
Genetic Etiology of Idiopathic Hypogonadotropic Hypogonadism

Abstract
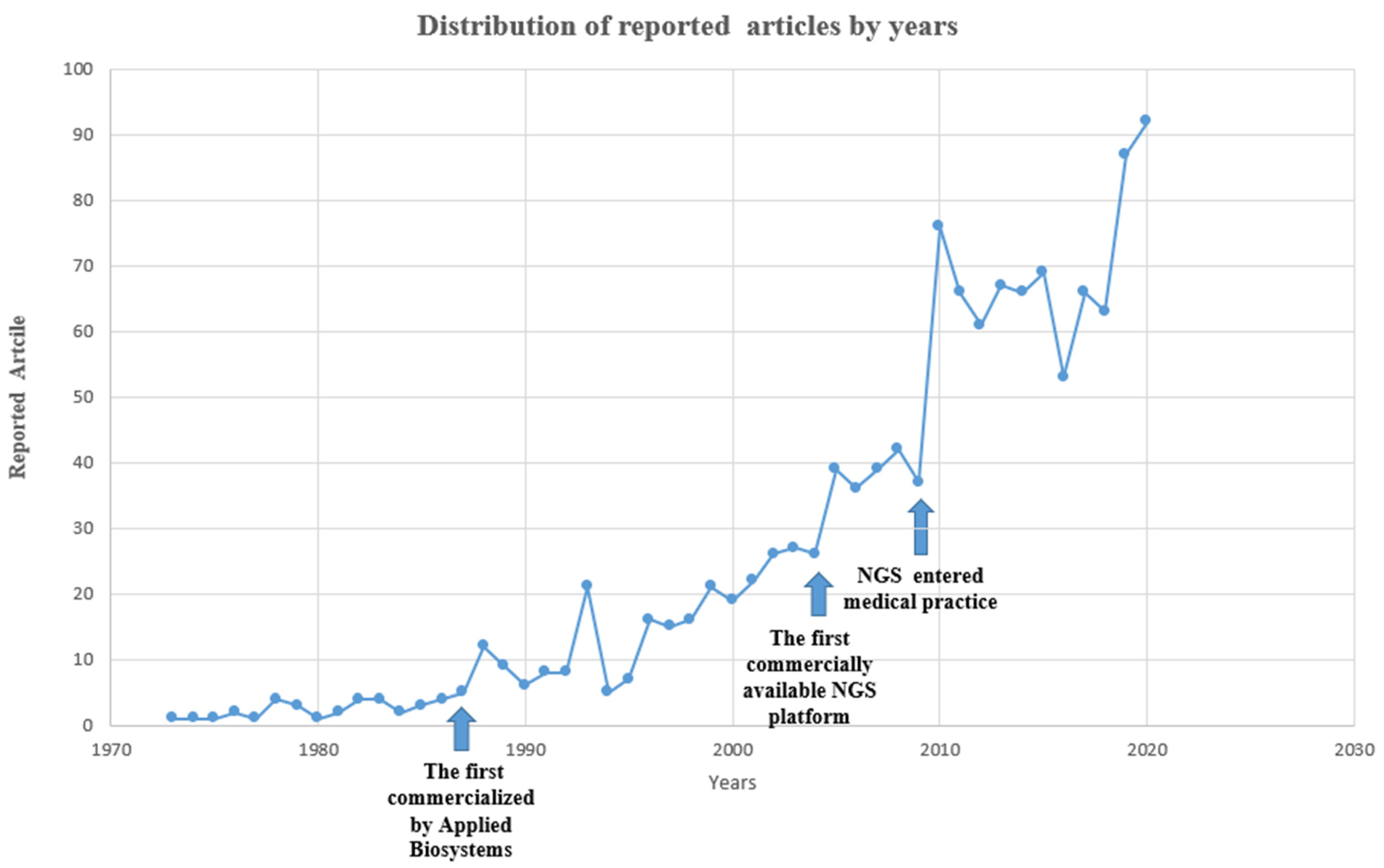
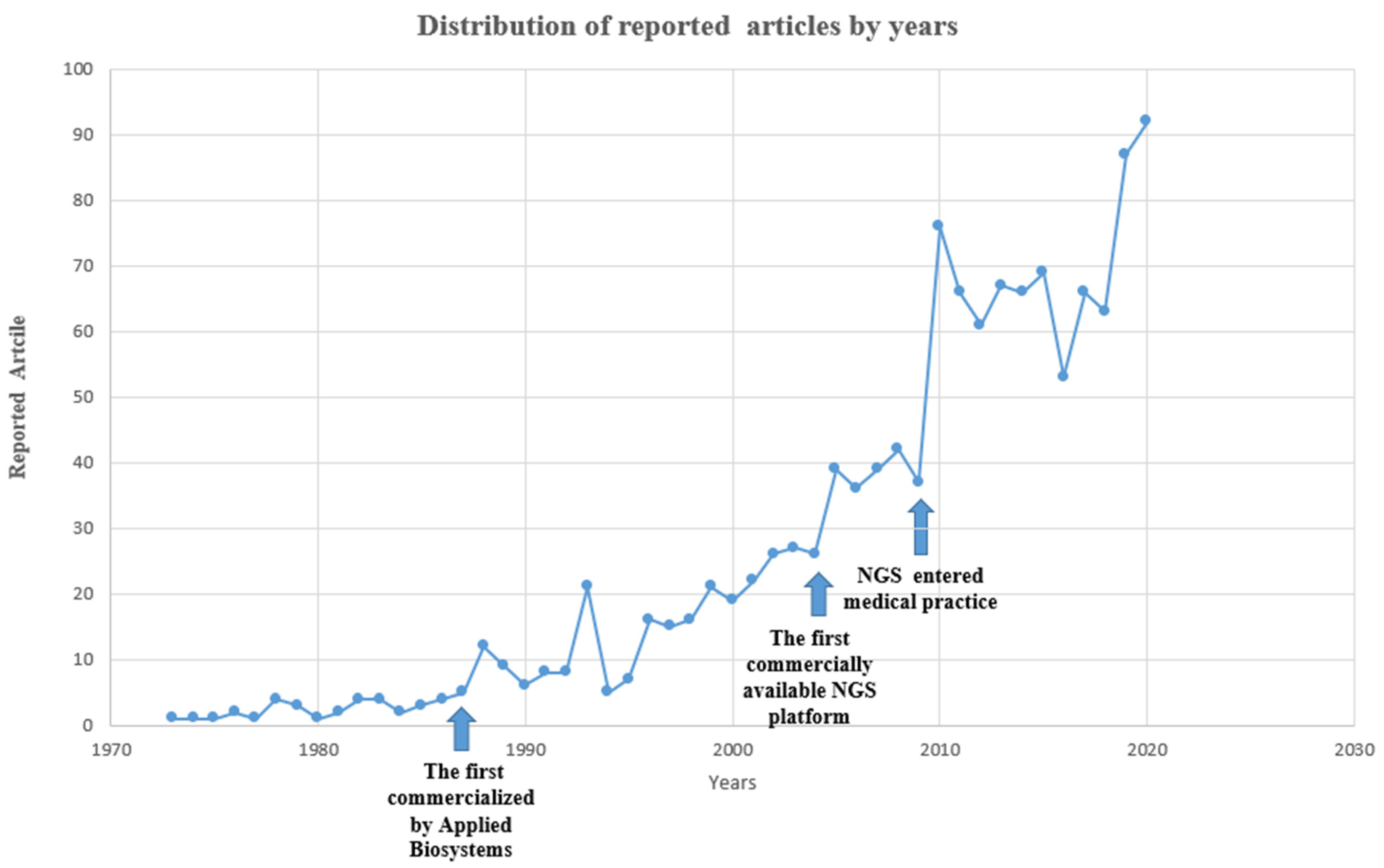
:1. Introduction
2. IHH-Associated Genes with OMIM Phenotype Number
3. The Genes Associated with IHH without Phenotype Numbers of OMIM
4. Concluding Remarks

{kind=link}
| Gene | HGNC ID | Clinical Phenotype | Gene Function | Phenotype Number of OMIM (or Ref.) |
|---|---|---|---|---|
| AMH | 464 | KS, nIHH | GnRH neuron migration | Malone et al. [100] |
| AMHR2 | 465 | nIHH | GnRH neuron migration | Malone et al. [100] |
| ANOS1 | 6211 | KS, nIHH | GnRH neuron migration | 308700 |
| AXL | 905 | KS, nIHH | GnRH neuron migration | 109135 |
| CCDC141 | 26821 | nIHH | GnRH neuron migration | Turan et al. [95] |
| CHD7 | 20626 | KS, nIHH, CHARGE | GnRH neuron migration | 612370 |
| CPE | 2303 | nIHH | Neuropeptide biosynthesis | Alsters et al. [78] |
| DCC | 2701 | KS, nIHH | GnRH neuron migration | Bouilly et al. [106]. |
| DLG2 | 2901 | DP | Neuroendocrine regulation | Jee et al. [107] |
| DMXL2 | 2938 | nIHH, PEPNS | ATPase regulation | 616113 |
| DUSP6 | 3072 | KS, nIHH | GnRH neuron migration | 615269 |
| FEZF1 | 22788 | KS | GnRH neuron migration | 616030 |
| FGF17 | 3673 | KS, nIHH, DWS | GnRH neuron development | 615270 |
| FGF8 | 3686 | KS, nIHH | GnRH neuron development | 612702 |
| FGFR1 | 3688 | KS, CPHD, SOD, SHFM, HS | Neuroendocrine regulation, Hypothalamus/pituitary development | 147950 |
| FLRT3 | 3762 | KS | GnRH neuron migration | 615271 |
| FSHB | 3964 | nIHH | Hypothalamus/pituitary development | 229070 |
| GNRH1 | 4419 | nIHH | Neuroendocrine regulation | 614841 |
| GNRHR | 4421 | nIHH | Neuroendocrine regulation | 146110 |
| HESX1 | 4877 | KS, CPHD, SOD | Hypothalamus/pituitary development | 182230 |
| HS6ST1 | 5201 | KS, nIHH | GnRH neuron migration | 614880 |
| IGSF10 | 26384 | DP | GnRH neuron migration | Howard et al. [91] |
| IL17RD | 17616 | KS, nIHH | GnRH neuron migration | 615267 |
| IRF2BPL | 14282 | DP | Ubiquitination | Mancini et al. [99] |
| KISS1 | 6341 | nIHH | Neuroendocrine regulation | 614842 |
| KISS1R | 4510 | nIHH | Neuroendocrine regulation | 614837 |
| KLB | 15527 | KS, nIHH | GnRH neuron development | Xu et al. [28] |
| LEP | 6553 | nIHH, Obesity | Neuroendocrine regulation | 614962 |
| LEPR | 6554 | nIHH, Obesity | Neuroendocrine regulation | 614963 |
| LHB | 6584 | nIHH | Hypothalamus/pituitary development | 228300 |
| NDNF | 26256 | KS | GnRH neuron migration | 618841 |
| NR0B1 | 7960 | nIHH, CAH | Hypothalamus/pituitary development | 300200 |
| NSMF | 29843 | KS | GnRH neuron migration | 614838 |
| NTN1 | 8029 | KS, nIHH | GnRH neuron migration | Bouilly et al. [106] |
| OTUD4 | 24949 | nIHH, GHS | Ubiquitination | 212840 |
| PCSK1 | 8743 | nIHH, Obesity | Hypothalamus/pituitary development | 600955, 162150 |
| PLXNA1 | 9099 | KS, nIHH | GnRH neuron migration | 601055 |
| PLXNA3 | 9101 | KS, nIHH | GnRH neuron migration | Kotan et al. [102] |
| PNPLA6 | 16268 | nIHH, GHS, BNS | Phospholipid homeostasis | 215470, 603197 |
| POLR3A | 30074 | 4H | DNA-dependent RNA polymerase | 607694 |
| POLR3B | 30348 | 4H | DNA-dependent RNA polymerase | 614381 |
| PROK2 | 18455 | KS, nIHH | GnRH neuron migration | 610628 |
| PROKR2 | 1836 | KS, nIHH, CPHD, MGS | GnRH neuron migration | 244200 |
| RAB18 | 14244 | WMS 3 | GTPase regulation | 614222 |
| RAB3GAP1 | 17063 | WMS 1 | GTPase regulation | 600118 |
| RAB3GAP2 | 17168 | MS | GTPase regulation | 212720 |
| RNF216 | 21698 | nIHH, GHS | Ubiquitination | 212840 |
| SEMA3A | 10723 | KS | GnRH neuron migration | 614897 |
| SEMA3E | 10727 | KS, nIHH | GnRH neuron migration | 608166 |
| SEMA3F | 10728 | GnRH neuron migration | Kotan et al. [102] | |
| SMCHD1 | 29090 | nIHH, CPHD, BAMS | DNA methylation | Shaw et al. [68] |
| SOX10 | 11190 | KS, WS | Hypothalamus/pituitary development | 613266 |
| SOX3 | 11199 | nIHH | Pituitary development | |
| SPRY4 | 15533 | KS | GnRH neuron migration | 615266 |
| SRA1 | 11281 | nIHH | Neuroendocrine regulation | Kotan et al. [93] |
| STUB1 | 11427 | Spinocerebellar ataxia | Ubiquitination | 615768 |
