
Chemical Stability of PFSA Membranes in Heavy-Duty Fuel Cells: Fluoride Emission Rate Model

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Theoretical
3. Results
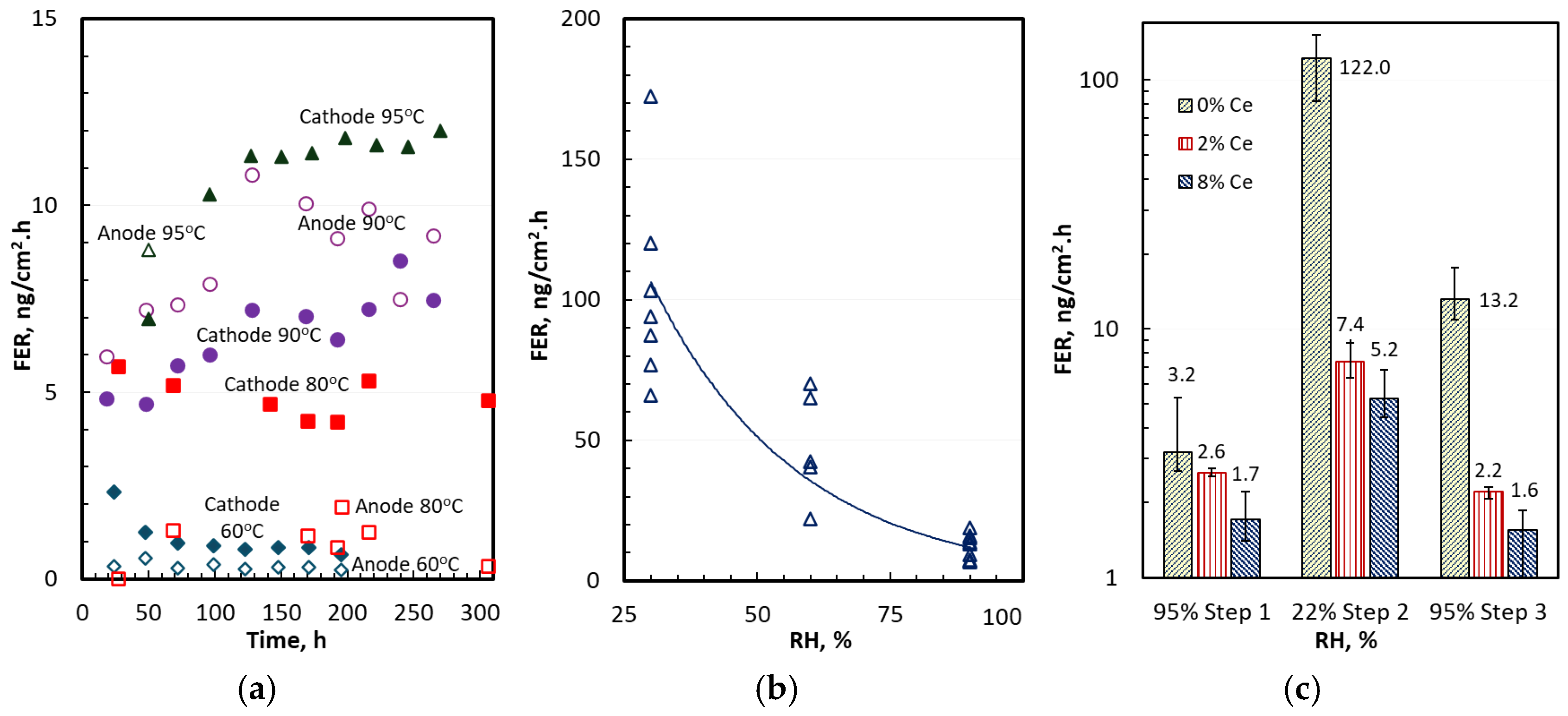
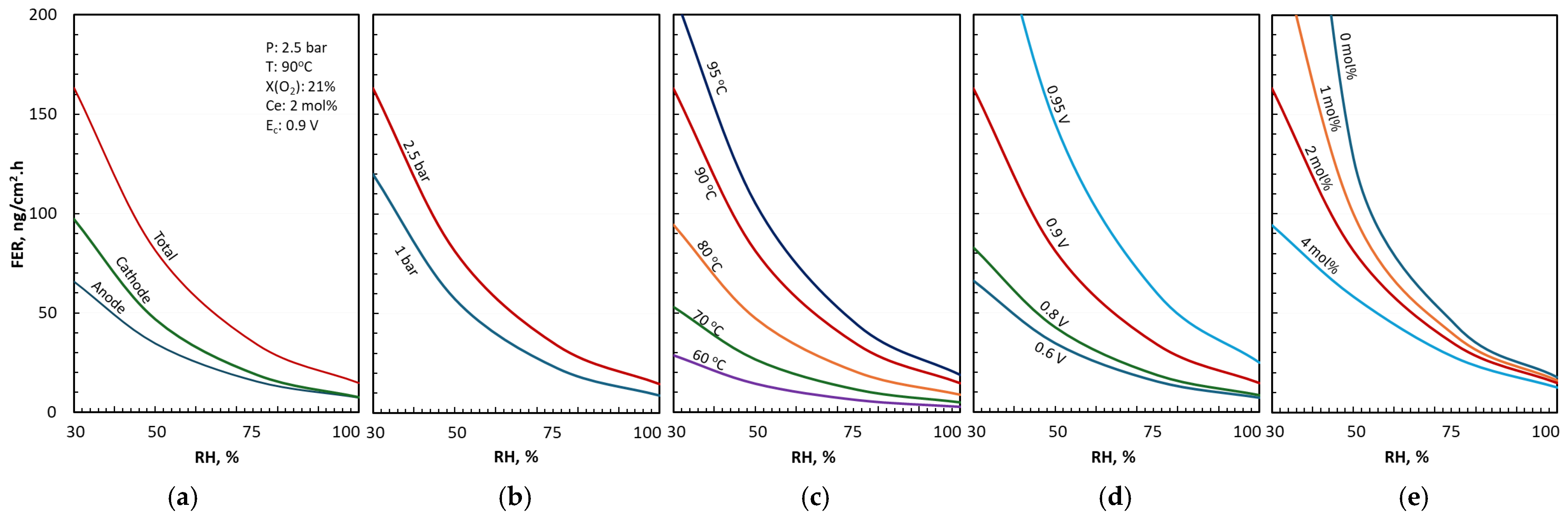
3.1. Anode and Cathode FERs
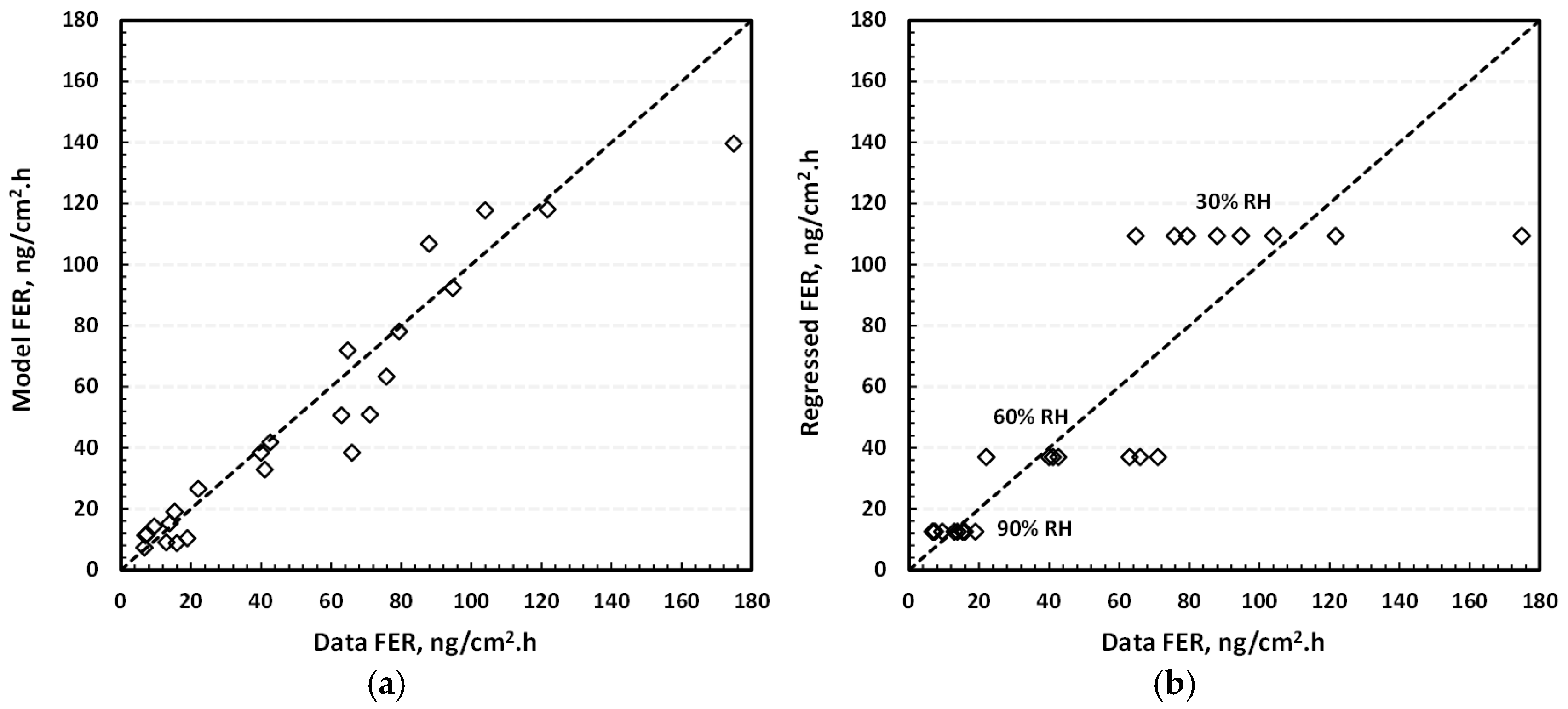
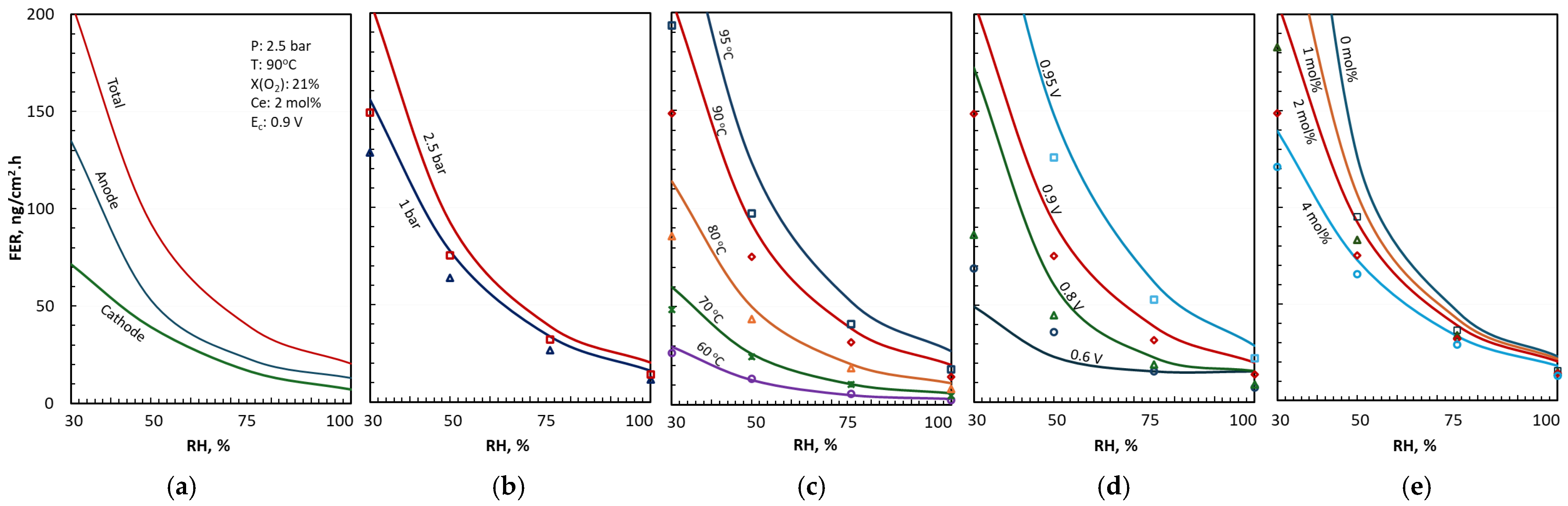
3.2. FER Correlation
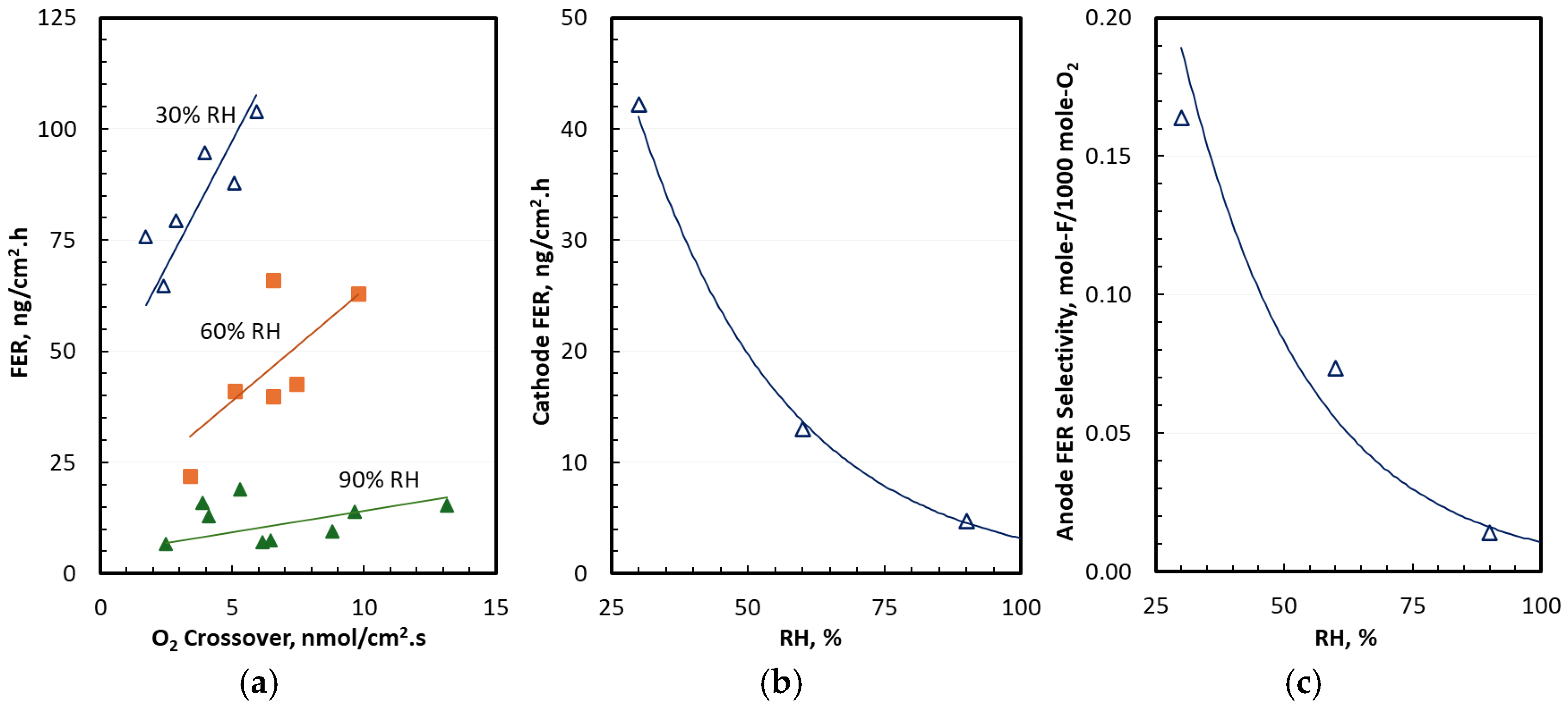
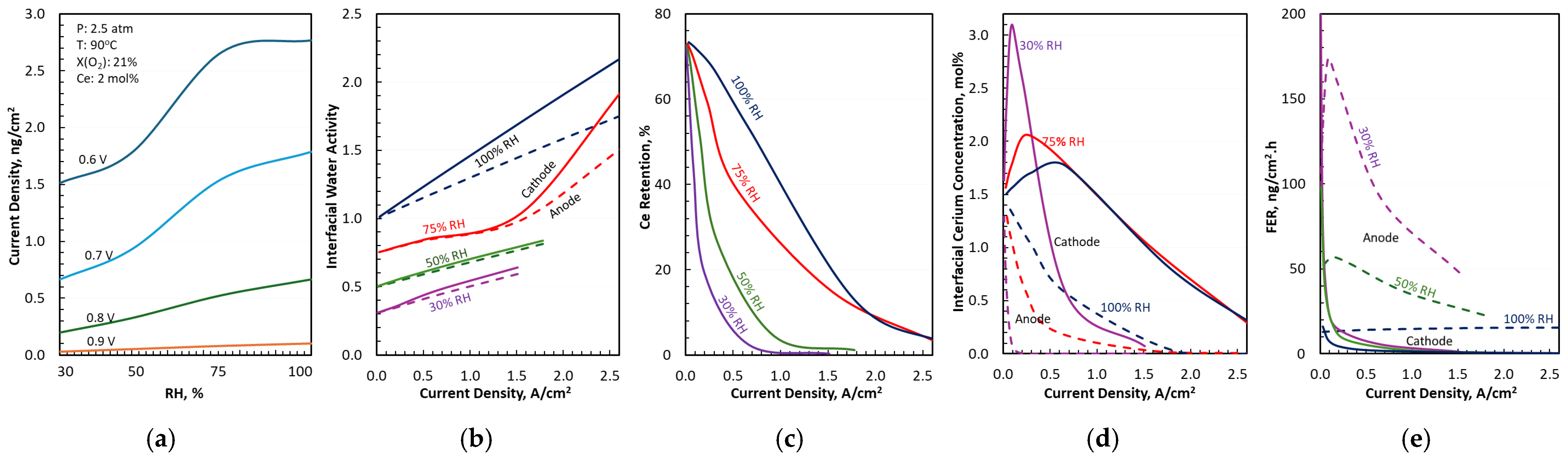
3.3. Water and Cerium Transport in Differential Cells
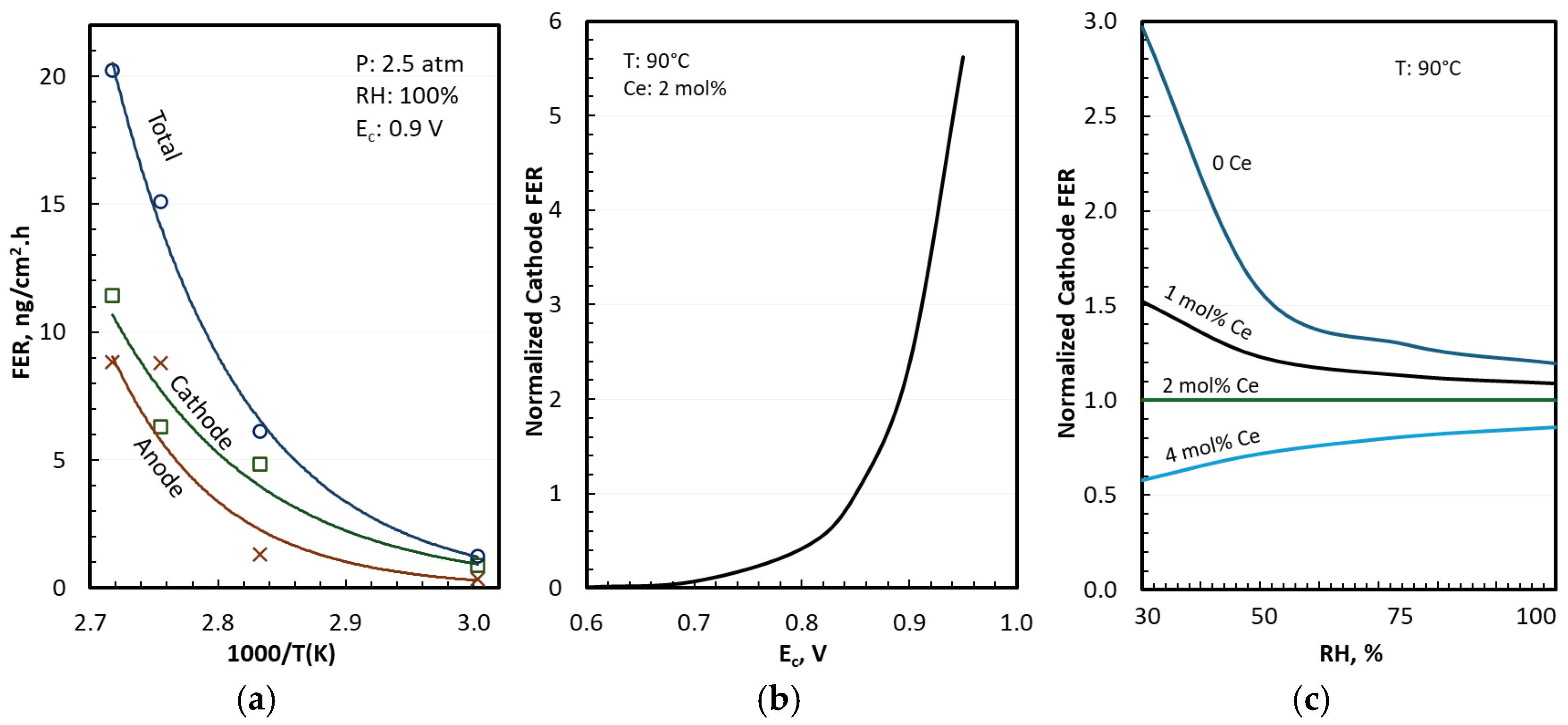
3.4. FER in Differential Cells
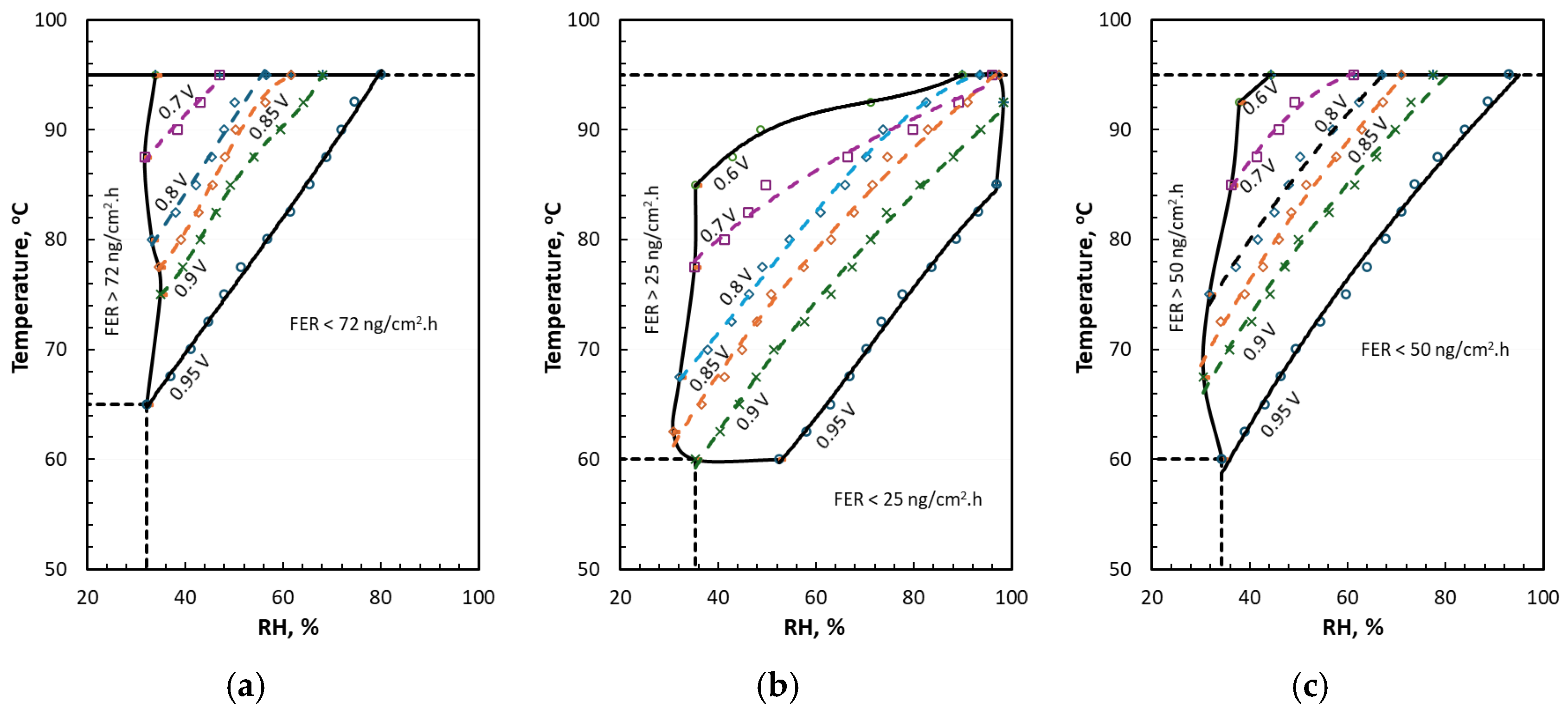
4. Discussion
5. Conclusions
- (a)
