Experimental Hypomagnesemia Induces Neurogenic Inflammation and Cardiac Dysfunction


Abstract
1. Introduction
2. Results and Discussion
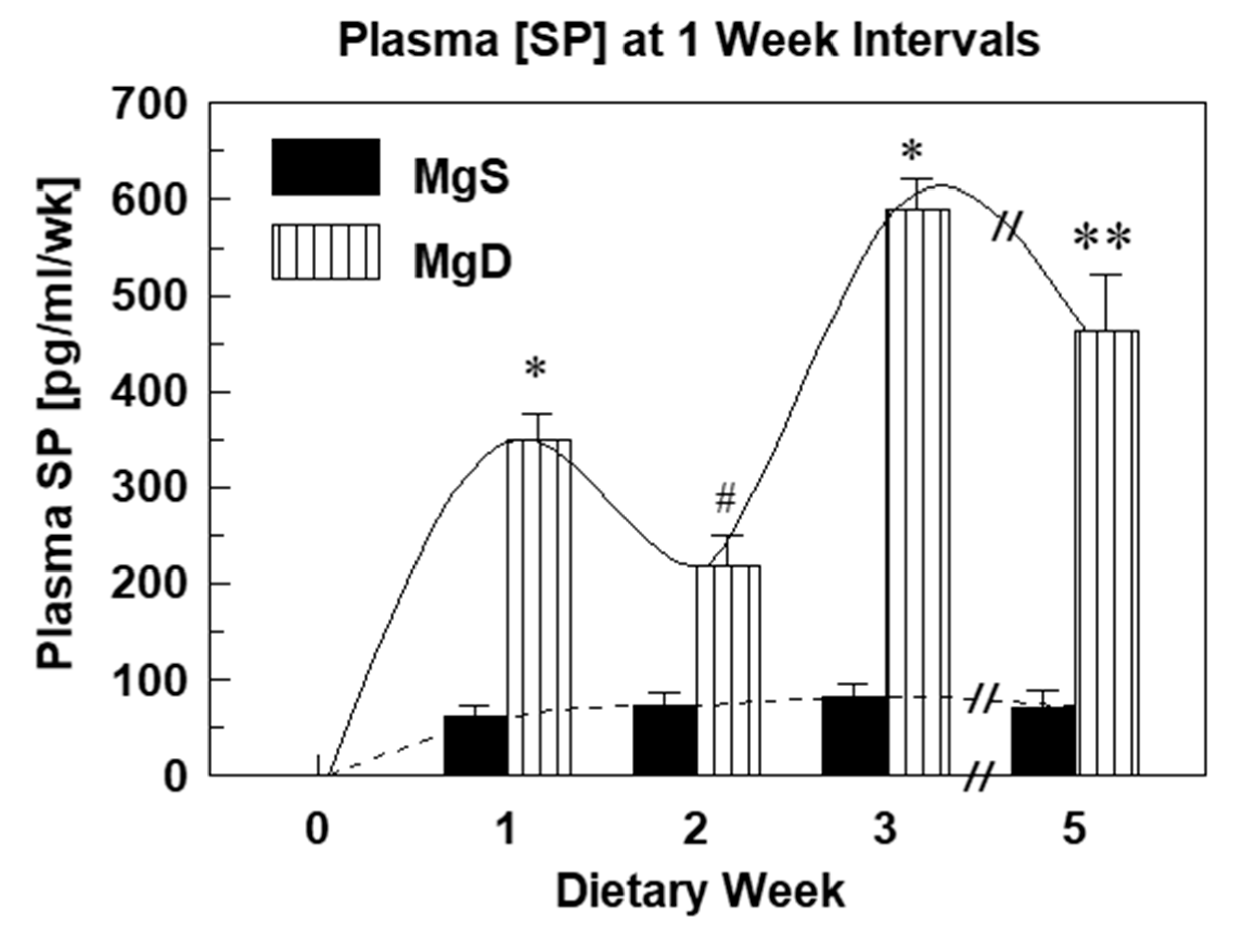
2.1. Substance P Elevations and Oxidative Stress During Experimental Mg Deficiency
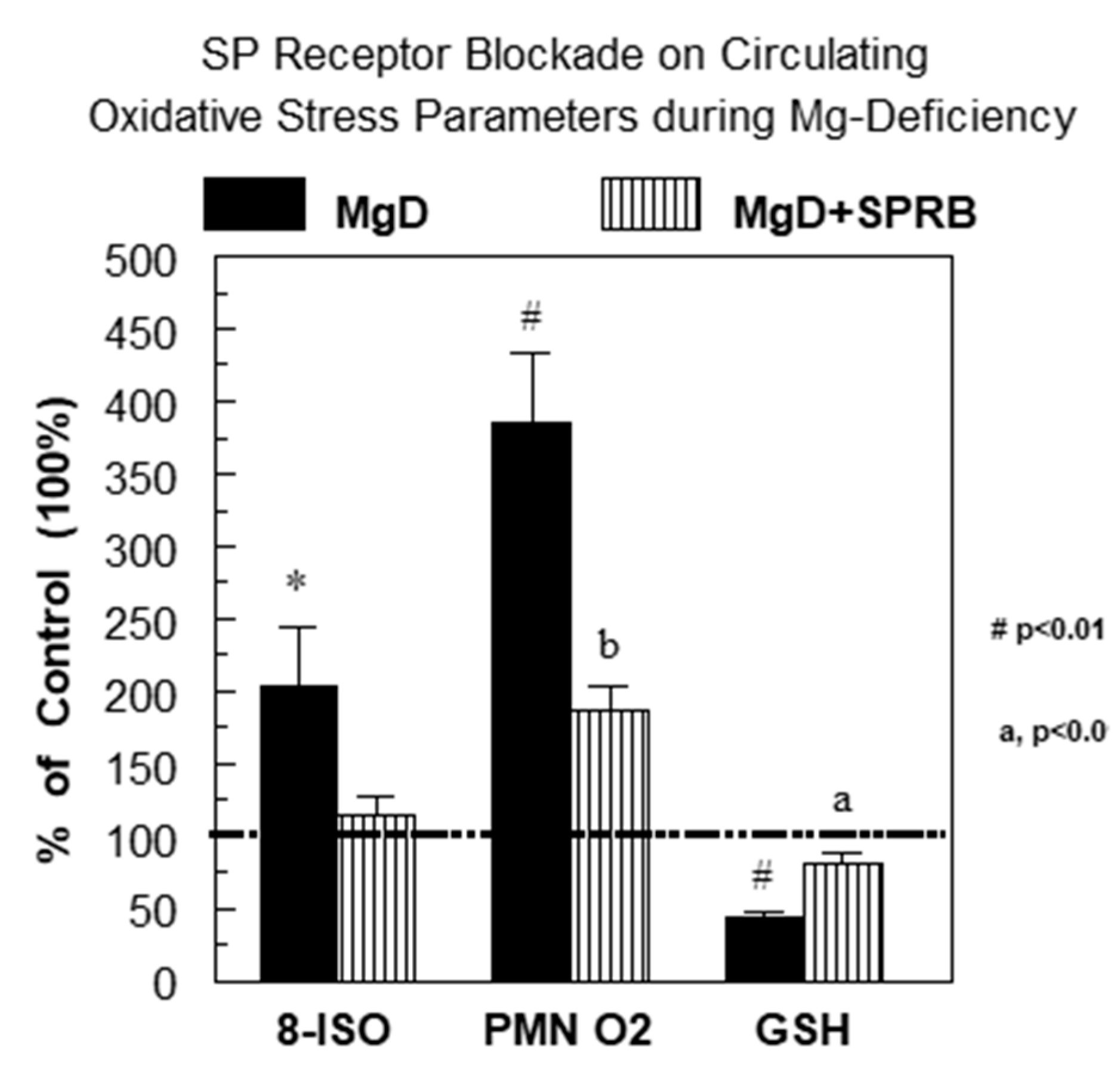
2.2. Substance P (Neurokinin-1 or NK-1) Receptor during Mg Deficiency
2.3. Substance P Receptor Blockade Attenuates Mg Deficiency-Induced Cardiac Dysfunction
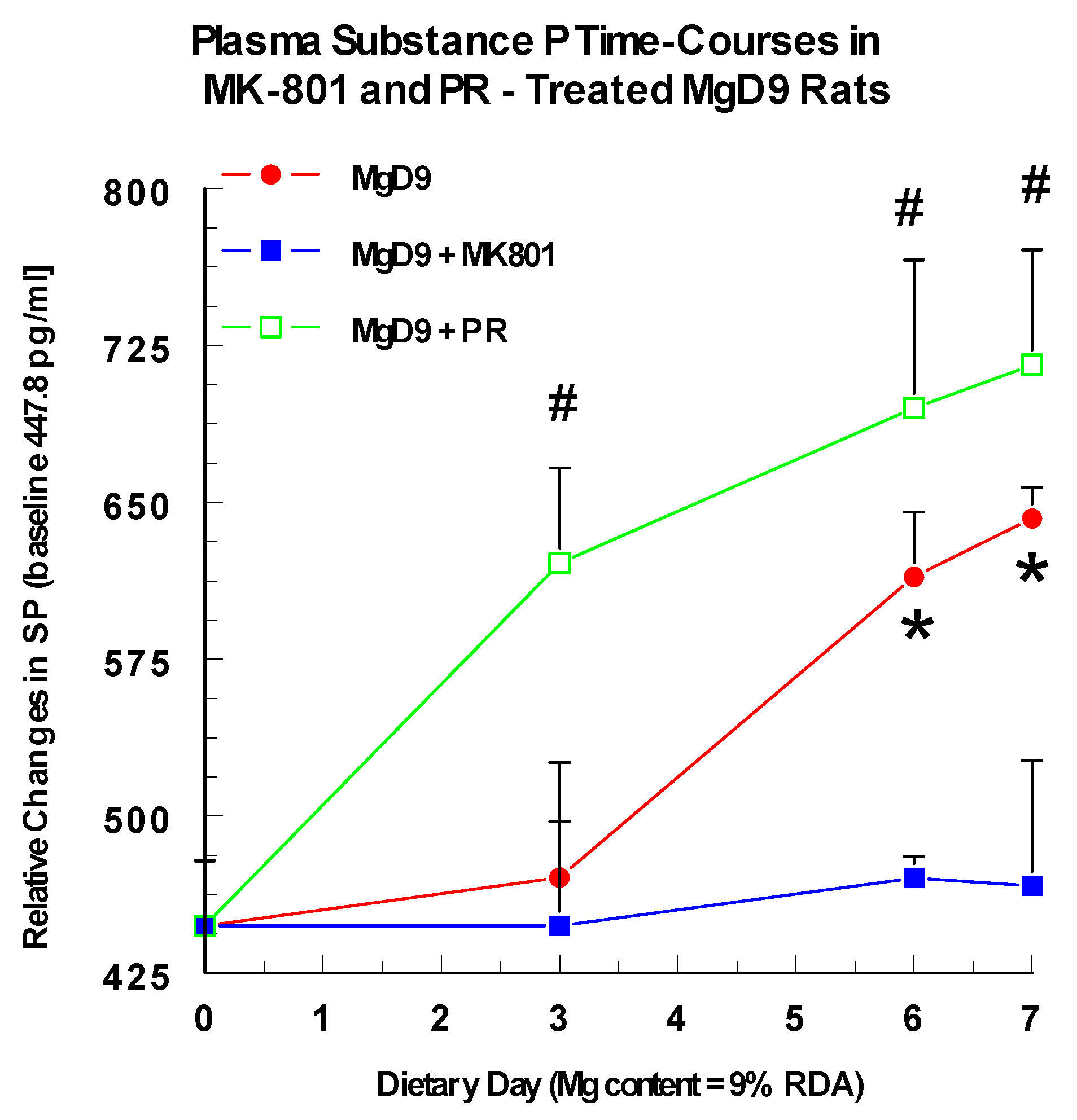
2.4. Modulation of Neuronal Substance P Release
2.5. Mg Deficiency-Mediated Cardiac Inflammation is Reduced by NMDA Receptor Blockade
2.6. Non-NMDA Receptor Modulation of Substance P Levels
2.7. Neutral Endopeptidase (NEP) Modulates Substance P Levels During Mg Deficiency
2.8. NEP Inhibition Modulates SP Bioavailability
2.9. NEP Modulates SP-Mediated Bioactivity and Neutrophil Activation During Mg Deficiency
2.10. SP Receptor Upregulation Linked to Enhanced PMN Responsiveness During Mg Deficiency
2.11. SP-Induced Elevation in NO• Partially Mediates Deleterious Events During Mg Deficiency
2.12. SP Receptor Blockade Protects Against Mg Deficiency-Induced Intolerance to Postischemic Stress
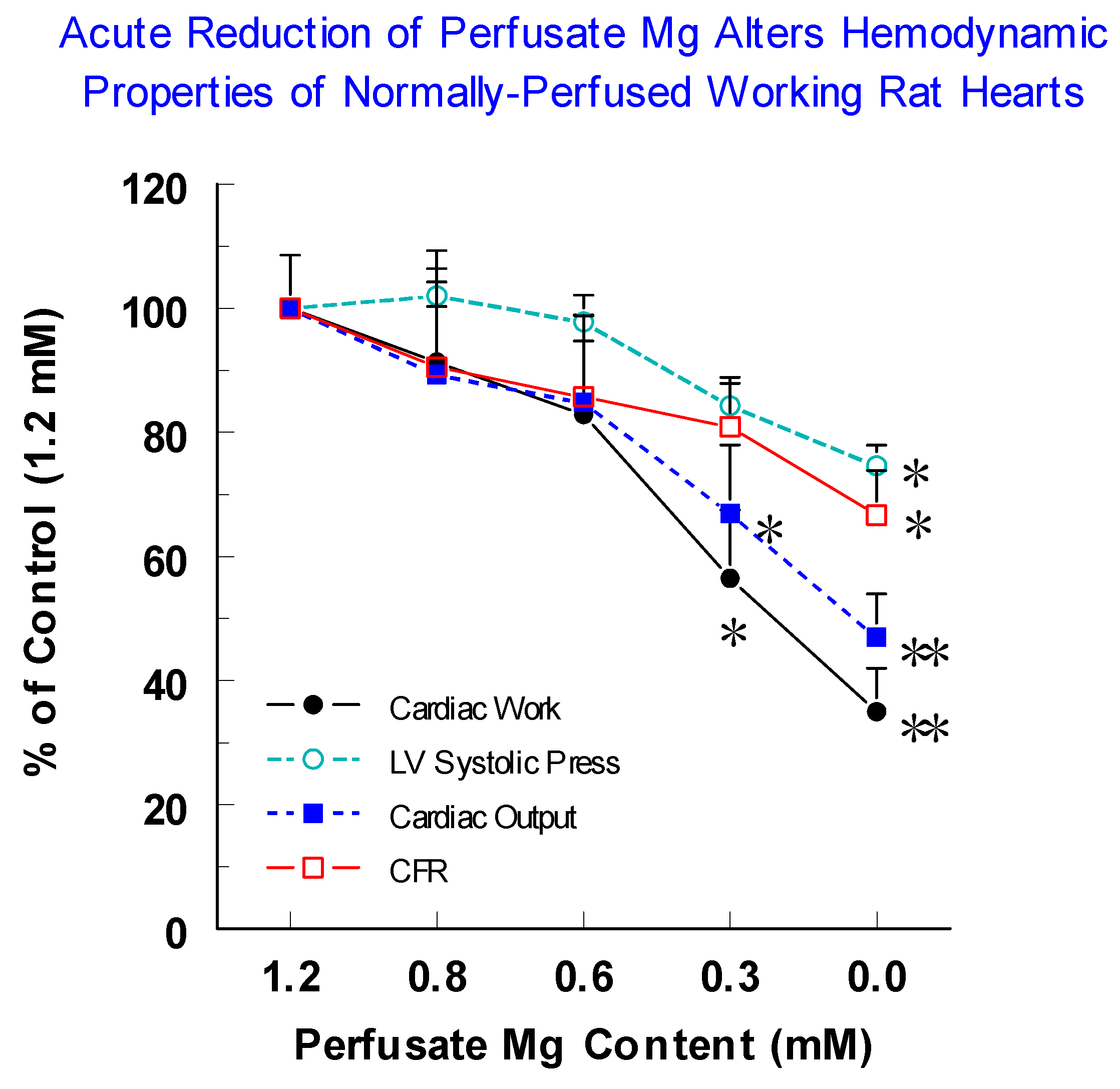
2.13. Acute Reduction in Perfusate Mg Modulates Neuropeptide and Cytokine Release from Normal Rat Hearts
3. Materials and Methods
3.1. Chemicals and Experimental Agents
3.2. Animal Assurance
3.3. Dietary Model
3.4. Blood Collection/Preparation
3.5. Plasma Magnesium
3.6. Plasma Substance P
3.7. Perfusate Neuropeptides and Inflammatory Cytokines
3.8. Red Blood Cell Glutathione
3.9. Plasma 8-Isoprostane
3.10. Neutrophil Basal and Stimulated Superoxide Generation
3.11. Non-Invasive Transthoracic Echocardiography
3.12. Perfused Heart Model
3.13. Statistical Approaches
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mak, I.T.; Kramer, J.H.; Chmielinska, J.J.; Spurney, C.F.; Weglicki, W.B. EGFR-TKI, erlotinib, causes hypomagnesemia, oxidative stress, and cardiac dysfunction: Attenuation by NK-1 receptor blockade. J. Cardiovasc. Pharmacol. 2015, 65, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.T.; Chmielinska, J.J.; Spurney, C.F.; Weglicki, W.B.; Kramer, J.H. Combination ART-Induced Oxidative/Nitrosative Stress, Neurogenic Inflammation and Cardiac Dysfunction in HIV-1 Transgenic (Tg) Rats: Protection by Mg. Int. J. Mol. Sci. 2018, 19, 2409. [Google Scholar] [CrossRef] [PubMed]
