–228. [Google Scholar]
- Hilczer, M.; Smyczyńska, J.; Lewiński, A. Monitoring and optimising of growth hormone (GH) therapy in GH-deficient children—The role of assessment of insulin-like growth factor-I (IGF-I) secretion and IGF-I/IGF binding protein-3 molar ratio. Med. Sci. Technol. 2006, 47, 219–223. [Google Scholar]
- Gaddas, M.; Périn, L.; Le Bouc, Y. Evaluation of IGF1/IGFBP3 molar ratio as an effective tool for assessing the safety of growth hormone therapy in small-for-gestational-age, growth hormone-deficient and prader-willi children. JCRPE J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 253–261. [Google Scholar] [CrossRef]
- Renes, J.S.; van Doorn, J.; Hokken-Koelega, A.C. Ternary complex formation and IGFBP-3 proteolytic activity during childhood: Age-dependent changes. J. Clin. Endocrinol. Metab. 2014, 99, E1988–E1996. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Renes, J.S.; van Doorn, J.; Hokken-Koelega, A.C.S. Current Insights into the Role of the Growth Hormone-Insulin-Like Growth Factor System in Short Children Born Small for Gestational Age. Horm. Res. Paediatr. 2019, 92, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Bøtkjær, J.A.; Noer, P.R.; Oxvig, C.; Andersen, C.Y. A common variant of the pregnancy-associated plasma protein-A (PAPPA) gene encodes a protein with reduced proteolytic activity towards IGF-binding proteins. Sci. Rep. 2019, 9, 13231. [Google Scholar] [CrossRef] [PubMed]
- Bang, P.; Thorell, A.; Carlsson-Skwirut, C.; Ljungqvist, O.; Brismar, K.; Nygren, J. Free dissociable IGF-I: Association with changes in IGFBP-3 proteolysis and insulin sensitivity after surgery. Clin. Nutr. 2016, 35, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Elmlinger, M.W.; Kühnel, W.; Weber, M.M.; Ranke, M.B. Reference ranges for two automated chemiluminescent assays for serum insulin-like growth factor I (IGF-I) and IGF-binding protein 3 (IGFBP-3). Clin. Chem. Lab. Med. 2004, 42, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.F.; Schweizer, R. Insulin-like growth factors and their binding proteins. In Diagnostics of Endocrine Function in Children and Adolescents; Ranke, M.B., Ed.; Karger: Basel, Switzerland, 2003; pp. 166–169. [Google Scholar]
- Siemens Healthcare Diagnostics Inc. Introducing the Restandardized Insulin-Like Growth Factor-I (IGF-I) Assay. Cust. Bull. 2016, 5, 1–12. [Google Scholar]
- Bang, P.; Bjerknes, R.; Dahlgren, J.; Dunkel, L.; Gustafsson, J.; Juul, A.; Kriström, B.; Tapanainen, P.; Åberg, V. A comparison of different definitions of growth response in short prepubertal children treated with growth hormone. Horm. Res. Paediatr. 2011, 75, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Bang, P.; Ahmed, S.F.; Argente, J.; Backeljauw, P.; Bettendorf, M.; Bona, G.; Coutant, R.; Rosenfeld, R.G.; Walenkamp, M.-J.; Savage, M.O. Identification and management of poor response to growth-promoting therapy in children with short stature. Clin. Endocrinol. 2012, 77, 169–181. [Google Scholar] [CrossRef]
- Straetemans, S.; De Schepper, J.; Thomas, M.; Tenoutasse, S.; Beauloye, V.; Rooman, R. Criteria for First-Year Growth Response to Growth Hormone Treatment in Prepubertal Children with Growth Hormone Deficiency: Do They Predict Poor Adult Height Outcome? Front. Endocrinol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Straetemans, S.; Thomas, M.; Craen, M.; Rooman, R.; De Schepper, J. Poor growth response during the first year of growth hormone treatment in short prepubertal children with growth hormone deficiency and born small for gestational age: A comparison of different criteria. Int. J. Pediatr. Endocrinol. 2018, 2018, 9. [Google Scholar] [CrossRef]
