Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy



Abstract
1. Introduction
2. Animal Models of Taurine Deficiency
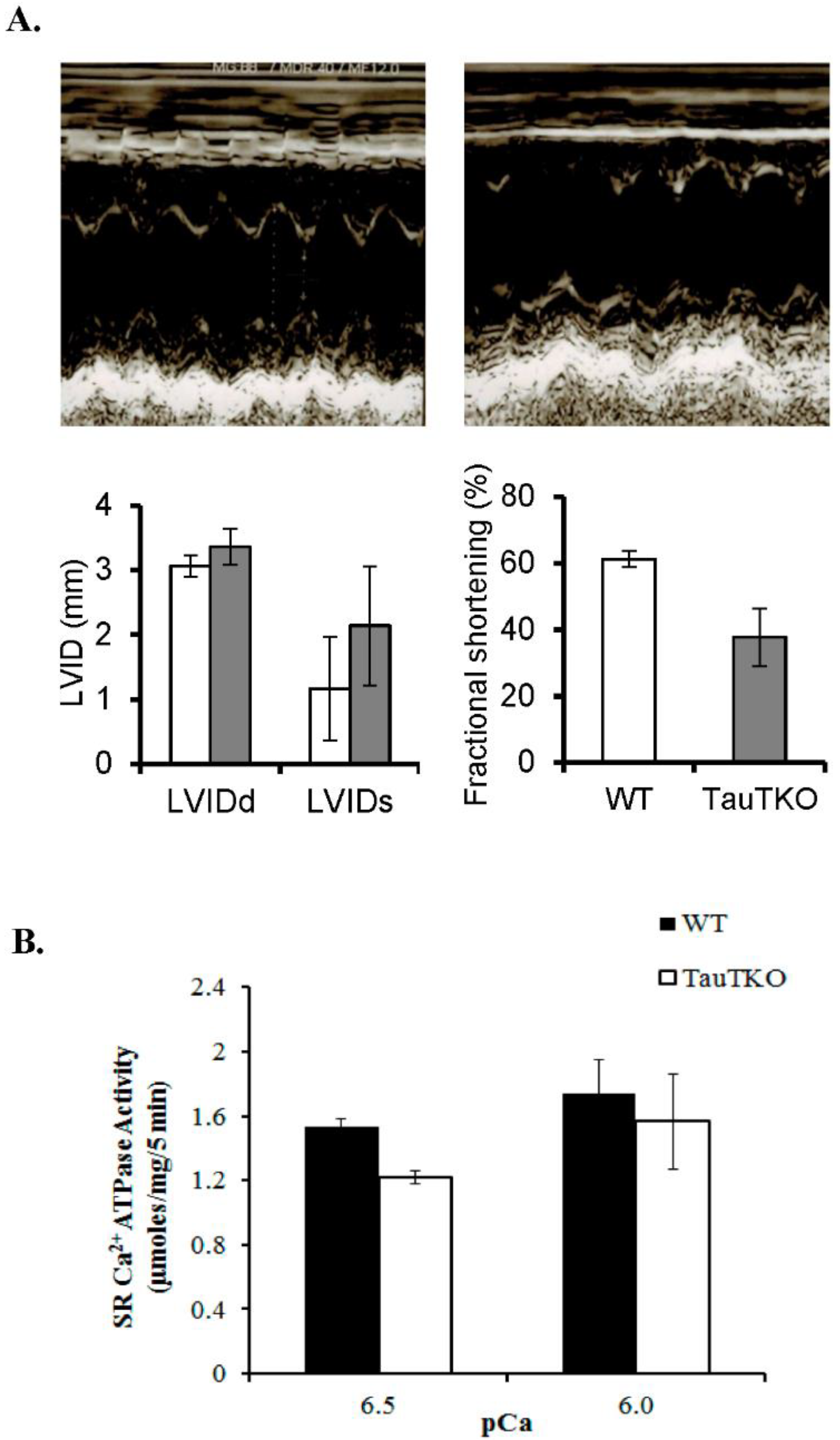
2.1. Impaired Calcium Handling Contributes to Cardiomyopathy
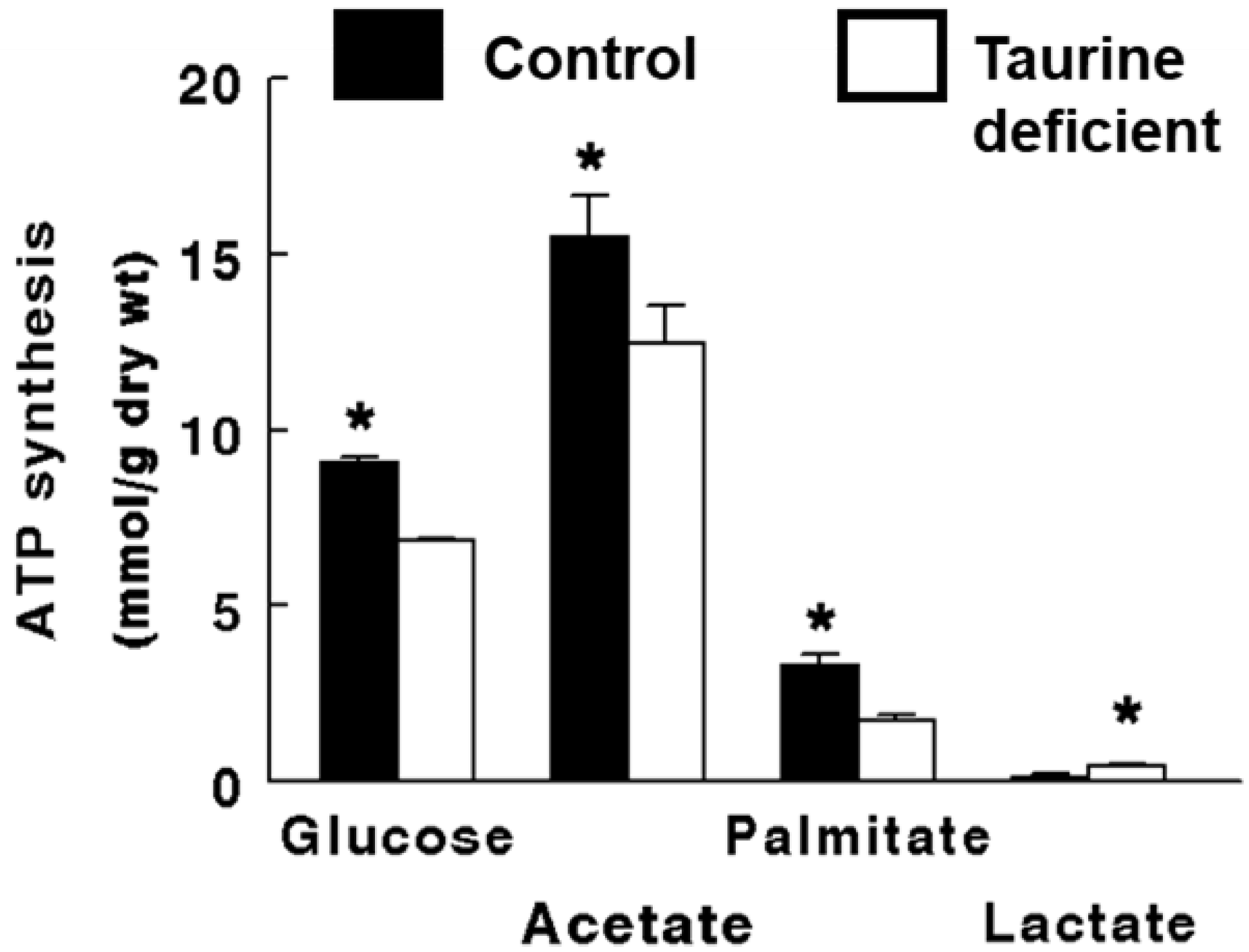
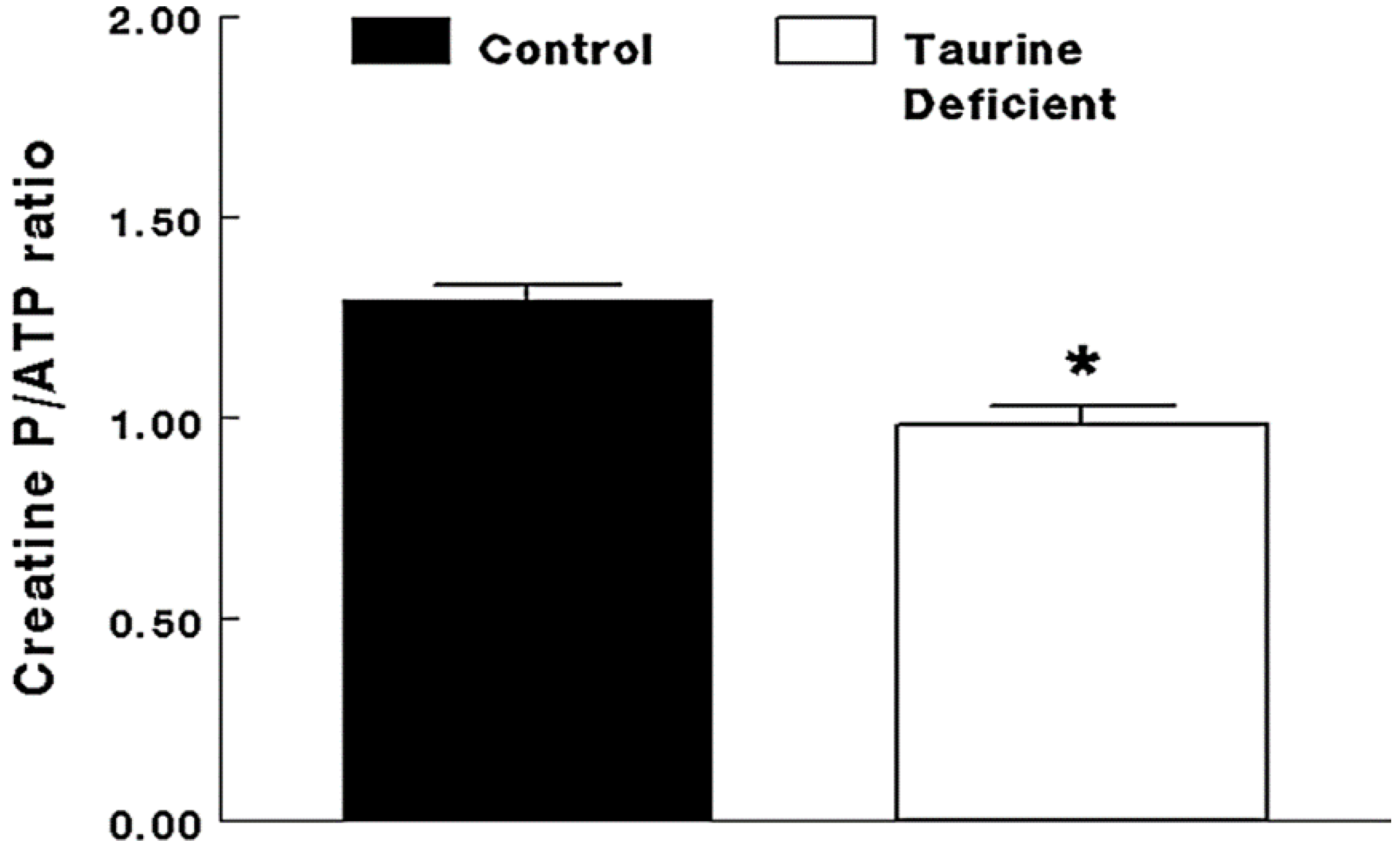
2.2. Taurine-Deficient Heart Is Energy-Deficient
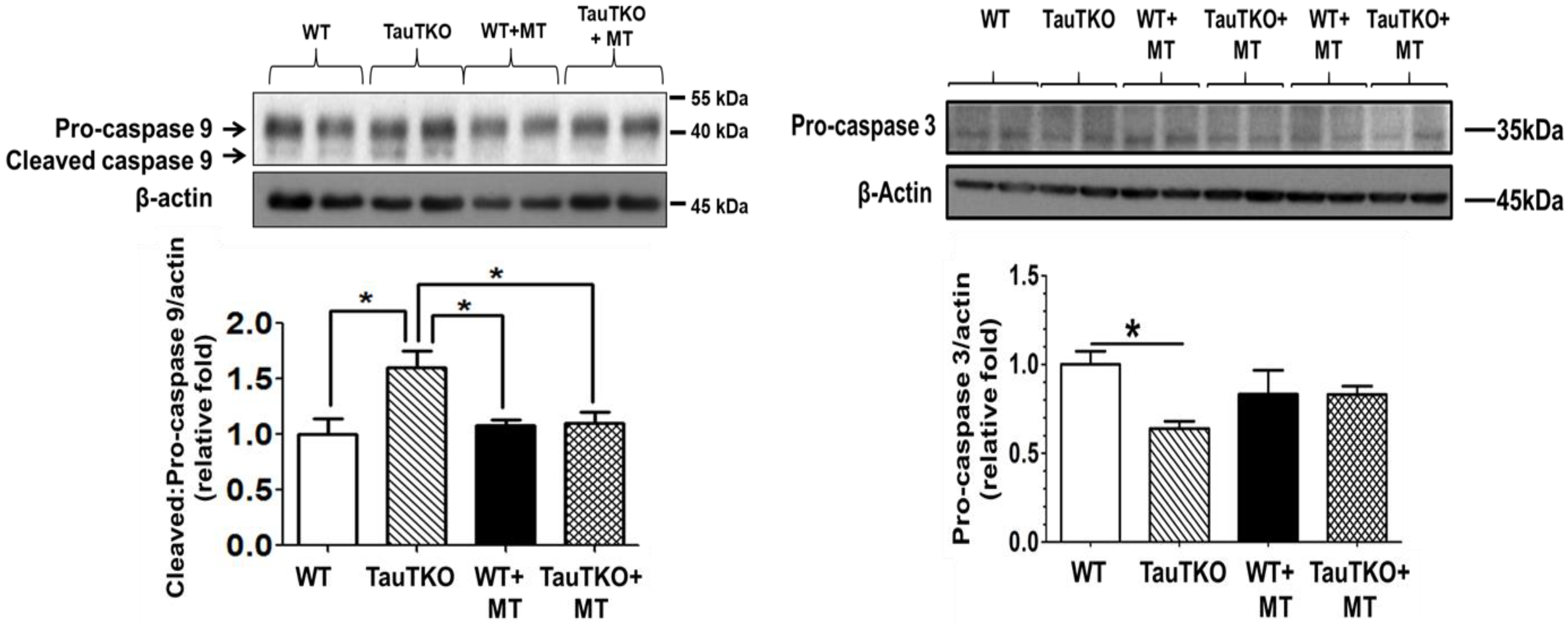
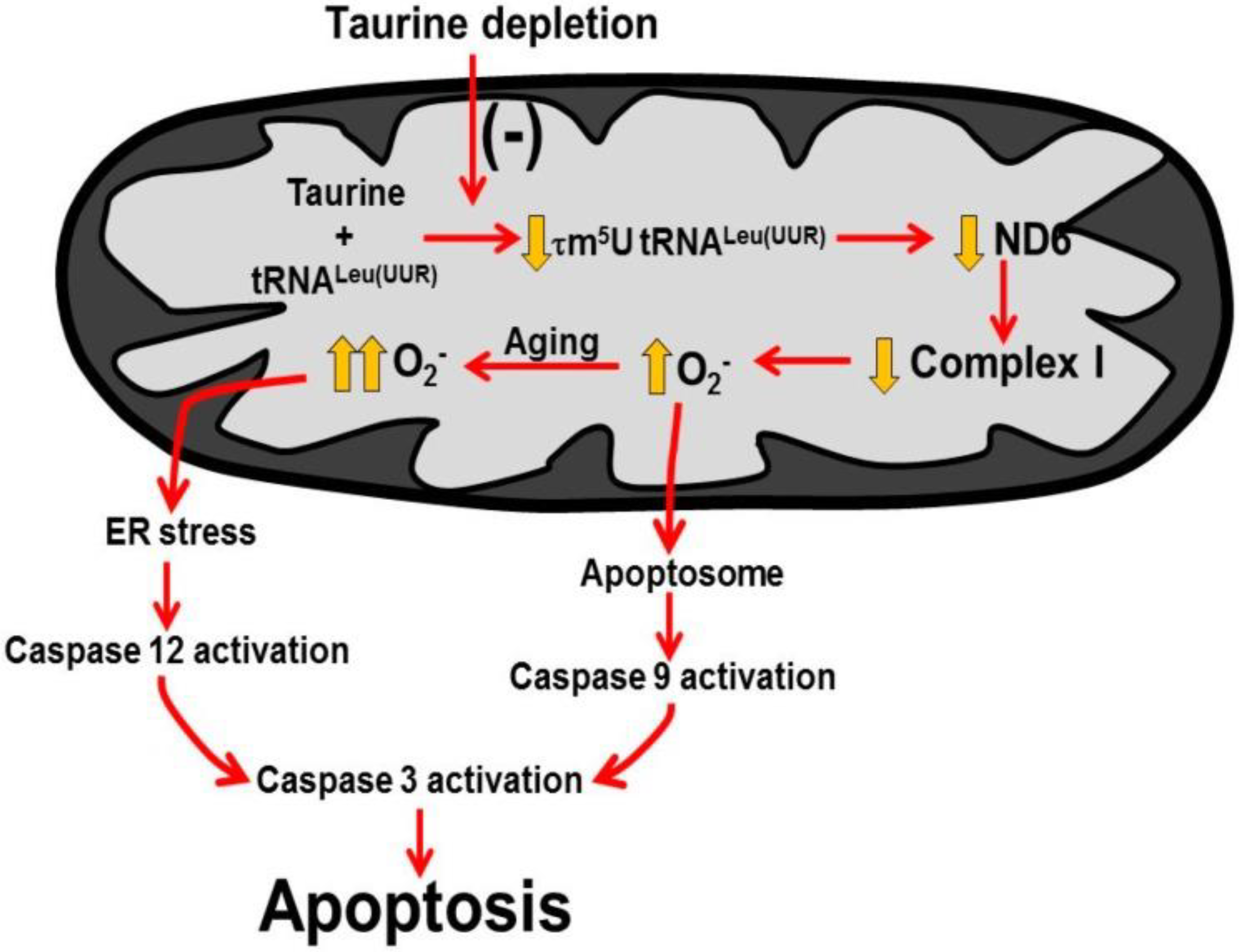
2.3. Role of Mitochondria, Oxidative Stress and Apoptosis in Development of Taurine-Deficient Cardiomyopathy
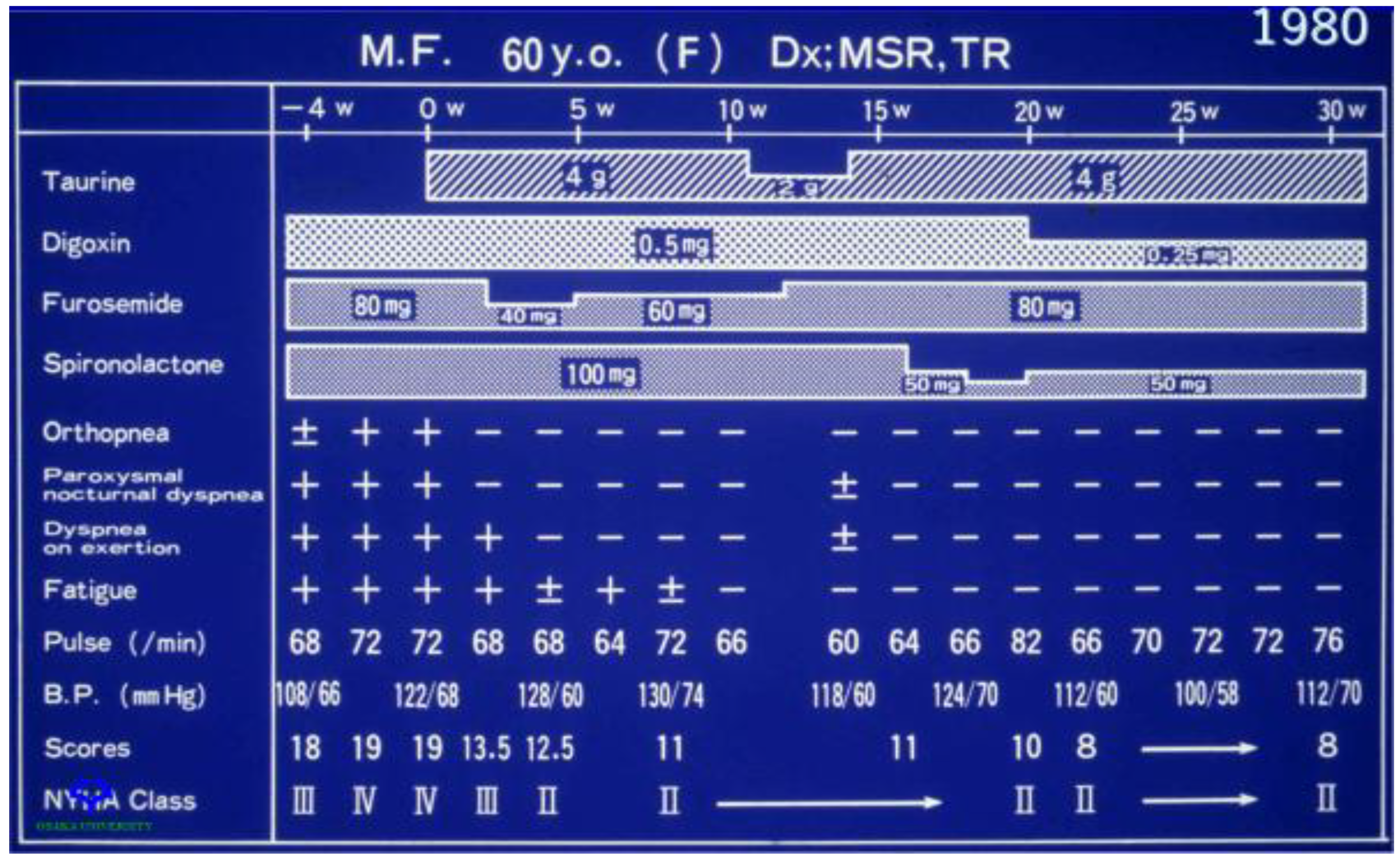
2.4. Clinical Importance of Taurine as Therapy in Treatment of Congestive Heart Failure
2.5. Taurine Involvement in the Disease, MELAS
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kramer, G.A.; Kittleson, M.D.; Fox, P.R.; Lewis, J.; Pion, P.D. Plasma taurine concentrations in normal dog and in dogs with heart disease. J. Vet. Intern. Med. 1995, 9, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Lubec, L.; Ya-hue, Z.; Pertti, T.; Kitzmueller, E.; Lubec, G. Distribution and disappearance of the radiolabeled carbon derived from -arginine and taurine in the mouse. Life Sci. 1997, 60, 2273–2381. [Google Scholar] [CrossRef]