- Seelig, M. Cardiovascular consequences of magnesium deficiency and loss: Pathogenesis, prevalence and manifestations—Magnesium and chloride loss in refractory potassium repletion. Am. J. Cardiol. 1989, 63, G4–G21. [Google Scholar] [CrossRef]
- Cheungpasitporn, W.; Thongprayoon, C.; Qian, Q. Dysmagnesemia in Hospitalized Patients: Prevalence and Prognostic Importance. Mayo Clin. Proc. 2015, 90, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Altura, B.M.; Altura, B.T. New perspective on the role of Mg in the pathophysiology of the cardiovascular system. 1. Clin. Asp. Magnes 1985, 4, 226–244. [Google Scholar]
- Severino, P.; Netti, L.; Mariani, M.V.; Maraone, A.; D’Amato, A.; Scarpati, R.; Infusino, F.; Pucci, M.; LaValle, C.; Maestrini, V.; et al. Prevention of Cardiovascular Disease: Screening for Magnesium Deficiency. Cardiol. Res. Pract. 2019, 2019, 4874921. [Google Scholar] [CrossRef]
- Heggtveit, H.A.; Herman, L.; Mishra, R.K. Cardiac necrosis and calcification in experimental magnesium deficiency. a light and electron microscopic study. Am. J. Pathol. 1964, 45, 757–782. [Google Scholar]
- Weglicki, W.B.; Bloom, S.; Cassidy, M.M.; Freedman, A.M.; Atrakchi, A.H.; Dickens, B.F. Antioxidants and the cardiomyopathy of Mg-deficiency. Am. J. Cardiovasc. Pathol. 1992, 4, 210–215. [Google Scholar]
- Itokawa, Y. Tissue minerals of magnesium-deficient rats with thiamine deficiency and excess. Magnesium 1987, 6, 48–54. [Google Scholar]
- Chang, C.; Varghese, P.J.; Downey, J.; Bloom, S. Magnesium deficiency and myocardial infarct size in the dog. J. Am. Coll. Cardiol. 1985, 5, 280–289. [Google Scholar] [CrossRef]
- Borchgrevink, P.C.; Jynge, P. Acquired magnesium deficiency and myocardial tolerance to ischemia. J. Am. Coll. Nutr. 1987, 6, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.H.; Mišík, V.; Weglicki, W.B. Magnesium deficiency potentiates free radical production associated with post ischemic injury to rat hearts: Vitamin E affords protection. Free Rad. Biol. Med. 1994, 16, 713–723. [Google Scholar] [CrossRef]
- Weglicki, W.B.; Phillips, T.M.; Mak, I.T.; Cassidy, M.M.; Dickens, B.F.; Stafford, R.; Kramer, J.H. Cytokines, neuropeptides, and reperfusion injury during magnesium deficiency. Ann. N.Y. Acad. Sci. 1994, 723, 246–257. [Google Scholar] [CrossRef]