- Cerium mobility, water transport, and FER under dynamically varying loads representative of duty cycles for different applications
- (b)
- Effect of Fe impurities on FER from Ce-doped membranes
- (c)
- Experimental measurements of anode and cathode FERs in operating cells at 0.6–0.85 V and 60–80 °C
- (d)
- Relationship between cumulative fluoride emission and membrane failure mode
- (e)
- Development of accelerated stress tests that can simulate membrane failure in a reasonable time without altering the degradation mechanism
- (f)
- Synergism between mechanical stresses and chemical degradation
- (g)
- Enhanced membrane stability by immobilization of cerium
- (h)
- Ionomer stability and its effect on ORR catalyst activity
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| activity | |
| diffusivity | |
| Faraday constant or fluorine | |
| current density (A/cm2) | |
| molecular or equivalent weight | |
| crossover flux | |
| fluoride release rate | |
| selectivity | |
| temperature | |
| through-plane | |
| ε | void volume fraction |
| η | overpotential |
| ω | for θ |
| σ | conductivity |
| current density (A/cm3) | |
| pressure | |
| gas constant | |
| saturation | |
| specific Pt area ( | |
| mobility | |
| δ | thickness |
| ionomer volume fraction | |
| θ | oxygen coverage |
| ρ | density |
| ψ | permeability |
| AST | Accelerated stress test |
| CFL | Cumulative fluoride release |
| DL | Design limit |
| EW | Equivalent weight |
| FER | Fluoride emission rate |