| TAC3 | 11521 | nIHH | Neuroendocrine regulation | 614839 |
| TACR3 | 11528 | nIHH | Neuroendocrine regulation | 614840 |
| TBC1D20 | 16133 | WMS 4 | Vesicle-mediated transport regulation | 615663 |
| WDR11 | 13831 | KS, CPHD | GnRH neuron migration | 614858 |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Topaloglu, A.K. Update on the Genetics of Idiopathic Hypogonadotropic Hypogonadism. J. Clin. Res. Pediatric Endocrinol. 2017, 9, 113–122. [Google Scholar] [CrossRef]
- Semple, R.K.; Topaloglu, A.K. The recent genetics of hypogonadotrophic hypogonadism—Novel insights and new questions. Clin. Endocrinol. 2010, 72, 427–435. [Google Scholar] [CrossRef]
- Gajdos, Z.K.; Henderson, K.D.; Hirschhorn, J.N.; Palmert, M.R. Genetic determinants of pubertal timing in the general population. Mol. Cell. Endocrinol. 2010, 324, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykiotis, G.P.; Plummer, L.; Hughes, V.A.; Au, M.; Durrani, S.; Nayak-Young, S.; Dwyer, A.A.; Quinton, R.; Hall, J.E.; Gusella, J.F.; et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 15140–15144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhoum, V.F.; Chan, Y.M.; Lippincott, M.F.; Balasubramanian, R.; Quinton, R.; Plummer, L.; Dwyer, A.; Pitteloud, N.; Hayes, F.J.; Hall, J.E.; et al. Reversal and relapse of hypogonadotropic hypogonadism: Resilience and fragility of the reproductive neuroendocrine system. J. Clin. Endocrinol. Metab. 2014, 99, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.B.; Nickerson, D.A.; Bamshad, M.J.; Shendure, J. Massively parallel sequencing and rare disease. Hum. Mol. Genet. 2010, 19, R119–R124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaynor, S.D.; Kim, H.G.; Cappello, E.M.; Williams, T.; Chorich, L.P.; Bick, D.P.; Sherins, R.J.; Layman, L.C. The prevalence of digenic mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil. Steril. 2011, 96, 1424–1430.e1426. [Google Scholar] [CrossRef] [Green Version]
- Pitteloud, N.; Quinton, R.; Pearce, S.; Raivio, T.; Acierno, J.; Dwyer, A.; Plummer, L.; Hughes, V.; Seminara, S.; Cheng, Y.Z.; et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Investig. 2007, 117, 457–463. [Google Scholar] [CrossRef]
- Boehm, U.; Bouloux, P.M.; Dattani, M.T.; De Roux, N.; Dode, C.; Dunkel, L.; Dwyer, A.A.; Giacobini, P.; Hardelin, J.P.; Juul, A.; et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism—Pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2015, 11, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.X.; Buckingham, K.J.; Jhangiani, S.N.; Boehm, C.; Sobreira, N.; Smith, J.D.; Harrell, T.M.; McMillin, M.J.; Wiszniewski, W.; Gambin, T.; et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 2015, 97, 199–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotan, L.D.; Isik, E.; Turan, I.; Mengen, E.; Akkus, G.; Tastan, M.; Gurbuz, F.; Yuksel, B.; Topaloglu, A.K. Prevalence and associated phenotypes of PLXNA1 variants in normosmic and anosmic idiopathic hypogonadotropic hypogonadism. Clin. Genet. 2019, 95, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Pitteloud, N.; Acierno, J.S., Jr.; Meysing, A.; Eliseenkova, A.V.; Ma, J.; Ibrahimi, O.A.; Metzger, D.L.; Hayes, F.J.; Dwyer, A.A.; Hughes, V.A.; et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2006, 103, 6281–6286. [Google Scholar] [CrossRef] [Green Version]
- Pitteloud, N.; Zhang, C.; Pignatelli, D.; Li, J.D.; Raivio, T.; Cole, L.W.; Plummer, L.; Jacobson-Dickman, E.E.; Mellon, P.L.; Zhou, Q.Y.; et al. Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2007, 104, 17447–17452. [Google Scholar] [CrossRef] [Green Version]
- Dode, C.; Levilliers, J.; Dupont, J.M.; De Paepe, A.; Le Du, N.; Soussi-Yanicostas, N.; Coimbra, R.S.; Delmaghani, S.; Compain-Nouaille, S.; Baverel, F.; et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003, 33, 463–465. [Google Scholar] [CrossRef] [Green Version]