- Kharb, S.; Singh, V. Magnesium deficiency potentiates free radical production associated with myocardial infarction. J. Assoc. Physicians India 2000, 48, 484–485. [Google Scholar] [PubMed]
- Weglicki, W.B.; Phillips, T.M.; Cassidy, M.M.; Mak, I.T.; Dickens, B.F.; Stafford, R.E.; Kramer, J.H. Pro oxidant stress in Mg Deficiency: Role of neuropeptides and cytokines. In The Oxygen Paradox; Davies, K.J.A., Ursini, F., Eds.; Cleop University Press: Padova, Italy, 1995; pp. 773–782. [Google Scholar]
- Weglicki, W.B.; Mak, I.T.; Chmielinska, J.J.; Tejero-Taldo, M.I.; Komarov, A.M.; Kramer, J.H. The role of magnesium deficiency in cardiovascular and intestinal inflammation. Magnes. Res. 2010, 23, S199–S206. [Google Scholar] [CrossRef] [PubMed]
- Weglicki, W.B.; Mak, I.T.; Stafford, R.E.; Dickens, B.F.; Cassidy, M.M.; Phillips, T.M. Neurogenic peptides and the cardiomyopathy of magnesium-deficiency: Effects of substance P-receptor inhibition. Mol. Cell. Biochem. 1994, 130, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Weglicki, W.B.; Mak, I.T.; Phillips, T.M. Blockade of cardiac inflammation in Mg2+ deficiency by substance P receptor inhibition. Circ. Res. 1994, 74, 1009–1013. [Google Scholar] [CrossRef]
- Weglicki, W.B.; Phillips, T.M. Pathobiology of magnesium deficiency: A cytokine/neurogenic inflammation hypothesis. Am. J. Physiol. Integr. Comp. Physiol. 1992, 263, R734–R737. [Google Scholar] [CrossRef]
- Weglicki, W.B.; Mak, I.T.; Kramer, J.H.; Dickens, B.F.; Cassidy, M.M.; Stafford, R.E.; Phillips, T.M. Role of free radicals and substance P in magnesium deficiency. Cardiovasc. Res. 1996, 31, 677–682. [Google Scholar] [CrossRef]
- Weglicki, W.B.; Chmielinska, J.J.; Tejero-Taldo, I.; Kramer, J.H.; Spurney, C.F.; Viswalingham, K.; Lu, B.; Mak, I.T. Neutral endopeptidase inhibition enhances substance P mediated inflammation due to hypomagnesemia. Magnes. Res. 2009, 22, 167S–173S. [Google Scholar] [CrossRef]
- Mak, I.T.; Chmielinska, J.J.; Kramer, J.H.; Spurney, C.F.; Weglicki, W.B. Loss of neutral endopeptidase activity contributes to neutrophil activation and cardiac dysfunction during chronic hypomagnesemia: Protection by substance P receptor blockade. Exp. Clin. Cardiol. 2011, 16, 121–124. [Google Scholar] [PubMed]