- Kocsis, J.J.; Kostos, V.J.; Baskin, S.I. Taurine levels in the heart tissues of various species. In Taurine; Huxtable, R., Barbeau, A., Eds.; Raven Press: New York, NY, USA, 1976; pp. 145–153. [Google Scholar]
- Schaffer, S.; Solodushko, V.; Azuma, J. Taurine-deficient cardiomyopathy: Role of phospholipids, calcium and osmotic stress. Adv. Expt. Med. Biol. 2000, 483, 57–69. [Google Scholar]
- Pion, P.D.; Kittleson, M.D.; Rogers, Q.R.; Morris, J.G. Myocardial failure in cats associated with low plasma taurine: A reversible cardiomyopathy. Science 1987, 237, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kimura, Y.; Uozumi, I.Y.; Takai, M.; Muraoka, S.; Matsuda, T.; Ueki, K.; Yoshiyama, M.; Ikawa, M.; Okabe, M.; et al. Taurine depletion caused by knocking out the taurine transporter gene leads to a cardiomyopathy with cardiac atrophy. J. Mol. Cell Cardiol. 2008, 44, 927–937. [Google Scholar] [CrossRef]
- Novotny, M.J.; Hogan, P.A.; Paley, D.M.; Adams, H.R. Systolic and diastolic dysfunction of the left ventricle induced by taurine deficiency in cats. Am. J. Physiol. 1991, 261, H121–H127. [Google Scholar] [CrossRef]
- Mihl, C.; Dassen, W.R.H.; Kuipers, H. Cardiac remodelling: Concentric versus eccentric hypertrophy in strength and endurance athletes. Neth. Heart J. 2008, 16, 129–133. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Copelas, L.; MacKinnon, R.; Schoen, F.J.; Feldman, M.D.; Grossman, W.; Morgan, J.P. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 1987, 61, 70–76. [Google Scholar] [CrossRef]