- Hardelin, J.P.; Dode, C. The complex genetics of Kallmann syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2, et al. Sex. Dev. 2008, 2, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, A.K.; Reimann, F.; Guclu, M.; Yalin, A.S.; Kotan, L.D.; Porter, K.M.; Serin, A.; Mungan, N.O.; Cook, J.R.; Imamoglu, S.; et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat. Genet. 2009, 41, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Franco, B.; Guioli, S.; Pragliola, A.; Incerti, B.; Bardoni, B.; Tonlorenzi, R.; Carrozzo, R.; Maestrini, E.; Pieretti, M.; Taillon-Miller, P.; et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991, 353, 529–536. [Google Scholar] [CrossRef]
- Pedersen-White, J.R.; Chorich, L.P.; Bick, D.P.; Sherins, R.J.; Layman, L.C. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol. Hum. Reprod. 2008, 14, 367–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, L.M.; Seminara, S.B.; Beranova, M.; Hayes, F.J.; Valkenburgh, S.B.; Schipani, E.; Costa, E.M.; Latronico, A.C.; Crowley, W.F., Jr.; Vallejo, M. The importance of autosomal genes in Kallmann syndrome: Genotype-phenotype correlations and neuroendocrine characteristics. J. Clin. Endocrinol. Metab. 2001, 86, 1532–1538. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.S.; Gill, J.C. Mechanisms of disease: Insights into X-linked and autosomal-dominant Kallmann syndrome. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 160–171. [Google Scholar] [CrossRef]
- Pitteloud, N.; Meysing, A.; Quinton, R.; Acierno, J.S.; Dwyer, A.A., Jr.; Plummer, L.; Fliers, E.; Boepple, P.; Hayes, F.; Seminara, S.; et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol. Cell. Endocrinol. 2006, 254–255, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Trarbach, E.B.; Costa, E.M.; Versiani, B.; De Castro, M.; Baptista, M.T.; Garmes, H.M.; De Mendonca, B.B.; Latronico, A.C. Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J. Clin. Endocrinol. Metab. 2006, 91, 4006–4012. [Google Scholar] [CrossRef] [Green Version]
- Pitteloud, N.; Acierno, J.S., Jr.; Meysing, A.U.; Dwyer, A.A.; Hayes, F.J.; Crowley, W.F., Jr. Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J. Clin. Endocrinol. Metab. 2005, 90, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Qin, Y.; Reindollar, R.H.; Tho, S.P.; McDonough, P.G.; Layman, L.C. A mutation in the fibroblast growth factor receptor 1 gene causes fully penetrant normosmic isolated hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2007, 92, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Raivio, T.; Sidis, Y.; Plummer, L.; Chen, H.; Ma, J.; Mukherjee, A.; Jacobson-Dickman, E.; Quinton, R.; Van Vliet, G.; Lavoie, H.; et al. Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2009, 94, 4380–4390. [Google Scholar] [CrossRef] [PubMed]
- Falardeau, J.; Chung, W.C.; Beenken, A.; Raivio, T.; Plummer, L.; Sidis, Y.; Jacobson-Dickman, E.E.; Eliseenkova, A.V.; Ma, J.; Dwyer, A.; et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Investig. 2008, 118, 2822–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miraoui, H.; Dwyer, A.A.; Sykiotis, G.P.; Plummer, L.; Chung, W.; Feng, B.; Beenken, A.; Clarke, J.; Pers, T.H.; Dworzynski, P.; et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am. J. Hum. Genet. 2013, 92, 725–743. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Messina, A.; Somm, E.; Miraoui, H.; Kinnunen, T.; Acierno, J., Jr.; Niederlander, N.J.; Bouilly, J.; Dwyer, A.A.; Sidis, Y.; et al. KLB, encoding beta-Klotho, is mutated in patients with congenital hypogonadotropic hypogonadism. EMBO Mol. Med. 2017, 9, 1379–1397. [Google Scholar] [CrossRef]
- Tornberg, J.; Sykiotis, G.P.; Keefe, K.; Plummer, L.; Hoang, X.; Hall, J.E.; Quinton, R.; Seminara, S.B.; Hughes, V.; Van Vliet, G.; et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc. Natl. Acad. Sci. USA 2011, 108, 11524–11529. [Google Scholar] [CrossRef] [Green Version]