- Kramer, J.H.; Mak, I.T.; Phillips, T.M.; Weglicki, W.B. Dietary Magnesium Intake Influences Circulating Pro-Inflammatory Neuropeptide Levels and Loss of Myocardial Tolerance to Postischemic Stress. Exp. Biol. Med. 2003, 228, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.H.; Spurney, C.; Iantorno, M.; Tziros, C.; Mak, I.-T.; Tejero-Taldo, M.I.; Chmielinska, J.J.; Komarov, A.M.; Weglicki, W.B. Neurogenic inflammation and cardiac dysfunction due to hypomagnesemia. Am. J. Med. Sci. 2009, 338, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Satake, H.; Kawada, T. Overview of the primary structure, tissue-distribution, and functions of tachykinins and their receptors. Curr. Drug Targets 2006, 7, 963–974. [Google Scholar] [CrossRef]
- Ho, W.-Z.; Douglas, S.D. Substance P and neurokinin-1 receptor modulation of HIV. J. Neuroimmunol. 2004, 157, 48–55. [Google Scholar] [CrossRef]
- Almeida, T.A.; Rojo, J.; Nieto, P.M.; Pinto, F.M.; Hernández, M.; Martin, J.D.; Candenas, M.L. Tachykinins and Tachykinin Receptors: Structure and Activity Relationships. Curr. Med. Chem. 2004, 11, 2045–2081. [Google Scholar] [CrossRef]
- Datar, P.; Srivastava, S.; Coutinho, E.; Govil, G. Substance P: Structure, function, and therapeutics. Curr. Top. Med. Chem. 2004, 4, 75–103. [Google Scholar] [CrossRef]
- Saria, A. The tachykinin NK1 receptor in the brain: Pharmacology and putative functions. Eur. J. Pharmacol. 1999, 375, 51–60. [Google Scholar] [CrossRef]
- Derose, V.; A Robbins, R.; Snider, R.M.; Spurzem, J.R.; Thiele, G.M.; I Rennard, S.; Rubinstein, I. Substance P increases neutrophil adhesion to bronchial epithelial cells. J. Immunol. 1994, 152, 1339–1346. [Google Scholar]
- Kubes, P.; Kanwar, S.; Niu, X.; Gaboury, J.P. Nitric oxide synthesis inhibition induces leukocyte adhesion via superoxide and mast cells. FASEB J. 1993, 7, 1293–1299. [Google Scholar] [CrossRef]
- Hartung, H.P.; Toyka, K.V. Activation of macrophages by substance P: Induction of oxidative burst and thromboxane release. Eur. J. Pharmacol. 1983, 89, 301–305. [Google Scholar] [CrossRef]
- Persson, M.G.; Hedqvist, P.; Gustafsson, L.E. Nerve induced tachykinin mediated vasodilation in skeletal muscle is dependent on nitric oxide formation. Eur. J. Pharmacol 1991, 205, 295–301. [Google Scholar] [CrossRef]
- Hafström, I.; Gyllenhammar, H.; Palmblad, J.; Ringertz, B. Substance P activates and modulates neutrophil oxidative metabolism and aggregation. J. Rheumatol. 1989, 16, 1033–1037. [Google Scholar] [PubMed]
