
The Stress Axis in Obesity and Diabetes Mellitus: An Update



{kind=link}
Abstract
1. Introduction
2. Methods
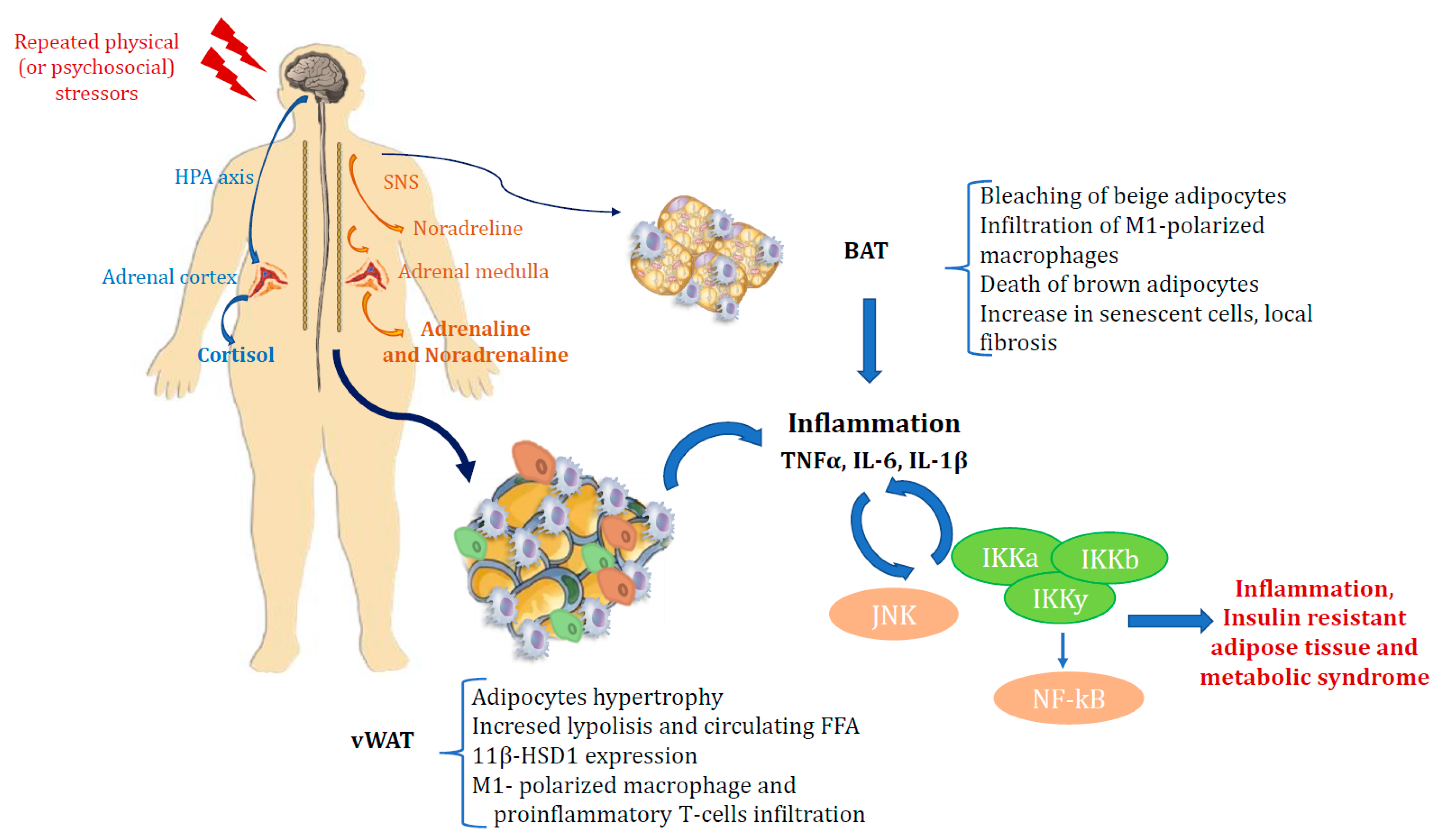
2.1. Stress and Adiposity: The Role of Cortisol in Increasing Visceral Fat Accumulation
2.2. Hpa Axis in Obesity and Diabetes
2.3. Tendency of GC Receptors and Their Polymorphisms to Modulate GC Sensitivity, Increasing Susceptibility to Likelihood of Adiposity and Insulin Resistance
2.4. Role of 11β-Hsd1 Enzyme Dysregulation in the Pathogenesis of Obesity and Insulin Resistance
2.5. Stress, Inflammation and Metabolic Syndrome
2.6. COVID-19, Stress and Diabetes: A Bidirectional Relationship
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Kyrou, I.; Chrousos, G.P.; Tsigos, C. Stress, visceral obesity and metabolic complications. Ann. N. Y. Acad. Sci. USA 2006, 1083, 77–110. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Olivan, B.; Laferre, B. Stress and obesity: The role of hypothalamic-pituitary adrenal axis in metabolic disease. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Anagnostis, P.; Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. The pathogenetic role of cortisol in the metabolic syndrome: A hypothesis. J. Clin. Endocrinol. Metab. 2009, 94, 2692–2701. [Google Scholar] [CrossRef]
- Reynolds, R.M. Glucocorticoid excess and the developmental origins of disease: Two decades of testing the hypothesis—2012 Curt Richter Award Winner. Psychoneuroendocrinology 2013, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fogelman, N.; Canli, T. Early life stress and cortisol: A meta-analysis. Horm. Behav. 2018, 98, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Gentile, S. Untreated depression during pregnancy: Short- and long-term effects in offspring. A systematic review. Neuroscience 2017, 342, 154–166. [Google Scholar] [CrossRef]
- Anekwe, C.H.; Jarrell, A.R.; Townsend, M.J.; Gaudier, G.I.; Hiserodt, J.M.; Stanford, C.S. Socioeconomics of obesity. Curr. Obes. Rep. 2020, 9, 272–279. [Google Scholar] [CrossRef]
- Hjelholt, A.; Høgild, M.; Bak, M.; Christiansen, M.; Bæk, A.A.-S.; Jessen, N.; Richelsen, B.; Bønløkke, S.; Møller, P.N.; Jørgensen, J.O.L. Growth hormone and obesity. Endocrinol. Metab. Clin. N. Am. 2020, 49, 239–250. [Google Scholar] [CrossRef]
- Van Der Valk, E.S.; Akker, E.L.V.D.; Savas, M.; Kleinendorst, L.; Visser, J.A.; Van Haelst, M.M.; Sharma, A.M.; Van Rossum, E.F. A comprehensive diagnostic approach to detect underlying causes of obesity in adults. Obes. Rev. 2019, 20, 795–804. [Google Scholar] [CrossRef]
- Björntorp, P. Do stress reactions cause abdominal obesity and comorbidities? Obes. Rev. 2001, 2, 73–86. [Google Scholar] [CrossRef]
- Rosmond, R.; Dallman, M.F.; Björntorp, P. Stress-related cortisol secretion in men: Relationships with abdominal obesity and en-docrine metabolic and hemodynamic abnormalities. J. Clin. Endocrinol. Metab. 1998, 83, 1853–1859. [Google Scholar] [PubMed]
- Scaroni, C.; Albiger, N.M.; Palmieri, S.; Iacuaniello, D.; Graziadio, C.; Damiani, L.; Zilo, M.; Stigliano, A.; Colao, A.M.; Pivonello, E.; et al. Approach to patients with pseudo-Cushing’s states. Endocr. Connect. 2020, 9, R1–R13. [Google Scholar] [CrossRef]