| FTIR | Fourier transform infrared spectroscopy |
| GrV | Graphitized Vulcan |
| HOR | Hydrogen oxidation reaction |
| I/C | Ionomer to carbon ratio |
| MEA | Membrane electrode assembly |
| OCV | Open circuit voltage |
| ORR | Oxygen reduction reaction |
| PEMFC | Polymer electrolyte membrane fuel cell |
| PFSA | Perfluorosulfonic acid |
| PTFE | Polytetrafluoroethylene (Teflon) |
| RRDE | Rotating ring disk electrode |
| RH | Relative humidity |
| SL | Safe limit |
| TFL | Theoretical failure limit |
| Subscripts and Superscripts | |
| anode | |
| effective | |
| ionomer or membrane | |
| reference | |
| water | |
| cathode | |
| ionic | |
| Nernst | |
| kinetic | |
| crossover | |
References
- Satyapal, S. Hydrogen and Fuel Cells Overview: Opportunities for Ports & Maritime Applications. In Proceedings of the H2@Ports Workshop, San Francisco, CA, USA, 10 September 2019. [Google Scholar]
- Ahluwalia, R.K.; Wang, X.; Star, A.G.; Papadias, D.D. Performance and Cost of Fuel Cells for Off-Road Heavy-Duty Vehicles. Int. J. Hydrogen Energy 2022, 47, 10990–11006. [Google Scholar] [CrossRef]
- Toyota, Kenworth Prove Fuel Cell Electric Truck Capabilities with Successful Completion of Truck Operations for ZANZEFF Project. Available online: https://pressroom.toyota.com/toyota-kenworth-prove-fuel-cell-electric-truck-capabilities-with-successful-completion-of-truck-operations-for-zanzeff-project/ (accessed on 12 June 2025).