- Mak, I.T.; Dickens, B.F.; Komarov, A.M.; Phillips, T.M.; Weglicki, W.B. Activation of the neutrophil and loss of plasma glutathione during Mg-deficiency—Modulation effect by NOS inhibition. Mol. Cell Biochem. 1997, 176, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Rude, R.K.; Singer, F.R.; Gruber, H.E. Skeletal and hormonal effects of magnesium deficiency. J. Am. Coll. Nutr. 2009, 28, 131–141. [Google Scholar] [CrossRef]
- Vink, R.; Donkin, J.J.; Cruz, M.I.; Nimmo, A.J.; Cernak, I. A substance P antagonist increases brain intracellular free magnesium concentration after diffuse traumatic brain injury in rats. J. Am. Coll. Nutr. 2004, 23, 538S–540S. [Google Scholar] [CrossRef]
- Grimaldi, B.L. The central role of magnesium deficiency in Tourette’s syndrome: Causal relationships between magnesium deficiency, altered biochemical pathways and symptoms relating to Tourette’s syndrome and several reported comorbid conditions. Med. Hypotheses 2002, 58, 47–60. [Google Scholar] [CrossRef]
- Feickert, M.; Burckhardt, B.B. Substance P in cardiovascular diseases-A bioanalytical review. Clin. Chim. Acta 2019, 495, 501–506. [Google Scholar] [CrossRef]
- Nordmann, R.; Ribière, C.; Rouach, H. Implication of free radical mechanisms in ethanol-induced cellular injury. Free Radic. Biol. Med. 1992, 12, 219–240. [Google Scholar] [CrossRef]
- Iversen, L.L.; Emson, P.C.; Lee, C.M.; Gilbert, R.F.; Hunt, S. Regulation of neuropeptide release. Proc. R. Soc. London Ser. B Biol. Sci. 1980, 210, 91–111. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and Modulation of Na+and Ca2+Channels. Ann. N.Y. Acad. Sci. 1993, 707, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R. The NF-κB Regulatory Network. Cardiovasc. Toxicol. 2006, 6, 111–130. [Google Scholar] [CrossRef]
- Furness, J.B.; Papka, R.E.; Della, N.G.; Costa, M.; Eskay, R.L. Substance P-like immunoreactivity in nerves associated with the vascular system of guinea-pigs. Neuroscience 1982, 7, 447–459. [Google Scholar] [CrossRef]
- Furness, J.; Costa, M.; Papka, R.E.; Della, N.G.; Murphy, R.; Delia, N.G. Neuropeptides Contained in Peripheral Cardiovascular Nerves. Clin. Exp. Hypertens. Part A Theory Pr. 1984, 6, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Coggeshall, R.E.; Carlton, S.M. Ultrastructural analysis of NMDA, AMPA, and kainate receptors on unmyelinated and myelinated axons in the periphery. J. Comp. Neurol. 1998, 391, 78–86. [Google Scholar] [CrossRef]