- Howard, S.R.; Oleari, R.; Poliandri, A.; Chantzara, V.; Fantin, A.; Ruiz-Babot, G.; Metherell, L.A.; Cabrera, C.P.; Barnes, M.R.; Wehkalampi, K.; et al. HS6ST1 Insufficiency Causes Self-Limited Delayed Puberty in Contrast With Other GnRH Deficiency Genes. J. Clin. Endocrinol. Metab. 2018, 103, 3420–3429. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Leslie, F.M.; Zhou, Q.Y. Expression of prokineticins and their receptors in the adult mouse brain. J. Comp. Neurol. 2006, 498, 796–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.; Balasubramanian, R.; Dwyer, A.A.; Au, M.G.; Sidis, Y.; Kaiser, U.B.; Seminara, S.B.; Pitteloud, N.; Zhou, Q.Y.; Crowley, W.F., Jr. The role of the prokineticin 2 pathway in human reproduction: Evidence from the study of human and murine gene mutations. Endocr. Rev. 2011, 32, 225–246. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, S.; Yamazaki, C.; Masumoto, K.H.; Nagano, M.; Naito, M.; Soga, T.; Hiyama, H.; Matsumoto, M.; Takasaki, J.; Kamohara, M.; et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc. Natl. Acad. Sci. USA 2006, 103, 4140–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, A.P.; Trarbach, E.B.; De Castro, M.; Frade Costa, E.M.; Versiani, B.; Matias Baptista, M.T.; Garmes, H.M.; Mendonca, B.B.; Latronico, A.C. Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4113–4118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, L.W.; Sidis, Y.; Zhang, C.; Quinton, R.; Plummer, L.; Pignatelli, D.; Hughes, V.A.; Dwyer, A.A.; Raivio, T.; Hayes, F.J.; et al. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: Molecular genetics and clinical spectrum. J. Clin. Endocrinol. Metab. 2008, 93, 3551–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissers, L.E.; Van Ravenswaaij, C.M.; Admiraal, R.; Hurst, J.A.; De Vries, B.B.; Janssen, I.M.; Van der Vliet, W.A.; Huys, E.H.; De Jong, P.J.; Hamel, B.C.; et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004, 36, 955–957. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.G.; Kurth, I.; Lan, F.; Meliciani, I.; Wenzel, W.; Eom, S.H.; Kang, G.B.; Rosenberger, G.; Tekin, M.; Ozata, M.; et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2008, 83, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Cassatella, D.; Van der Sloot, A.M.; Quinton, R.; Hauschild, M.; De Geyter, C.; Fluck, C.; Feller, K.; Bartholdi, D.; Nemeth, A.; et al. Evaluating CHARGE syndrome in congenital hypogonadotropic hypogonadism patients harboring CHD7 variants. Genet. Med. 2018, 20, 872–881. [Google Scholar] [CrossRef]
- Layman, W.S.; Hurd, E.A.; Martin, D.M. Reproductive dysfunction and decreased GnRH neurogenesis in a mouse model of CHARGE syndrome. Hum. Mol. Genet. 2011, 20, 3138–3150. [Google Scholar] [CrossRef] [Green Version]
- De Roux, N.; Young, J.; Misrahi, M.; Genet, R.; Chanson, P.; Schaison, G.; Milgrom, E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N. Engl. J. Med. 1997, 337, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Beranova, M.; Oliveira, L.M.; Bedecarrats, G.Y.; Schipani, E.; Vallejo, M.; Ammini, A.C.; Quintos, J.B.; Hall, J.E.; Martin, K.A.; Hayes, F.J.; et al. Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2001, 86, 1580–1588. [Google Scholar] [CrossRef]
- Beneduzzi, D.; Trarbach, E.B.; Min, L.; Jorge, A.A.; Garmes, H.M.; Renk, A.C.; Fichna, M.; Fichna, P.; Arantes, K.A.; Costa, E.M.; et al. Role of gonadotropin-releasing hormone receptor mutations in patients with a wide spectrum of pubertal delay. Fertil. Steril. 2014, 102, 838–846. [Google Scholar] [CrossRef] [Green Version]
- King, J.A.; Millar, R.P. Evolutionary aspects of gonadotropin-releasing hormone and its receptor. Cell. Mol. Neurobiol. 1995, 15, 5–23. [Google Scholar] [CrossRef]
- Bouligand, J.; Ghervan, C.; Tello, J.A.; Brailly-Tabard, S.; Salenave, S.; Chanson, P.; Lombes, M.; Millar, R.P.; Guiochon-Mantel, A.; Young, J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N. Engl. J. Med. 2009, 360, 2742–2748. [Google Scholar] [CrossRef]
- Chan, Y.M.; De Guillebon, A.; Lang-Muritano, M.; Plummer, L.; Cerrato, F.; Tsiaras, S.; Gaspert, A.; Lavoie, H.B.; Wu, C.H.; Crowley, W.F., Jr.; et al. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2009, 106, 11703–11708. [Google Scholar] [CrossRef] [Green Version]
- Mengen, E.; Tunc, S.; Kotan, L.D.; Nalbantoglu, O.; Demir, K.; Gurbuz, F.; Turan, I.; Seker, G.; Yuksel, B.; Topaloglu, A.K. Complete Idiopathic Hypogonadotropic Hypogonadism due to Homozygous GNRH1 Mutations in the Mutational Hot Spots in the Region Encoding the Decapeptide. Horm. Res. Paediatr. 2016, 85, 107–111. [Google Scholar] [CrossRef]