- Rosmond, R.; Radulovic, V.; Holm, G. A brief update of glucocorticoid receptor variants and obesity risk. Ann. N. Y. Acad. Sci. USA 2006, 1083, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Stalder, T.; Kirschbaum, C.; Kudielka, B.M.; Adam, E.K.; Pruessner, J.; Wüst, S.; Dockray, S.; Smyth, N.; Evans, P.; Hellhammer, D.H.; et al. Assessment of the cortisol awakening response: Expert consensus guidelines. Psychoneuroendocrinology 2015, 63, 414–432. [Google Scholar] [CrossRef] [PubMed]
- Fries, E.; Dettenborn, L.; Kirschbaum, C. The cortisol awakening response (CAR): Facts and future directions. Int. J. Psychophysiol. 2009, 72, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Rodrigueza, A.C.I.; Epelb, E.S.; Whitea, M.L.; Standena, E.C.; Secklc, J.R.; Tomiyamaa, A.J. Hypothalamic-pituitary-adrenal axis dysregulation and cortisol activity in obesity: A systematic review. Psychoneuroendocrinology 2015, 62, 301–308. [Google Scholar] [CrossRef]
- Champaneri, S.; Xu, X.; Carnethon, M.; Bertoni, A.; Seeman, T.; DeSantis, A.S.; Roux, A.D.; Shrager, S.; Golden, S.H. Diurnal salivary cortisol is associated with body mass index and waist circumference: The multiethnic study of atherosclerosis. Obesity 2012, 21, E56–E63. [Google Scholar] [CrossRef]
- Stone, A.A.; Schwartz, J.E.; Smyth, J.; Kirschbaum, C.; Cohen, S.; Hellhammer, D.; Grossman, S. Individual differences in the diurnal cycle of salivary freecortisol: A replication of flattened cycles for some individuals. Psychoneuroendocrinology 2001, 26, 295–306. [Google Scholar] [CrossRef]
- Pruessner, J.C.; Kirschbaum, C.; Meinlschmid, G.; Hellhammer, D.H. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology 2003, 28, 916–931. [Google Scholar] [CrossRef]
- Rosmond, R.; Bjorntorp, P. The hypothalamic-pituitary-adrenal axis activity as a predictor of cardiovascular disease, type 2 diabetes and stroke. J. Intern. Med. 2000, 247, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, R.; Ambrosi, B.; Armanini, D.; Cavagnini, F.; Uberti, E.D.; Rio, G.D.; de Pergola, G.; Maccario, M.; Mantero, F.; Marugo, M.; et al. Cortisol and ACTH response to oral dexamethasone in obesity and effectsof sex, body fat distribution, and dexamethasone concentrations: Adose-response study. J. Clin. Endocrinol. Metab. 2002, 87, 166–175. [Google Scholar] [CrossRef]
- Manenschijn, L.; Akker, E.L.T.V.D.; Lamberts, S.W.J.; Van Rossum, E.F.C. Clinical features associated with glucocorticoid receptor polymorphisms. Ann. N. Y. Acad. Sci. USA 2009, 1179, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, E.F.; Koper, J.W.; Van Den Beld, A.W.; Uitterlinden, A.G.; Arp, P.; Ester, W.; Janssen, J.A.M.J.L.; Brinkmann, A.O.; De Jong, F.H.; Grobbee, D.E.; et al. Identification of the BclI polymorphism in the glucocorticoid receptor gene: Association with sensitivity to glucocorticoids in vivo and body mass index. Clin. Endocrinol. 2003, 59, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.U.; Hitman, G.A.; Kopelman, P.G. An association between a BclI restriction fragment length polymorphism of the gluco-corticoid receptor locus and hyperinsulinaemia in obese women. J. Mol. Endocrinol. 1992, 9, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Geelen, C.C.; van Greevenbroek, M.M.; van Rossum, E.F.; Schaper, N.C.; Nijpels, G.; Hart, L.M.; Schalkwijk, C.G.; Ferreira, I.; van der Kallen, C.J.; Sauerwein, H.P.; et al. BclI glucocorticoid receptor polymorphism is associated with greater body fatness: The hoorn and CODAM studies. J. Clin. Endocrinol. Metab. 2013, 98, E595–E599. [Google Scholar] [CrossRef][Green Version]