- Lin, Y.J.; Bovetto, S.; Carver, J.M.; Giordano, T. Cloning of the cDNA for the human NMDA receptor NR2C subunit and its expression in the central nervous system and periphery. Mol. Brain Res. 1996, 43, 57–64. [Google Scholar] [CrossRef]
- Mishina, M.; Mori, H.; Araki, K.; Kushiya, E.; Meguro, H.; Kutsuwada, T.; Kashiwabuchi, N.; Ikeda, K.; Nagasawa, M.; Yamazaki, M.; et al. Molecular and Functional Diversity of the NMDA Receptor Channel. Ann. N.Y. Acad. Sci. 1993, 707, 136–152. [Google Scholar] [CrossRef]
- Masu, M.; Nakajima, Y.; Moriyoshi, K.; Ishii, T.; Akazawa, C.; Nakanashi, S. Molecular Characterization of NMDA and Metabotropic Glutamate Receptors. Ann. N.Y. Acad. Sci. 1993, 707, 153–164. [Google Scholar] [CrossRef]
- Tejero-Taldo, M.I.; Chmielinska, J.J.; Gonzalez, G.; Mak, I.T.; Weglicki, W.B. N-Methyl-d-aspartate Receptor Blockade Inhibits Cardiac Inflammation in the Mg2+-Deficient Rat. J. Pharmacol. Exp. Ther. 2004, 311, 8–13. [Google Scholar] [CrossRef]
- Tejero-Taldo, M.I.; Chmielinska, J.J.; Weglicki, W.B. Decreased VHL results in VEGF up-regulation in the magnesium deficient rat heart (abstract). J. Mol. Cell Cardiol. 2006, 40, 904. [Google Scholar] [CrossRef]
- Chmielinska, J.J.; Tejero-Taldo, M.I.; Mak, I.T.; Weglicki, W.B. Intestinal and cardiac inflammatory response shows enhanced endotoxin receptor (CD14) expression in magnesium deficiency. Mol. Cell. Biochem. 2005, 278, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Tejero-Taldo, M.I.; Chmielinska, J.J.; Weglicki, W.B. Magnesium deficiency stimulates angiogenesis in the rat heart (abstract). J. Mol. Cell Cardiol. 2004, 36, 631. [Google Scholar]
- Tejero-Taldo, M.I.; Chmielinska, J.J.; Weglicki, W.B. Chronic dietary Mg2+ deficiency induces cardiac apoptosis in the rat heart. Magnes. Res. 2007, 20, 208–212. [Google Scholar] [PubMed]
- Ho, W.-Z.; Lai, J.P.; Zhu, X.H.; Uvaydova, M.; Douglas, S.D. Human monocytes and macrophages express substance P and neurokinin-1 receptor. J. Immunol. 1997, 159, 5654–5660. [Google Scholar]
- Lai, J.-P.; Douglas, S.D.; Ho, W.-Z. Human lymphocytes express substance P and its receptor. J. Neuroimmunol. 1998, 86, 80–86. [Google Scholar] [CrossRef]
- Khare, V.K.; Albino, A.P.; Reed, J.A. The neuropeptide/mast cell secretagogue substance P is expressed in cutaneous melanocytic lesions. J. Cutan. Pathol. 1998, 25, 2–10. [Google Scholar] [CrossRef]