- Ahluwalia, R.K.; Wang, X.; Papadias, D.D.; Star, A.G. Performance and Total Cost of Ownership of a Fuel Cell Hybrid Mining Truck. Energies 2023, 16, 286. [Google Scholar] [CrossRef]
- Anglo American Unveils a Prototype of the World’s Largest Hydrogen-Powered Mine Haul Truck—A Vital Step towards Reducing Carbon Emissions over Time 2022. Available online: https://www.angloamerican.com/media/press-releases/2022/06-05-2022 (accessed on 12 June 2025).
- Ahluwalia, R.K.; Papadias, D.; Peng, J.-K.; Krause, T.; Chan, S.; Devlin, P. Total Cost of Ownership for Line Haul, Yard Switchers and Regional Passenger Locomotives–Preliminary Results. In Proceedings of the H2@Ports Workshop, Lansing, MI, USA, 26–27 March 2019. [Google Scholar]
- Marcinkoski, J.; Vijayagopal, R.; Adams, J.; James, B.; Kopasz, J.; Ahluwalia, R.K. Hydrogen Class 8 Long Haul Truck Targets. DOE Advanced Truck Technologies. December 2019. Available online: https://www.hydrogen.energy.gov/pdfs/19006_hydrogen_class8_long_haul_truck_targets.pdf (accessed on 11 June 2025).
- Padgett, E.; Kleen, G.; Mukundan, R.; Wang, X.; Ahluwalia, R.; Papageorgopoulos, D. Development of Targets for Heavy Duty Fuel Cell Vehicles with Application-Driven System Modelling. In Proceedings of the ECS/PRIME 2020, Honolulu, HI, USA, 4–9 October 2020. [Google Scholar]
- Wang, X.; Hu, L.; Neyerlin, K.C.; Ahluwalia, R. Baselining Activity and Stability of ORR Catalysts and Electrodes for Proton Exchange Membrane Fuel Cells for Class-8 Heavy-Duty Applications. J. Electrochem. Soc. 2023, 170, 024503–024512. [Google Scholar] [CrossRef]
- Ahluwalia, R.K.; Wang, X. Performance and Durability of Hybrid Fuel Cell Systems for Class-8 Long Haul Trucks for Heavy-Duty Applications. J. Electrochem. Soc. 2024, 171, 034507–034521. [Google Scholar] [CrossRef]
- Ahluwalia, R.K.; Wang, X.; Chen, K.; Wang, X.; Spendelow, J.S. Performance and Durability of Heavy-Duty Fuel Cell Systems with an Advanced Ordered Intermetallic ORR Alloy Catalyst and Novel Support. J. Electrochem. Soc. 2024, 171, 114512–114523. [Google Scholar] [CrossRef]
- Babic, U.; Suermann, M.; Büchi, F.N.; Gubler, L.; Schmidt, T.J. Critical Review—Identifying Critical Gaps for Polymer Electrolyte Water Electrolysis Development. J. Electrochem. Soc. 2017, 164, F387. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Mathias, M.F. Fundamental Research and Development Challenges in Polymer Electrolyte Fuel Cell Technology. ECS Proc. Vol. 2005, 2002, 1. [Google Scholar] [CrossRef]
- LaConti, A.B.; Hamdan, M.; McDonald, R.C. Mechanisms of Membrane Degradation. In Handbook of Fuel Cells; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Borup, R.; Meyers, J.; Pivovar, B.; Kim, Y.S.; Mukundan, R.; Garland, N.; Myers, D.; Wilson, M.; Garzon, F.; Wood, D.; et al. Scientific Aspects of Polymer Electrolyte Fuel Cell Durability and Degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef]
- Dow Chemical Company. Nafion® Membranes: An Introduction and Overview; Dow Chemical Company: Midland, MI, USA, 1996. [Google Scholar]
- Kolde, J.A. Advanced Composite Polymer Electrolyte Fuel Cell Membranes. ECS Proc. Vol. 1995, 23, 193–201. [Google Scholar] [CrossRef]
- Penner, R.M.; Martin, C.R. Ion Transporting Composite Membranes: I. Nafion-Impregnated Gore-Tex. J. Electrochem. Soc. 1985, 132, 514–515. [Google Scholar] [CrossRef]
- Shah, A.A.; Ralph, T.R.; Walsh, F.C. Modeling and Simulation of the Degradation of Perfluorinated Ion-Exchange Membranes in PEM Fuel Cells. J. Electrochem. Soc. 2009, 156, B465. [Google Scholar] [CrossRef]
- Ehlinger, V.M.; Crothers, A.R.; Kusoglu, A.; Weber, A.Z. Modeling Proton-Exchange-Membrane Fuel Cell Performance/Degradation tradeoffs with Chemical Scavengers. J. Phys. Energy 2020, 2, 044006. [Google Scholar] [CrossRef]