- Seminara, S.B.; Messager, S.; Chatzidaki, E.E.; Thresher, R.R.; Acierno, J.S., Jr.; Shagoury, J.K.; Bo-Abbas, Y.; Kuohung, W.; Schwinof, K.M.; Hendrick, A.G.; et al. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 2003, 349, 1614–1627. [Google Scholar] [CrossRef] [Green Version]
- Cerrato, F.; Shagoury, J.; Kralickova, M.; Dwyer, A.; Falardeau, J.; Ozata, M.; Van Vliet, G.; Bouloux, P.; Hall, J.E.; Hayes, F.J.; et al. Coding sequence analysis of GNRHR and GPR54 in patients with congenital and adult-onset forms of hypogonadotropic hypogonadism. Eur. J. Endocrinol. 2006, 155, S3–S10. [Google Scholar] [CrossRef]
- Navarro, V.M.; Tena-Sempere, M. Neuroendocrine control by kisspeptins: Role in metabolic regulation of fertility. Nat. Rev. Endocrinol. 2011, 8, 40–53. [Google Scholar] [CrossRef]
- Gottsch, M.L.; Cunningham, M.J.; Smith, J.T.; Popa, S.M.; Acohido, B.V.; Crowley, W.F.; Seminara, S.; Clifton, D.K.; Steiner, R.A. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology 2004, 145, 4073–4077. [Google Scholar] [CrossRef] [Green Version]
- Topaloglu, A.K.; Tello, J.A.; Kotan, L.D.; Ozbek, M.N.; Yilmaz, M.B.; Erdogan, S.; Gurbuz, F.; Temiz, F.; Millar, R.P.; Yuksel, B. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N. Engl. J. Med. 2012, 366, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Acierno, J.S., Jr.; Seminara, S.B. Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH). J. Hum. Genet. 2004, 49, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, P.R.; Wray, S. Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons. Genes Dev. 2000, 14, 1824–1834. [Google Scholar] [CrossRef]
- Goodman, R.L.; Lehman, M.N.; Smith, J.T.; Coolen, L.M.; De Oliveira, C.V.; Jafarzadehshirazi, M.R.; Pereira, A.; Iqbal, J.; Caraty, A.; Ciofi, P.; et al. Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology 2007, 148, 5752–5760. [Google Scholar] [CrossRef]
- Gianetti, E.; Tusset, C.; Noel, S.D.; Au, M.G.; Dwyer, A.A.; Hughes, V.A.; Abreu, A.P.; Carroll, J.; Trarbach, E.; Silveira, L.F.; et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J. Clin. Endocrinol. Metab. 2010, 95, 2857–2867. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Botstein, D. Homozygosity mapping: A way to map human recessive traits with the DNA of inbred children. Science 1987, 236, 1567–1570. [Google Scholar] [CrossRef] [Green Version]
- Francou, B.; Bouligand, J.; Voican, A.; Amazit, L.; Trabado, S.; Fagart, J.; Meduri, G.; Brailly-Tabard, S.; Chanson, P.; Lecomte, P.; et al. Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: Characterization of neuroendocrine phenotypes and novel mutations. PLoS ONE 2011, 6, e25614. [Google Scholar]
- Kim, H.G.; Ahn, J.W.; Kurth, I.; Ullmann, R.; Kim, H.T.; Kulharya, A.; Ha, K.S.; Itokawa, Y.; Meliciani, I.; Wenzel, W.; et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2010, 87, 465–479. [Google Scholar] [CrossRef] [Green Version]
- McCormack, S.E.; Li, D.; Kim, Y.J.; Lee, J.Y.; Kim, S.H.; Rapaport, R.; Levine, M.A. Digenic Inheritance of PROKR2 and WDR11 Mutations in Pituitary Stalk Interruption Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 2501–2507. [Google Scholar] [CrossRef] [Green Version]
- Cariboni, A.; Davidson, K.; Rakic, S.; Maggi, R.; Parnavelas, J.G.; Ruhrberg, C. Defective gonadotropin-releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: Implications for the aetiology of hypogonadotropic hypogonadism. Hum. Mol. Genet. 2011, 20, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Hanchate, N.K.; Giacobini, P.; Lhuillier, P.; Parkash, J.; Espy, C.; Fouveaut, C.; Leroy, C.; Baron, S.; Campagne, C.; Vanacker, C.; et al. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 2012, 8, e1002896. [Google Scholar] [CrossRef]
- Young, J.; Metay, C.; Bouligand, J.; Tou, B.; Francou, B.; Maione, L.; Tosca, L.; Sarfati, J.; Brioude, F.; Esteva, B.; et al. SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum. Reprod. 2012, 27, 1460–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariboni, A.; Andre, V.; Chauvet, S.; Cassatella, D.; Davidson, K.; Caramello, A.; Fantin, A.; Bouloux, P.; Mann, F.; Ruhrberg, C. Dysfunctional SEMA3E signaling underlies gonadotropin-releasing hormone neuron deficiency in Kallmann syndrome. J. Clin. Investig. 2015, 125, 2413–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansakoski, J.; Fagerholm, R.; Laitinen, E.M.; Vaaralahti, K.; Hackman, P.; Pitteloud, N.; Raivio, T.; Tommiska, J. Mutation screening of SEMA3A and SEMA7A in patients with congenital hypogonadotropic hypogonadism. Pediatr. Res. 2014, 75, 641–644. [Google Scholar] [CrossRef]