- Cioni, C.; Renzi, D.; Calabrò, A.; Annunziata, P. Enhanced secretion of substance P by cytokine-stimulated rat brain endothelium cultures. J. Neuroimmunol. 1998, 84, 76–85. [Google Scholar] [CrossRef]
- Scholzen, T.E.; Steinhoff, M.; Bonaccorsi, P.; Klein, R.; Amadesi, S.; Geppetti, P.; Lu, B.; Gerard, N.P.; Olerud, J.E.; Luger, T.A.; et al. Neutral endopeptidase terminates substance P-induced inflammation in allergic contact dermatitis. J. Immunol. 2001, 166, 1285–1291. [Google Scholar] [CrossRef]
- Wang, L.; Sadoun, E.; Stephens, R.E.; Ward, P.E. Metabolism of substance P and neurokinin A by human vascular endothelium and smooth muscle. Peptides 1994, 15, 497–503. [Google Scholar] [CrossRef]
- Nadel, J.A. Peptidase modulation of neurogenic inflammation. In Neurogenic Inflammation; Geppetti, P., Holzer, P., Eds.; CRC Press: Boca Raton, FL, USA, 1996; pp. 115–127. [Google Scholar]
- Lesser, M.; Fung, K.; Choi, H.S.; Yoo, O.H.; Cardozo, C. Identification of two zinc metalloendopeptidases in alveolar macrophages of rats, guinea pigs, and human beings. J. Lab. Clin. Med. 1992, 120, 597–603. [Google Scholar]
- Iwamoto, I.; Kimura, A.; Ochiai, K.; Tomioka, H.; Yoshida, S. Distribution of NEP activity in human blood leukocytes. J. Leukad. Biol. 1991, 49, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Lauderback, C.M.; Hackett, J.M.; Huang, F.F.; Keller, J.N.; Szweda, L.I.; Butterfield, D.A. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: The role of Abeta1-42. J. Neurochem. 2001, 78, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-S.; Iwata, N.; Hama, E.; Saido, T.C.; Dickson, D.W. Oxidized neprilysin in aging and Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 2003, 310, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.T.; Kramer, J.H.; Chmielinska, J.J.; Khalid, M.H.; Landgraf, K.M.; Weglicki, W.B. Inhibition of neutral endopeptidase potentiates neutrophil activation during Mg-deficiency in the rat. Inflamm. Res. 2008, 57, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.T.; Kramer, J.H.; Weglicki, W.B. Suppression of neutrophil and endothelial activation by substance P receptor blockade in the Mg-deficient rat. Magnes. Res. 2003, 16. [Google Scholar]
- Mak, I.T.; Komarov, A.M.; Wagner, T.L.; Stafford, R.E.; Dickens, B.F.; Weglicki, W.B. Enhanced NO production during Mg deficiency and its role in mediating red blood cell glutathione loss. Am. J. Physiol. Physiol. 1996, 271, C385–C390. [Google Scholar] [CrossRef]