- Chandesris, M.; Vincent, R.; Guetaz, L.; Roch, J.S.; Thoby, D.; Quinaud, M. Membrane Degradation in PEM Fuel Cells: From Experimental Results to Semi-Empirical Degradation Laws. Int. J. Hydrogen Energy 2017, 42, 8139–8149. [Google Scholar] [CrossRef]
- Spears, A.J.; Rockward, T.; Mukundan, R.; Garzon, F. Investigation of Membrane Chemical Degradation as a Function of Catalyst Platinum Loading. J. Electrochem. Soc. 2021, 168, 064503. [Google Scholar] [CrossRef]
- Alavijeh, S.A.; Goulet, M.A.; Khorasany, R.M.H.; Ghataurah, J.; Lim, C.; Lauritzen, M.; Kjeang, E.; Wang, G.G.; Rajapakse, R.K.N.D. Decay in Mechanical Properties of Catalyst Coated Membranes Subjected to Combined Chemical and Mechanical Membrane Degradation. Fuel Cells 2015, 15, 204–213. [Google Scholar] [CrossRef]
- Khorasany, R.M.H.; Goulet, M.A.; Alavijeh, A.S.; Kjeang, E.; Wang, G.G.; Rajapakse, R.K.N.D. On the Constitutive Relations for Catalyst Coated Membrane Applied to In-Situ Fuel Cell Modeling. J. Power Sources 2014, 252, 138–143. [Google Scholar] [CrossRef]
- Venkatesan, S.V.; El Hannach, M.; Holdcroft, S.; Kjeang, E. Probing Nanoscale Membrane Degradation in Fuel Cells through Electron Tomography. J. Memb. Sci. 2017, 539, 138–143. [Google Scholar] [CrossRef]
- Venkatesan, S.V.; Lim, C.; Holdcroft, S.; Kjeang, E. Progression in the Morphology of Fuel Cell Membranes upon Conjoint Chemical and Mechanical Degradation. J. Electrochem. Soc. 2016, 163, F637. [Google Scholar] [CrossRef]
- Singh, Y.; Orfino, F.P.; Dutta, M.; Kjeang, E. 3D Failure Analysis of Pure Mechanical and Pure Chemical Degradation in Fuel Cell Membranes. J. Electrochem. Soc. 2017, 164, F1331. [Google Scholar] [CrossRef]
- Madhav, D.; Wang, J.; Keloth, R.; Mus, J.; Buysschaert, F.; Vandeginste, V. A Review of Proton Exchange Membrane Degradation Pathways, Mechanisms, and Mitigation Strategies in a Fuel Cell. Energies 2024, 17, 998. [Google Scholar] [CrossRef]
- Chen, C.; Fuller, T.F. The Effect of Humidity on the Degradation of Nafion® Membrane. Polym. Degrad. Stab. 2009, 94, 1436–1447. [Google Scholar] [CrossRef]
- Coms, F.D. The Chemistry of Fuel Cell Membrane Chemical Degradation. ECS Trans. 2008, 16, 235–255. [Google Scholar] [CrossRef]
- Healy, J.; Hayden, C.; Xie, T.; Olson, K.; Waldo, R.; Brundage, M.; Gasteiger, H.; Abbott, J. Aspects of the Chemical Degradation of PFSA Ionomers Used in PEM Fuel Cells. Fuel Cells 2005, 5, 302–308. [Google Scholar] [CrossRef]
- Rodgers, M.P.; Bonville, L.J.; Kunz, H.R.; Slattery, D.K.; Fenton, J.M. Fuel Cell Perfluorinated Sulfonic Acid Membrane Degradation Correlating Accelerated Stress Testing and Lifetime. Chem. Rev. 2012, 112, 6075–6103. [Google Scholar] [CrossRef] [PubMed]
- Chandesris, M.; Médeau, V.; Guillet, N.; Chelghoum, S.; Thoby, D.; Fouda-Onana, F. Membrane Degradation in PEM Water Electrolyzer: Numerical Modeling and Experimental Evidence of the Influence of Temperature and Current Density. Int. J. Hydrogen Energy 2015, 40, 1353–1366. [Google Scholar] [CrossRef]
- Xie, T.; Hayden, C.A. A Kinetic Model for the Chemical Degradation of Perfluorinated Sulfonic Acid Ionomers: Weak End Groups versus Side Chain Cleavage. Polymer 2007, 48, 5497–5506. [Google Scholar] [CrossRef]
- Frühwirt, P.; Kregar, A.; Törring, J.T.; Katrašnik, T.; Gescheidt, G. Holistic Approach to Chemical Degradation of Nafion Membranes in Fuel Cells: Modelling and Predictions. Phys. Chem. Chem. Phys. 2020, 22, 5647–5666. [Google Scholar] [CrossRef]
- Wong, K.H.; Kjeang, E. Macroscopic In-Situ Modeling of Chemical Membrane Degradation in Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 2014, 161, F823–F832. [Google Scholar] [CrossRef]
- Trogadas, P.; Parrondo, J.; Ramani, V. Degradation Mitigation in Polymer Electrolyte Membranes Using Cerium Oxide as a Regenerative Free-Radical Scavenger. Electrochem. Solid-State Lett. 2008, 11, B113. [Google Scholar] [CrossRef]