- Tomlinson, J.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11β-Hydroxysteroid Dehydrogenase Type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Bujalska, I.J.; Kumar, S.; Stewart, P.M. Does central obesity reflect “Cushing’s disease of the omentum”? Lancet 1997, 349, 1210–1213. [Google Scholar] [CrossRef]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 324, E2482–E2491. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A Transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef]
- Moreno-Díaz, H.; Villalobos-Molina, R.; Ortiz-Andrade, R.; Díaz-Coutiño, D.; Medina-Franco, J.L.; Webster, S.; Binnie, M.; Estrada-Soto, S.; Ibarra-Barajas, M.; León-Rivera, I.; et al. Antidiabetic activity of N-(6-substituted-1,3-benzothiazol-2-yl)benzenesulfonamides. Bioorganic Med. Chem. Lett. 2008, 18, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Z.; Yang, H.; Chen, J.; Feng, Y.; Du, L.; Leng, Y.; Shen, J. 4-(Phenylsulfonamidomethyl)benzamides as potent and selective inhibitors of the 11β-hydroxysteroid dehydrogenase type 1 with efficacy in diabetic ob/ob mice. Bioorganic Med. Chem. Lett. 2009, 19, 4455–4458. [Google Scholar] [CrossRef]
- Siu, M.; Johnson, T.O.; Wang, Y.; Nair, S.K.; Taylor, W.D.; Cripps, S.J.; Matthews, J.J.; Edwards, M.P.; Pauly, T.A.; Ermolieff, J.; et al. N-(Pyridin-2-yl) arylsulfonamide inhibitors of 11β-hydroxysteroid dehydrogenase type 1: Discovery of PF-915275. Bioorganic Med. Chem. Lett. 2009, 19, 3493–3497. [Google Scholar] [CrossRef]
- Bhat, B.G.; Hosea, N.; Fanjul, A.; Herrera, J.; Chapman, J.; Thalacker, F.; Stewart, P.M.; Rejto, P.A. Demonstration of proof of mechanism and pharmacokinetics and pharmacodynamic relationship with 4′-Cyano-biphenyl-4-sulfonic Acid (6-Amino-pyridin-2-yl)-amide (PF-915275), an Inhibitor of 11β-Hydroxysteroid Dehydrogenase Type 1, in Cynomolgus Monkeys. J. Pharmacol. Exp. Ther. 2007, 324, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.D.; Azevedo, I.; Monteiro, R.; Martins, M.J. 11β-Hydroxysteroid dehydrogenase type 1: Relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes. Metab. 2012, 14, 869–881. [Google Scholar] [CrossRef]
- Fraser, R.; Ingram, M.C.; Anderson, N.H.; Morrison, C.; Davies, E.; Connell, J.M. Connell cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension 1999, 33, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Rask, E.; Olsson, T.; Soderberg, S.; Andrew, R.; Livingstone, D.E.; Johnson, O.; Walker, B.R. Tissue-specific dysregulation of cortisol metabolism in human obesity. J. Clin. Endocrinol. Metab. 2001, 86, 1418–1421. [Google Scholar] [CrossRef]
- Rask, E.; Walker, B.R.; Söderberg, S.; Livingstone, D.E.W.; Eliasson, M.; Johnson, O.; Andrew, R.; Olsson, T. Tissue-specific changes in peripheral cortisol metabolism in obese women: Increased adipose 11b-hydroxysteroid dehydrogenase type 1 activity. J. Clin. Endocrinol. Metab. 2002, 87, 3330–3336. [Google Scholar]
- Tomlinson, J.W.; Sinha, B.; Bujalska, I.; Hewison, M.; Stewart, P.M. Expression of 11b-hydroxysteroiddehydrogenase type 1 in adipose tissue is not increased in human obesity. J. Clin. Endocrinol. Metab. 2002, 87, 5630–5635. [Google Scholar] [CrossRef]