- Tejero-Taldo, M.I.; Kramer, J.H.; Mak, I.T.; Komarov, A.M.; Weglicki, W.B. The nerve-heart connection in the pro-oxidant response to Mg-deficiency. Heart Fail. Rev. 2006, 11, 35–44. [Google Scholar] [CrossRef]
- Kramer, J.H.; Phillips, T.M.; Weglicki, W.B. Magnesium-deficiency-enhanced Post-ischemic Myocardial Injury is Reduced by Substance P Receptor Blockade. J. Mol. Cell. Cardiol. 1997, 29, 97–110. [Google Scholar] [CrossRef]
- Mak, I.T.; Kramer, J.H.; Chen, X.; Chmielinska, J.J.; Spurney, C.F.; Weglicki, W.B. Mg supplementation attenuates ritonavir-induced hyperlipidemia, oxidative stress, and cardiac dysfunction in rats. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R1102–R1111. [Google Scholar] [CrossRef]
- Deheinzelin, D.; Negri, E.; Tucci, M.; Salem, M.; Da Cruz, V.; Oliveira, R.; Nishimoto, I.; Hoelz, C. Hypomagnesemia in critically ill cancer patients: A prospective study of predictive factors. Braz. J. Med Biol. Res. 2000, 33, 1443–1448. [Google Scholar] [CrossRef][Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inflammatory Mediator | MgS Heart Effluent Levels [pg/g prot/ml] per gm Wet wt Tissue |
|---|---|
| IL-1 | 5.03 ± 0.23 |
| IL-6 | 3.13 ± 0.26 |
| TNFα | 6.74 ± 0.22 |
| SP | 5.09 ± 0.21 |
| CGRP | 4.33 ± 0.20 |
| VIP | 2.95 ± 0.22 |
| NY | 3.62 ± 0.52 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kramer, J.H.; Mak, I.T.; Chmielinska, J.J.; Spurney, C.F.; Phillips, T.M.; Weglicki, W.B. Experimental Hypomagnesemia Induces Neurogenic Inflammation and Cardiac Dysfunction. Hearts 2020, 1, 99-116. https://doi.org/10.3390/hearts1020011
Kramer JH, Mak IT, Chmielinska JJ, Spurney CF, Phillips TM, Weglicki WB. Experimental Hypomagnesemia Induces Neurogenic Inflammation and Cardiac Dysfunction. Hearts. 2020; 1(2):99-116. https://doi.org/10.3390/hearts1020011
Chicago/Turabian StyleKramer, Jay H., I. Tong Mak, Joanna J. Chmielinska, Christopher F. Spurney, Terry M. Phillips, and William B. Weglicki. 2020. "Experimental Hypomagnesemia Induces Neurogenic Inflammation and Cardiac Dysfunction" Hearts 1, no. 2: 99-116. https://doi.org/10.3390/hearts1020011
APA StyleKramer, J. H., Mak, I. T., Chmielinska, J. J., Spurney, C. F., Phillips, T. M., & Weglicki, W. B. (2020). Experimental Hypomagnesemia Induces Neurogenic Inflammation and Cardiac Dysfunction. Hearts, 1(2), 99-116. https://doi.org/10.3390/hearts1020011