- Straetemans, S.; Rooman, R.; De Schepper, J. Is a Two-Year Growth Response to Growth Hormone Treatment a Better Predictor of Poor Adult Height Outcome Than a First-Year Growth Response in Prepubertal Children with Growth Hormone Deficiency? Front. Endocrinol. 2021, 12, 678094. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Ranke, M.B.; Albertsson-Wikland, K.; Carrascosa, A.; Rosenfeld, R.G.; Van Buuren, S.; Kristrom, B.; Schoenau, E.; Audi, L.; Hokken-Koelega, A.C.S.; et al. Personalized Approach to Growth Hormone Treatment: Clinical Use of Growth Prediction Models. Horm. Res. Paediatr. 2013, 79, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Hindmarsh, P.C.; Cole, T.J. Evidence-based growth hormone therapy prediction models. J. Pediatr. Endocrinol. Metab. 2000, 13 (Suppl. S6), 1359–1364. [Google Scholar] [CrossRef]
- Smyczyńska, U.; Smyczyńska, J.; Tadeusiewicz, R. Neural modelling of growth hormone therapy for the prediction of therapy results. Bio-Algorithms Med-Syst. 2015, 11, 33–45. [Google Scholar] [CrossRef]
- Carel, J.C.; Ecosse, E.; Nicolino, M.; Tauber, M.; Leger, J.; Cabrol, S.; Bastie-Sigeac, I.; Chaussain, J.L.; Coste, J. bservational follow up study of the FAdult height after long term treatment with recombinant growth hormone for idiopathic isolated growth hormone deficiency: Orench population based registry. BMJ 2002, 325, 70. [Google Scholar] [CrossRef]
- De Ridder, M.A.J.; Stijnen, T.; Hokken-Koelega, A.C.S. Prediction of adult height in growth-hormone-treated children with growth hormone deficiency. J. Clin. Endocrinol. Metab. 2007, 92, 925–931. [Google Scholar] [CrossRef]
- Ranke, M.B.; Lindberg, A.; Chatelain, P.; Wilton, P.; Cutfield, W.; Albertsson-Wikland, K.; Price, D.A. Derivation and Validation of a Mathematical Model for Predicting the Response to Exogenous Recombinant Human Growth Hormone (GH) in Prepubertal Children with Idiopathic GH Deficiency. J. Clin. Endocrinol. Metab. 1999, 84, 1174–1183. [Google Scholar] [CrossRef]
- Ranke, M.B.; Lindberg, A.; Martin, D.D.; Bakker, B.; Wilton, P.; Albertsson-Wikland, K.; Cowell, C.T.; Price, D.A.; Reiter, E.O. The mathematical model for total pubertal growth in idiopathic growth hormone (GH) deficiency suggests a moderate role of GH dose. J. Clin. Endocrinol. Metab. 2003, 88, 4748–4753. [Google Scholar] [CrossRef]
- Smyczyńska, U.; Smyczyńska, J.; Hilczer, M.; Stawerska, R.; Tadeusiewicz, R.; Lewiński, A. Pre-treatment growth and IGF-I deficiency as main predictors of response to growth hormone therapy in neural models. Endocr. Connect. 2018, 7, 239–249. [Google Scholar] [CrossRef]
- Attanasio, A.; Attie, K.; Baxter, R.; Bengtsson, B.-A.; Black, A.; Blethen, S.; Carlsson, L.; Casaneuva, F.; Chipman, J.; Christiansen, J.S.; et al. Consensus guidelines for the diagnosis and treatment of adults with growth hormone deficiency: Summary statement of the growth hormone research society workshop on adult growth hormone deficiency. J. Clin. Endocrinol. Metab. 1998, 83, 379–381. [Google Scholar] [CrossRef]
- Zucchini, S.; Pirazzoli, P.; Baronio, F.; Gennari, M.; Bal, M.O.; Balsamo, A.; Gualandi, S.; Cicognani, A. Effect on adult height of pubertal growth hormone retesting and withdrawal of therapy in patients with previously diagnosed growth hormone deficiency. J. Clin. Endocrinol. Metab. 2006, 91, 4271–4276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Penta, L.; Cofini, M.; Lucchetti, L.; Zenzeri, L.; Leonardi, A.; Lanciotti, L.; Galeazzi, D.; Verrotti, A.; Esposito, S. Growth hormone (GH) therapy during the transition period: Should we think about early retesting in patients with idiopathic and isolated GH deficiency? Int. J. Environ. Res. Public Health 2019, 16, 307. [Google Scholar] [CrossRef] [PubMed]
- Krukowska-Andrzejczyk, B.J.; Kalina, M.; Kalina-Faska, B.; Małecka-Tendera, E. Growth hormone therapy in children with partial growth hormone deficiency. Are we treating the right patients? Pediatr. Endocrinol. Diabetes Metab. 2020, 26, 65–72. [Google Scholar] [CrossRef]