- Chapman, R.A.; Suleiman, M.-S.; Earm, Y.E. Taurine and the heart. Cardiovasc. Res. 1993, 27, 358–363. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Solodushko, V.; Kakhniashvili, D. Beneficial effect of taurine depletion on osmotic sodium and calcium loading during chemical hypoxia. Am. J. Physiol. 2002, 282, C1113–C1120. [Google Scholar] [CrossRef]
- Jong, C.J.; Ito, T.; Mozaffari, M.; Azuma, J.; Schaffer, S. Effect of β-alanine treatment on mitochondrial taurine level and 5-taurinomethyluridine content. J. Biomed. Sci. 2011, 17 (Suppl. S1), S25. [Google Scholar] [CrossRef] [PubMed]
- Kurata, S.; Ohtsuki, T.; Wada, T.; Kirino, Y.; Takai, K.; Saigo, K.; Watanabe, K.; Suzuki, T. Decoding property of C5 uridine modification at the wobble position of tRNA anticodon. Nucleic Acids Res. 2003, 3, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.W.; Jong, C.J.; Ito, T.; Azuma, J. Role of taurine in the pathogenesis of MELAS and MERRF. Amino Acids 2014, 46, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Jong, C.J.; Ito, T.; Prentice, H.; Wu, J.-Y.; Schaffer, S.W. Role of Mitochondria and Endoplasmic Reticulum in Taurine-Deficiency-Mediated Apoptosis. Nutrients 2017, 9, 795. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Shimada-Takaura, K.; Jong, C.J.; Ito, T.; Takahashi, K. Impaired energy metabolism of the taurine-deficient heart. Amino. Acids 2016, 48, 549–558. [Google Scholar] [CrossRef]
- Shetewy, A.; Shimada-Takaura, K.; Warner, D.; Jong, C.J.; Mehdi, A.B.; Alexeyev, M.; Takahashi, K.; Schaffer, S.W. Mitochondrial defects associated with B-alanine toxicity: Relevance to hyper-beta-alaninemia. Mol. Cell Biochem. 2016, 416, 11–16. [Google Scholar] [CrossRef]
- Radad, K.; Rausch, W.-D.; Gille, G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem. Int. 2006, 49, 379–386. [Google Scholar] [CrossRef]
- Patel, M.S.; Korotchkina, L.G. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 2006, 34, 217–222. [Google Scholar] [CrossRef]
- Hansson, A.; Hance, N.; Dufour, E.; Rantanen, A.; Hultenby, K.; Clayton, D.A.; Wibom, R.; Larsson, N.G. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc. Natl. Acad. Sci. USA 2004, 101, 3136–3141. [Google Scholar] [CrossRef]