- Kotan, L.D.; Hutchins, B.I.; Ozkan, Y.; Demirel, F.; Stoner, H.; Cheng, P.J.; Esen, I.; Gurbuz, F.; Bicakci, Y.K.; Mengen, E.; et al. Mutations in FEZF1 cause Kallmann syndrome. Am. J. Hum. Genet. 2014, 95, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Eckler, M.J.; McKenna, W.L.; Taghvaei, S.; McConnell, S.K.; Chen, B. Fezf1 and Fezf2 are required for olfactory development and sensory neuron identity. J. Comp. Neurol. 2011, 519, 1829–1846. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Pulli, K.; Santini, S.; Acierno, J.; Kansakoski, J.; Cassatella, D.; Xu, C.; Casoni, F.; Malone, S.A.; Ternier, G.; et al. Neuron-Derived Neurotrophic Factor Is Mutated in Congenital Hypogonadotropic Hypogonadism. Am. J. Hum. Genet. 2020, 106, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Shaw, N.D.; Brand, H.; Kupchinsky, Z.A.; Bengani, H.; Plummer, L.; Jones, T.I.; Erdin, S.; Williamson, K.A.; Rainger, J.; Stortchevoi, A.; et al. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet. 2017, 49, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinjo, K.; Nagasaki, K.; Muroya, K.; Suzuki, E.; Ishiwata, K.; Nakabayashi, K.; Hattori, A.; Nagao, K.; Nozawa, R.S.; Obuse, C.; et al. Rare variant of the epigenetic regulator SMCHD1 in a patient with pituitary hormone deficiency. Sci. Rep. 2020, 10, 10985. [Google Scholar] [CrossRef]
- Pingault, V.; Bodereau, V.; Baral, V.; Marcos, S.; Watanabe, Y.; Chaoui, A.; Fouveaut, C.; Leroy, C.; Verier-Mine, O.; Francannet, C.; et al. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am. J. Hum. Genet. 2013, 92, 707–724. [Google Scholar] [CrossRef] [Green Version]
- Strobel, A.; Issad, T.; Camoin, L.; Ozata, M.; Strosberg, A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998, 18, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Wangensteen, T.; Collins, S.; Kimber, W.; Matarese, G.; Keogh, J.M.; Lank, E.; Bottomley, B.; Lopez-Fernandez, J.; Ferraz-Amaro, I.; et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N. Engl. J. Med. 2007, 356, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauters, M.; Considine, R.V.; Van Gaal, L.F. Human leptin, from an adipocyte hormone to an endocrine mediator. Eur. J. Endocrinol. 2000, 143, 293–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef]
- Ahima, R.S.; Prabakaran, D.; Mantzoros, C.; Qu, D.; Lowell, B.; Maratos-Flier, E.; Flier, J.S. Role of leptin in the neuroendocrine response to fasting. Nature 1996, 382, 250–252. [Google Scholar] [CrossRef]
- Ahima, R.S.; Dushay, J.; Flier, S.N.; Prabakaran, D.; Flier, J.S. Leptin accelerates the onset of puberty in normal female mice. J. Clin. Investig. 1997, 99, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Muscatelli, F.; Strom, T.M.; Walker, A.P.; Zanaria, E.; Recan, D.; Meindl, A.; Bardoni, B.; Guioli, S.; Zehetner, G.; Rabl, W.; et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 1994, 372, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Alsters, S.I.; Goldstone, A.P.; Buxton, J.L.; Zekavati, A.; Sosinsky, A.; Yiorkas, A.M.; Holder, S.; Klaber, R.E.; Bridges, N.; Van Haelst, M.M.; et al. Truncating Homozygous Mutation of Carboxypeptidase E (CPE) in a Morbidly Obese Female with Type 2 Diabetes Mellitus, Intellectual Disability and Hypogonadotrophic Hypogonadism. PLoS ONE 2015, 10, e0131417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durmaz, A.; Aykut, A.; Atik, T.; Ozen, S.; Ayyildiz Emecen, D.; Ata, A.; Isik, E.; Goksen, D.; Cogulu, O.; Ozkinay, F. A New Cause of Obesity Syndrome Associated with a Mutation in the Carboxypeptidase Gene Detected in Three Siblings with Obesity, Intellectual Disability and Hypogonadotropic Hypogonadism. J. Clin. Res. Pediatr. Endocrinol. 2021, 13, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Newbern, K.; Natrajan, N.; Kim, H.G.; Chorich, L.P.; Halvorson, L.M.; Cameron, R.S.; Layman, L.C. Identification of HESX1 mutations in Kallmann syndrome. Fertil. Steril. 