- Lutz, S.Z.; Peter, A.; Machicao, F.; Lamprinou, A.; Machann, J.; Schick, F.; Königsrainer, I.; Königsrainer, A.; Fritsche, A.; Staiger, H.; et al. Genetic Variation in the 11β-hydroxysteroid-dehydrogenase 1 Gene Determines NAFLD and Visceral Obesity. J. Clin. Endocrinol. Metab. 2016, 101, 4743–4751. [Google Scholar] [CrossRef]
- Mai, K.; Andres, J.; Bobbert, T.; Maser-Gluth, C.; Möhlig, M.; Bähr, V.; Pfeiffer, A.F.H.; Spranger, J.; Diederich, S. Rosiglitazone decreases 11beta-hydroxysteroid dehydrogenase type 1 in subcutaneous adipose tissue. Clin. Endocrinol. 2007, 67, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Wake, D.J.; Stimson, R.H.; Tan, G.D. Effects of peroxisome proliferator-activated receptor-alpha and -gamma agonists on 11beta-hydroxysteroid dehydrogenase type 1 in subcutaneous adipose tissue in men. J. Clin. Endocrinol. Metab. 2007, 92, 1848–1856. [Google Scholar] [CrossRef][Green Version]
- Anagnostis, P.; Katsiki, N.; Adamidou, F.; Athyros, V.G.; Karagiannis, A.; Kita, M.; Mikhailidis, D.P. 11beta-Hydroxysteroid dehydrogenase type 1 inhibitors: Novel agents for the treatment of metabolic syndrome and obesity-related disorders? Metabolism 2012, 62, 21–33. [Google Scholar] [CrossRef]
- Heise, T.; Morrow, L.; Hompesch, M.; Häring, H.-U.; Kapitza, C.; Abt, M.; Ramsauer, M.; Magnone, M.-C.; Fuerst-Recktenwald, S. Safety, efficacy and weight effect of two 11β-HSD1 inhibitors in metformin-treated patients with type 2 diabetes. Diabetes Obes. Metab. 2014, 16, 1070–1077. [Google Scholar] [CrossRef]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Ferrante, A.W. Macrophages, fat, and the emergence of immunometabolism. J. Clin. Investig. 2013, 123, 4992–4993. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose tissue: Physiology to metabolic dysfunction. In The Insulin Receptor and Its Signal Transduction Network; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Eds.; Endotext [Internet] MD Text.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar] [PubMed]
- Kumari, M.; Heeren, J.; Scheja, L. Regulation of immunometabolism in adipose tissue. Semin. Immunopathol. 2017, 40, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cohen, P.; Spiegelman, B.M. Adaptive thermogenesis in adipocytes: Is beige the new brown? Genes Dev. 2013, 27, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Brandão, B.; Poojari, A.; Rabiee, A. Thermogenic fat: Development, physiological function, and therapeutic potential. Int. J. Mol. Sci. 2021, 22, 5906. [Google Scholar] [CrossRef]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and importance of brown ad-ipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Rothwell, N.J.; Stock, M.J. Surgical removal of brown fat results in rapid and complete compensation by other depots. Am. J. Physiol. Integr. Comp. Physiol. 1989, 257, R253–R258. [Google Scholar] [CrossRef]
- Rogers, N.H. Brown adipose tissue during puberty and with aging. Ann. Med. 2014, 47, 142–149. [Google Scholar] [CrossRef]