- Ranke, M.B.; Schweizer, R.; Binder, G. Basal characteristics and first year responses to human growth hormone (GH) vary according to diagnostic criteria in children with non-acquired GH deficiency (naGHD): Observations from a single center over a period of five decades. J. Pediatr. Endocrinol. Metab. 2018, 31, 1257–1266. [Google Scholar] [CrossRef]
- Felício, J.S.; Janaú, L.C.; Moraes, M.A.; Zahalan, N.A.; de Souza Resende, F.; de Lemos, M.N.; de Souza Neto, N.J.K.; Farias de Franco, I.I.; Leitão, L.T.C.; Silva, L.D.S.D.A.; et al. Diagnosis of Idiopathic GHD in Children Based on Response to rhGH Treatment: The Importance of GH Provocative Tests and IGF-1. Front. Endocrinol. 2019, 10, 638. [Google Scholar] [CrossRef]
- Cohen, P.; Rogol, A.D.; Deal, C.L.; Saenger, P.; Reiter, E.O.; Ross, J.L.; Chernausek, S.D.; Savage, M.O.; Wit, J.M. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: A summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J. Clin. Endocrinol. Metab. 2008, 93, 4210–4217. [Google Scholar] [CrossRef]
- Henry, R.K. Childhood growth hormone deficiency, a diagnosis in evolution: The intersection of growth hormone history and ethics. Growth Horm. IGF Res. 2020, 55, 101358. [Google Scholar] [CrossRef]
- Rodari, G.; Profka, E.; Giacchetti, F.; Cavenaghi, I.; Arosio, M.; Giavoli, C. Influence of biochemical diagnosis of growth hormone deficiency on replacement therapy response and retesting results at adult height. Sci. Rep. 2021, 11, 14553. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Problems related to direct assessment of GH secretion |
| 1. Arbitrarily defined subnormal GH peak in stimulation tests. 2. Disregarding factors influencing GH secretion in interpreting the results of stimulation tests. 3. Insufficient diagnostic accuracy and poor reproducibility of GH stimulation tests. 4. Discrepancies between spontaneous and stimulated GH secretion. 5. Neurosecretory dysfunction of GH secretion as a clinical entity. |
| Problems related to diagnosing GHD as secondary IGF-1 deficiency |
| 1. Discrepancies between IGF-1 levels and the results of GH stimulation tests. 2. Significance of direct confirmation of secondary IGF-1 deficiency for the diagnosis of GHD. 3. Re-standardization of assays and the need for validation of IGF-1 reference ranges. |
| Problems related to the duration of rhGH therapy in children with idiopathic GHD |
| 1. Discrepancies between IGF-1 levels and the results of GH stimulation tests. 2. Optimal time for rhGH therapy withdrawal and retesting of GH secretion. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyczyńska, J. Inclusion and Withdrawal Criteria for Growth Hormone (GH) Therapy in Children with Idiopathic GH Deficiency—Towards Following the Evidence but Still with Unresolved Problems. Endocrines 2022, 3, 55-75. https://doi.org/10.3390/endocrines3010006
Smyczyńska J. Inclusion and Withdrawal Criteria for Growth Hormone (GH) Therapy in Children with Idiopathic GH Deficiency—Towards Following the Evidence but Still with Unresolved Problems. Endocrines. 2022; 3(1):55-75. https://doi.org/10.3390/endocrines3010006
Chicago/Turabian StyleSmyczyńska, Joanna. 2022. "Inclusion and Withdrawal Criteria for Growth Hormone (GH) Therapy in Children with Idiopathic GH Deficiency—Towards Following the Evidence but Still with Unresolved Problems" Endocrines 3, no. 1: 55-75. https://doi.org/10.3390/endocrines3010006
APA StyleSmyczyńska, J. (2022). Inclusion and Withdrawal Criteria for Growth Hormone (GH) Therapy in Children with Idiopathic GH Deficiency—Towards Following the Evidence but Still with Unresolved Problems. Endocrines, 3(1), 55-75. https://doi.org/10.3390/endocrines3010006