2013, 99, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Tata, B.; Huijbregts, L.; Jacquier, S.; Csaba, Z.; Genin, E.; Meyer, V.; Leka, S.; Dupont, J.; Charles, P.; Chevenne, D.; et al. Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol. 2014, 12, e1001952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolin, D.H.; Kousi, M.; Chan, Y.M.; Lim, E.T.; Schmahmann, J.D.; Hadjivassiliou, M.; Hall, J.E.; Adam, I.; Dwyer, A.; Plummer, L.; et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 2013, 368, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.H.; Schisler, J.C.; Rubel, C.E.; Tan, S.; Song, B.; McDonough, H.; Xu, L.; Portbury, A.L.; Mao, C.Y.; True, C.; et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum. Mol. Genet. 2014, 23, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topaloglu, A.K.; Lomniczi, A.; Kretzschmar, D.; Dissen, G.A.; Kotan, L.D.; McArdle, C.A.; Koc, A.F.; Hamel, B.C.; Guclu, M.; Papatya, E.D.; et al. Loss-of-function mutations in PNPLA6 encoding neuropathy target esterase underlie pubertal failure and neurological deficits in Gordon Holmes syndrome. J. Clin. Endocrinol. Metab. 2014, 99, E2067–E2075. [Google Scholar] [CrossRef] [Green Version]
- Synofzik, M.; Gonzalez, M.A.; Lourenco, C.M.; Coutelier, M.; Haack, T.B.; Rebelo, A.; Hannequin, D.; Strom, T.M.; Prokisch, H.; Kernstock, C.; et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014, 137, 69–77. [Google Scholar] [CrossRef]
- Dumay-Odelot, H.; Durrieu-Gaillard, S.; Da Silva, D.; Roeder, R.G.; Teichmann, M. Cell growth- and differentiation-dependent regulation of RNA polymerase III transcription. Cell Cycle 2010, 9, 3687–3699. [Google Scholar] [CrossRef] [Green Version]
- Bernard, G.; Chouery, E.; Putorti, M.L.; Tetreault, M.; Takanohashi, A.; Carosso, G.; Clement, I.; Boespflug-Tanguy, O.; Rodriguez, D.; Delague, V.; et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Tetreault, M.; Choquet, K.; Orcesi, S.; Tonduti, D.; Balottin, U.; Teichmann, M.; Fribourg, S.; Schiffmann, R.; Brais, B.; Vanderver, A.; et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 652–655. [Google Scholar] [CrossRef] [Green Version]
- Izumi, Y.; Suzuki, E.; Kanzaki, S.; Yatsuga, S.; Kinjo, S.; Igarashi, M.; Maruyama, T.; Sano, S.; Horikawa, R.; Sato, N.; et al. Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil. Steril. 2014, 102, 1130–1136.e1133. [Google Scholar] [CrossRef]
- Salian-Mehta, S.; Xu, M.; Knox, A.J.; Plummer, L.; Slavov, D.; Taylor, M.; Bevers, S.; Hodges, R.S.; Crowley, W.F., Jr.; Wierman, M.E. Functional consequences of AXL sequence variants in hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2014, 99, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Howard, S.R.; Guasti, L.; Ruiz-Babot, G.; Mancini, A.; David, A.; Storr, H.L.; Metherell, L.A.; Sternberg, M.J.; Cabrera, C.P.; Warren, H.R.; et al. IGSF10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol. Med. 2016, 8, 626–642. [Google Scholar] [CrossRef]
- Chooniedass-Kothari, S.; Emberley, E.; Hamedani, M.K.; Troup, S.; Wang, X.; Czosnek, A.; Hube, F.; Mutawe, M.; Watson, P.H.; Leygue, E. The steroid receptor RNA activator is the first functional RNA encoding a protein. FEBS Lett. 2004, 566, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Kotan, L.D.; Cooper, C.; Darcan, S.; Carr, I.M.; Ozen, S.; Yan, Y.; Hamedani, M.K.; Gurbuz, F.; Mengen, E.; Turan, I.; et al. Idiopathic Hypogonadotropic Hypogonadism Caused by Inactivating Mutations in SRA1. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 125–134. [Google Scholar] [CrossRef]
- Marcos, S.; Monnier, C.; Rovira, X.; Fouveaut, C.; Pitteloud, N.; Ango, F.; Dode, C.; Hardelin, J.P. Defective signaling through plexin-A1 compromises the development of the peripheral olfactory system and neuroendocrine reproductive axis in mice. Hum. Mol. Genet. 2017, 26, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Turan, I.; Hutchins, B.I.; Hacihamdioglu, B.; Kotan, L.D.; Gurbuz, F.; Ulubay, A.; Mengen, E.; Yuksel, B.; Wray, S.; Topaloglu, A.K. CCDC141 Mutations in Idiopathic Hypogonadotropic Hypogonadism. J. Clin. Endocrinol. Metab. 2017, 102, 1816–1825. [Google Scholar] [CrossRef]