- Ingwall, J.S.; Weiss, R.G. Is the failing heart energy starved? Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1199. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorium, P.; Farber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased Rates of Substrate Oxidation Ex Vivo Predict the Onset of Heart Failure and Contractile in Rats With Pressure Overload Dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail Rev. 2013, 18, 1007. [Google Scholar] [CrossRef] [PubMed]
- Grivernnikova, V.G.; Vinogradov, A.D. Generation of superoxide by the mitochondrial complex I. Biochim. Biophys. Acta 2006, 1757, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Drose, B. Superoxide generation by complex III. From mechanistic rationales to functional consequences. Biochem. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Jong, C.J.; Azuma, J.; Schaffer, S. Mechanism underlying the antioxidant activity of taurine: Prevention of mitochondrial oxidant production. Amino Acids 2012, 42, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef]
- Suen, D.-F.; Norris, K.L.; Youle, R.J. Mitochondria dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef]
- Henry-Mowatt, J.; Dive, C.; Martinou, J.-C.; James, D. Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene 2004, 23, 2850–2860. [Google Scholar] [CrossRef]
- Jong, C.J.; Azuma, J.; Schaffer, S.W. Role of Mitochondrial Permeability Transition in Taurine Deficiency-Induced Apoptosis. Exp. Clin. Cardiol. 2011, 16, 125–128. [Google Scholar]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that dividend fuse mitochondria. Annu. Rev. Biochem. 2007, 7, 751–780. [Google Scholar] [CrossRef] [PubMed]
- Azuma, J.; Hasegawa, H.; Sawamura, A.; Awata, N.; Harada, H.; Ogura, K.; Kishimoto, S. Taurine for treatment of congestive heart failure. Int. J. Cardiol. 1982, 2, 303–304. [Google Scholar] [CrossRef]
- Takihara, K.; Azuma, J.; Awata, N.; Ohta, H.; Hamaguchi, T.; Sawamura, A.; Tanaka, Y.; Kishimoto, S.; Sperelakis, N. Beneficial effect of taurine in rabbits with chronic congestive heart failure. Am. Heart J. 1986, 112, 11278–11284. [Google Scholar] [CrossRef]
- Ohta, H.; Azuma, J.; Onishi, S.; Awata, N.; Takihara, K.; Kishimoto, S. Protective effect of taurine against isoprenaline-induced myocardial damage. Basic Res. Cardiol. 1986, 81, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Azuma, J.; Awata, N.; Hamaguchi, T.; Tanaka, Y.; Sawamura, A.; Kishimoto, S.; Sperelakis, N. Mechanism of the protective action of taurine against isoprenaline induced myocardial damage. Cardiovasc. Res. 1988, 22, 407–413. [Google Scholar] [CrossRef]