- Hibi, M.; Oishi, S.; Matsushita, M.; Yoneshiro, T.; Yamaguchi, T.; Usui, C.; Yasunaga, K.; Katsuragi, Y.; Kubota, K.; Tanaka, S.; et al. Brown adipose tissue is involved in diet-induced thermogenesis and whole-body fat utilization in healthy humans. Int. J. Obes. 2016, 40, 1655–1661. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Brown and beige adipose tissues in health and disease. Compr. Physiol. 2017, 7, 1281–1306. [Google Scholar] [CrossRef] [PubMed]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2015, 125, 259–266. [Google Scholar] [CrossRef] [PubMed]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef]
- Boutens, L.; Stienstra, R. Adipose tissue macrophages: Going off track during obesity. Diabetologia 2016, 59, 879–894. [Google Scholar] [CrossRef]
- Glass, C.K.; Olefsky, J.M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012, 15, 635–645. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the cen-tral regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell. Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell. 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Carey, A.L.; Formosa, M.F.; Van Every, B.; Bertovic, D.; Eikelis, N.; Lambert, G.W.; Kalff, V.; Duffy, S.J.; Cherk, M.H.; Kingwell, B.A. Ephedrine activates brown adipose tissue in lean but not obese humans. Diabetologia 2013, 56, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Zylan, K.D.; Carlisle, H.J. Effect of ambient temperature on the paradoxical metabolic responses to norepinephrine. Pharmacol. Biochem. Behav. 1992, 43, 577–582. [Google Scholar] [CrossRef]
- Kotzbeck, P.; Giordano, A.; Mondini, E.; Murano, I.; Severi, I.; Venema, W.; Cecchini, M.P.; Kershaw, E.E.; Barbatelli, G.; Haemmerle, G.; et al. Brown adipose tissue whitening leads to brown adi-pocyte death and adipose tissue inflammation. J. Lipid Res. 2018, 59, 784–794. [Google Scholar] [CrossRef]
- van den Beukel, J.C.; Grefhorst, A.; Quarta, C.; Steenbergen, J.; Mastroberardino, P.G.; Lombès, M.; Delhanty, P.J.; Mazza, R.; Pagotto, U.; van der Lely, A.J.; et al. Direct activating effects of adrenocorticotropic hormone (ACTH) on brown adipose tissue are attenuated by corticosterone. FASEB J. Off. Public Fed. Am. Soc. Exp. Biol. 2014, 28, 4857–4867. [Google Scholar]
- Ramage, L.E.; Akyol, M.; Fletcher, A.M.; Forsythe, J.; Nixon, M.; Carter, R.N.; van Beek, E.J.; Morton, N.M.; Walker, B.R.; Stimson, R.H. Glucocorticoids acutely increase brown adipose tissue activity in humans, revealing species-specific differences in UCP-1 regulation. Cell Metab. 2016, 24, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yu, X.; Chen, Y. Recruitment of thermogenic fat: Trigger of fat burning. Front. Endocrinol. 2021, 12, 696505. [Google Scholar] [CrossRef]
- Solinas, G.; Karin, M. JNK1 and IKKbeta: Molecular links between obesity and metabolic dysfunction. FASEB J. Off. Public. Fed. Am. Soc. Exp. Biol. 2010, 24, 2596–2611. [Google Scholar]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Speaker, K.J.; Fleshner, M. Interleukin-1 beta: A potential link between stress and the development of visceral obesity. BMC Physiol. 2012, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Moore, J.; Cooper, M.S.; Bujalska, I.; Shahmanesh, M.; Burt, C.; Strain, A.; Hewison, M.; Stewart, P.M. Regulation of expression of 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue: Tissue-specific induction by cytokines. Endocrinology 2001, 142, 1982–1989. [Google Scholar] [CrossRef]