- Hutchins, B.I.; Kotan, L.D.; Taylor-Burds, C.; Ozkan, Y.; Cheng, P.J.; Gurbuz, F.; Tiong, J.D.; Mengen, E.; Yuksel, B.; Topaloglu, A.K.; et al. CCDC141 Mutation Identified in Anosmic Hypogonadotropic Hypogonadism (Kallmann Syndrome) Alters GnRH Neuronal Migration. Endocrinology 2016, 157, 1956–1966. [Google Scholar] [CrossRef] [Green Version]
- Hou, Q.; Wu, J.; Zhao, Y.; Wang, X.; Jiang, F.; Chen, D.N.; Zheng, R.; Men, M.; Li, J.D. Genotypic and phenotypic spectrum of CCDC141 variants in a Chinese cohort with congenital hypogonadotropic hypogonadism. Eur. J. Endocrinol. 2020, 183, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Heger, S.; Mastronardi, C.; Dissen, G.A.; Lomniczi, A.; Cabrera, R.; Roth, C.L.; Jung, H.; Galimi, F.; Sippell, W.; Ojeda, S.R. Enhanced at puberty 1 (EAP1) is a new transcriptional regulator of the female neuroendocrine reproductive axis. J. Clin. Investig. 2007, 117, 2145–2154. [Google Scholar] [CrossRef]
- Mancini, A.; Howard, S.R.; Cabrera, C.P.; Barnes, M.R.; David, A.; Wehkalampi, K.; Heger, S.; Lomniczi, A.; Guasti, L.; Ojeda, S.R.; et al. EAP1 regulation of GnRH promoter activity is important for human pubertal timing. Hum. Mol. Genet. 2019, 28, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Malone, S.A.; Papadakis, G.E.; Messina, A.; Mimouni, N.E.H.; Trova, S.; Imbernon, M.; Allet, C.; Cimino, I.; Acierno, J.; Cassatella, D.; et al. Defective AMH signaling disrupts GnRH neuron development and function and contributes to hypogonadotropic hypogonadism. Elife 2019, 8, e47198. [Google Scholar] [CrossRef]
- Oleari, R.; Andre, V.; Lettieri, A.; Tahir, S.; Roth, L.; Paganoni, A.; Eberini, I.; Parravicini, C.; Scagliotti, V.; Cotellessa, L.; et al. A Novel SEMA3G Mutation in Two Siblings Affected by Syndromic GnRH Deficiency. Neuroendocrinology 2021, 111, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Kotan, L.D.; Ternier, G.; Cakir, A.D.; Emeksiz, H.C.; Turan, I.; Delpouve, G.; Kardelen, A.D.; Ozcabi, B.; Isik, E.; Mengen, E.; et al. Loss-of-function variants in SEMA3F and PLXNA3 encoding semaphorin-3F and its receptor plexin-A3 respectively cause idiopathic hypogonadotropic hypogonadism. Genet. Med. 2021, 23, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Borck, G.; Wunram, H.; Steiert, A.; Volk, A.E.; Korber, F.; Roters, S.; Herkenrath, P.; Wollnik, B.; Morris-Rosendahl, D.J.; Kubisch, C. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum. Genet. 2011, 129, 45–50. [Google Scholar] [CrossRef]
- Handley, M.T.; Aligianis, I.A. RAB3GAP1, RAB3GAP2 and RAB18: Disease genes in Micro and Martsolf syndromes. Biochem. Soc. Trans. 2012, 40, 1394–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handley, M.; Sheridan, E. RAB18 deficiency. In GeneReviews ((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Bouilly, J.; Messina, A.; Papadakis, G.; Cassatella, D.; Xu, C.; Acierno, J.S.; Tata, B.; Sykiotis, G.; Santini, S.; Sidis, Y.; et al. DCC/NTN1 complex mutations in patients with congenital hypogonadotropic hypogonadism impair GnRH neuron development. Hum. Mol. Genet. 2018, 27, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Jee, Y.H.; Won, S.; Lui, J.C.; Jennings, M.; Whalen, P.; Yue, S.; Temnycky, A.G.; Barnes, K.M.; Cheetham, T.; Boden, M.G.; et al. DLG2 variants in patients with pubertal disorders. Genet. Med. 2020, 22, 1329–1337. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Topaloglu, A.K.; Turan, I. Genetic Etiology of Idiopathic Hypogonadotropic Hypogonadism. Endocrines 2022, 3, 1-15. https://doi.org/10.3390/endocrines3010001
Topaloglu AK, Turan I. Genetic Etiology of Idiopathic Hypogonadotropic Hypogonadism. Endocrines. 2022; 3(1):1-15. https://doi.org/10.3390/endocrines3010001
Chicago/Turabian StyleTopaloglu, Ali Kemal, and Ihsan Turan. 2022. "Genetic Etiology of Idiopathic Hypogonadotropic Hypogonadism" Endocrines 3, no. 1: 1-15. https://doi.org/10.3390/endocrines3010001
APA StyleTopaloglu, A. K., & Turan, I. (2022). Genetic Etiology of Idiopathic Hypogonadotropic Hypogonadism. Endocrines, 3(1), 1-15. https://doi.org/10.3390/endocrines3010001