- Azuma, J.; Sawamura, A.; Awata, N.; Ohta, H.; Hamaguchi, T.; Harada, H.; Takihara, K.; Hasegawa, H.; Yamagami, T.; Ishiyama, T.; et al. Therapeutic effect of taurine in congestive heart failure: A double blind crossover trial. Clin. Cardiol. 1985, 8, 276–282. [Google Scholar] [CrossRef]
- Ito, T.; Schaffer, S.; Azuma, J. The effect of taurine on chronic heart failure: Actions of taurine against catecholamine and angiotensin II. Amino Acids 2013, 46, 111–119. [Google Scholar] [CrossRef]
- Jeejeebhoy, F.; Keith, M.; Freeman, M.; Barr, A.; McCall, M.; Kurian, R.; Mazer, D.; Errett, L. Nutritional supplementation with MyoVive repletes essential cardiac myocyte nutrients and reduces left ventricular size in patients with left ventricular dysfunction. Am. Heart. J. 2002, 143, 1092–1100. [Google Scholar] [CrossRef]
- Brody, J.I.; Soltys, H.D.; Zinsser, H.F. Folic acid deficiency in congestive heart failure. Brit. Heart J. 1969, 31, 741–745. [Google Scholar] [CrossRef][Green Version]
- Kirino, Y.; Goto, Y.-I.; Campos, Y.; Aremos, J.; Suzuki, T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl. Acad. Sci. USA 2005, 1029, 7127–7132. [Google Scholar] [CrossRef]
- Rikimaru, M.; Ohsawa, Y.; Wolf, A.M.; Ichimiya, H.; Kamimura, N.; Nishimatsu, S.; Ohta, S.; Sunada, Y. Taurine ameliorates impaired mitochondrial function and prevents stroke-like episodes in patients with MELAS. Intern. Med. 2012, 51, 3351–3357. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Respiration of Beta-Alanine Treated and Untreated Permeabilized Cardiomyocytes | ||||