- Jager, J.; Grémeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.-F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef]
- Old, L.J. Tumor necrosis factor. Sci. Am. 1988, 258, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.T.; Ree, D.; Kolls, J.K.; Fuselier, J.; Coy, D.H.; Bryer-Ash, M. An in vivo model for elucidation of the mechanism of tumor necrosis factor-α (TNF-α)-induced insulin resistance: Evidence for differential regulation of insulin signaling by TNF-α. Endocrinology 1998, 139, 4928–4935. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Dobrescu, C.; Bagby, G.J. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 1992, 130, 43–52. [Google Scholar] [CrossRef]
- Borst, S.E. The role of TNF-α in insulin resistance. Endocrine 2004, 23, 177–182. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis fac-tor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Hotamisligil, G.; Shargill, N.; Spiegelman, B. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Fleischman, A.; Shoelson, S.E.; Bernier, R.; Goldfine, A.B. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 2007, 31, 289–294. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.A.; Vølund, A.; Ehses, J.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1–Receptor Antagonist in Type 2 Diabetes Mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Nakatani, Y.; Miyatsuka, T.; Kawamori, D.; Matsuoka, T.-A.; Matsuhisa, M.; Kajimoto, Y.; Ichijo, H.; Yamasaki, Y.; Hori, M. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat. Med. 2004, 10, 1128–1132. [Google Scholar] [CrossRef]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Bhadada, S.K. COVID-19 and diabetes mellitus: An unholy interaction of two pandemics. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 513–517. [Google Scholar] [CrossRef]
- Fadini, G.P.; Morieri, M.L.; Longato, E.; Avogaro, A. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J. Endocrinol. Investig. 2020, 43, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Morieri, M.L.; Boscari, F.; Fiorettoa, P.; Marana, A.; Busettoa, L.; Bonoraa, B.M.; Selmina, E.; Arcidiacono, G.; Pinelli, S.; et al. Newly diagnosed diabetes and admission hyperglicemia predict COVID severity by ag-gravating repiratory dysfunction. Diabetes Res. Clin. Prat. 2020, 168, 108374. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Zheng, M.-H.; Targher, G. Diabetes as a risk factor for greater COVID-19 severity and in-hospital death: A meta-analysis of observational studies. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1236–1248. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; She, Z.-G.; Cheng, X.; Qin, J.-J.; Zhang, X.-J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077.e3. [Google Scholar] [CrossRef]
- Steenblock, C.; Todorov, V.; Kanczkowski, W.; Eisenhofer, G.; Schedl, A.; Wong, M.-L.; Licinio, J.; Bauer, M.; Young, A.; Gainetdinov, R.R.; et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the neuroendocrine stress axis. Mol. Psychiatry 2020, 25, 1611–1617. [Google Scholar] [CrossRef]
- Cuschieri, S.; Grech, S. COVID-19 and diabetes: The why, the what and the how. J. Diabetes Complicat. 2020, 34, 107637. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, Y.; Gou, X.; Puab, K.; Chen, Z.; Guo, Q.; Jia, R.; Wang, H.; Wang, Y.; Zhou, Y.; et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Type 2 diabetes mellitus is associated with multiple cardiometabolic risk factors. Clin. Cornerstone 2007, 8, 53–68. [Google Scholar] [CrossRef]