|---|---|---|---|---|
| Condition | Substrate | State 3 rate | State 4 rate | RCR |
| Control | Glu-Mal | 1.0 (43.61) | 1.0 (11.03) | 1.0 |
| B-alanine | Glu-Mal | 0.78 ± 0.08 * | 0.9 ± 0.09 | 0.8 |
| Control | Succinate | 1.0 (65.4) | 1.0 (15.8) | 1.0 |
| B-alanine | Succinate | 0.98 ± 0.09 | 1.2 ± 0.1 | 0.95 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaffer, S.W.; Ito, T.; Azuma, J.; Jong, C.J.; Kramer, J.H. Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy. Hearts 2020, 1, 86-98. https://doi.org/10.3390/hearts1020010
Schaffer SW, Ito T, Azuma J, Jong CJ, Kramer JH. Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy. Hearts. 2020; 1(2):86-98. https://doi.org/10.3390/hearts1020010
Chicago/Turabian StyleSchaffer, Stephen W., Takashi Ito, Junichi Azuma, Chian Ju Jong, and Jay H. Kramer. 2020. "Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy" Hearts 1, no. 2: 86-98. https://doi.org/10.3390/hearts1020010
APA StyleSchaffer, S. W., Ito, T., Azuma, J., Jong, C. J., & Kramer, J. H. (2020). Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy. Hearts, 1(2), 86-98. https://doi.org/10.3390/hearts1020010