- Drucker, D.J. Coronavirus infections and type 2 diabetes—Shared pathways with therapeutic implications. Endocr. Rev. 2020, 41, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Malavazos, A.E.; Ferreira, T. COVID-19 rise in younger adults with obesity: Visceral adiposity can predict the risk. Obesity 2020, 28, 1795. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef]
- Eketunde, A.O.; Mellacheruvu, S.P.; Oreoluwa, P. A review of postmortem findings in patients with COVID-19. Cureus 2020, 12, 9438. [Google Scholar] [CrossRef]
- Guo, W.; Li, M.; Dong, Y.; Zhou, H.; Zhang, Z.; Tian, C.; Qin, R.; Wang, H.; Shen, Y.; Du, K.; et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab. Res. Rev. 2020, 36, e3319. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-K.; Feng, Y.; Yuan, M.Y.; Fu, H.J.; Wu, B.Y.; Sun, G.Z.; Yang, G.R.; Zhang, X.L.; Wang, L.; Xu, X.P.; et al. Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabet. Med. 2006, 23, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Morra, M.E.; Thanh, L.; Kamel, M.G.; Ghazy, A.A.; Altibi, A.M.; Dat, L.M.; Thy, T.N.X.; Vuong, N.L.; Mostafa, M.R.; Ahmed, S.I.; et al. Clinical outcomes of current medical approaches for Middle East respiratory syndrome: A systematic review and meta-analysis. Rev. Med. Virol. 2018, 28, e1977. [Google Scholar] [CrossRef] [PubMed]
- Philips, B.J.; Meguer, J.-X.; Redman, J.; Baker, E. Factors determining the appearance of glucose in upper and lower respiratory tract secretions. Intensiv. Care Med. 2003, 29, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Holman, N.; Knighton, P.; Kar, P.; O’Keefe, J.; Curley, M.; Weaver, A.; Barron, E.; Bakhai, C.; Khunti, K.; Wareham, N.J.; et al. Risk factors for COVID-19-related mortality in people with type 1 and type 2 diabetes in England: A population-based cohort study. Lancet Diabetes Endocrinol. 2020, 8, 823–833. [Google Scholar] [CrossRef]
- le Roux, C.W. COVID-19 alters thinking and management in metabolic diseases. Nat. Rev. Endocrinol. 2020, 17, 71–72. [Google Scholar] [CrossRef]
- Landstra, C.P.; de Koning, J.P. COVID-19 and diabetes: Understanding the interrelationship and risks for a severe course. Front. Endocrinol. 2021, 12, 599. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianotti, L.; Belcastro, S.; D’Agnano, S.; Tassone, F. The Stress Axis in Obesity and Diabetes Mellitus: An Update. Endocrines 2021, 2, 334-347. https://doi.org/10.3390/endocrines2030031
Gianotti L, Belcastro S, D’Agnano S, Tassone F. The Stress Axis in Obesity and Diabetes Mellitus: An Update. Endocrines. 2021; 2(3):334-347. https://doi.org/10.3390/endocrines2030031
Chicago/Turabian StyleGianotti, Laura, Sara Belcastro, Salvatore D’Agnano, and Francesco Tassone. 2021. "The Stress Axis in Obesity and Diabetes Mellitus: An Update" Endocrines 2, no. 3: 334-347. https://doi.org/10.3390/endocrines2030031
APA StyleGianotti, L., Belcastro, S., D’Agnano, S., & Tassone, F. (2021). The Stress Axis in Obesity and Diabetes Mellitus: An Update. Endocrines, 2(3), 334-347. https://doi.org/10.3390/endocrines2030031