- Pearman, B.P.; Mohajeri, N.; Brooker, R.P.; Rodgers, M.P.; Slattery, D.K.; Hampton, M.D.; Cullen, D.A.; Seal, S. The Degradation Mitigation Effect of Cerium Oxide in Polymer Electrolyte Membranes in Extended Fuel Cell Durability Tests. J. Power Sources 2013, 225, 75–83. [Google Scholar] [CrossRef]
- Gubler, L.; Koppenol, W.H. Kinetic Simulation of the Chemical Stabilization Mechanism in Fuel Cell Membranes Using Cerium and Manganese Redox Couples. J. Electrochem. Soc. 2011, 159, B211. [Google Scholar] [CrossRef]
- Xiao, S.; Zhang, H.; Bi, C.; Zhang, Y.; Zhang, Y.; Dai, H.; Mai, Z.; Li, X. Degradation Location Study of Proton Exchange Membrane at Open Circuit Operation. J. Power Sources 2010, 195, 5305–5311. [Google Scholar] [CrossRef]
- Wang, L.; Advani, S.G.; Prasad, A.K. Self-Hydrating Pt/CeO2-Nafion Composite Membrane for Improved Durability and Performance. ECS Electrochem. Lett. 2014, 3, F30. [Google Scholar] [CrossRef]
- Lim, C.; Alavijeh, A.S.; Lauritzen, M.; Kolodziej, J.; Knights, S.; Kjeanga, E. Fuel Cell Durability Enhancement with Cerium Oxide under Combined Chemical and Mechanical Membrane Degradation. ECS Electrochem. Lett. 2015, 4, F29. [Google Scholar] [CrossRef]
- Coms, F.D.; McQuarters, A.B.; Marks, A. PFSA Membrane Thickness Impact on Chemical Degradation Rates. J. Electrochem. Soc. 2025, 172, 054501. [Google Scholar] [CrossRef]
- Kumaraguru, S. Durable High Power Membrane Electrode Assembly with Low Pt Loading. Energy. Hydrog. Fuel Cells Progr. Annu. Merit Rev. 2018, 1, 1–9. [Google Scholar]
- M2FCT, Heavy-Duty Fuel Cell Catalyst AST Protocol. Available online: https://millionmilefuelcelltruck.org/ast-protocols (accessed on 9 April 2025).
- Ahluwalia, R.K.; Wang, X. Rapid Self-Start of Polymer Electrolyte Fuel Cell Stacks from Subfreezing Temperatures. J. Power Sources 2006, 162, 502–512. [Google Scholar] [CrossRef]
- Baker, A.M.; Babu, S.K.; Mukundan, R.; Advani, S.G.; Prasad, A.K.; Spernjak, D.; Borup, R.L. Cerium Ion Mobility and Diffusivity Rates in Perfluorosulfonic Acid Membranes Measured via Hydrogen Pump Operation. J. Electrochem. Soc. 2017, 164, F1272–F1278. [Google Scholar] [CrossRef]
- COMSOL AB. COMSOL Multiphysics 6.0; COMSOL AB: Stockholm, Sweden; Available online: www.comsol.com (accessed on 13 November 2024).
- Sethuraman, V.A.; Weidner, J.W.; Haug, A.T.; Motupally, S.; Protsailo, L.V. Hydrogen Peroxide Formation Rates in a PEMFC Anode and Cathode. J. Electrochem. Soc. 2008, 155, B50. [Google Scholar] [CrossRef]
- Ahluwalia, R.K.; Wang, X.; Peng, J.-K.; Konduru, V.; Arisetty, S.; Ramaswamy, N.; Kumaraguru, S. Achieving 5000-h and 8000-h Low-PGM Electrode Durability on Automotive Drive Cycles. J. Electrochem. Soc. 2021, 168, 044518. [Google Scholar] [CrossRef]
- Kundu, S.; Fowler, M.W.; Simon, L.C.; Grot, S. Morphological Features (Defects) in Fuel Cell Membrane Electrode Assemblies. J. Power Sources 2006, 157, 650–656. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Symbol | Symbol | |||
|---|---|---|---|---|---|
| 248 | 1.25 | 30.2 kJ/mol | |||
| 22.6 mV | |||||
| 3.1 | 0.65 | 28.1 kJ/mol | |||
| 60.8 mV | 2.9 kJ/mol | ||||
| ε | 0.4 | 0.26 | β | 0.9 | |
| 51.6 kJ/mol | 46.5 kJ/mol | ||||
| 123.3 ng/cm2·h | 3.7 | 71 kJ/mol | |||
| 0.54 | 0.85 V | 363 K | |||
| 4.1 | 78 kJ/mol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, L.R.; Wang, X.; Quesada, C.; Wang, X.; Mukundan, R.; Ahluwalia, R. Chemical Stability of PFSA Membranes in Heavy-Duty Fuel Cells: Fluoride Emission Rate Model. Electrochem 2025, 6, 25. https://doi.org/10.3390/electrochem6030025
Johnson LR, Wang X, Quesada C, Wang X, Mukundan R, Ahluwalia R. Chemical Stability of PFSA Membranes in Heavy-Duty Fuel Cells: Fluoride Emission Rate Model. Electrochem. 2025; 6(3):25. https://doi.org/10.3390/electrochem6030025
Chicago/Turabian StyleJohnson, Luke R., Xiaohua Wang, Calita Quesada, Xiaojing Wang, Rangachary Mukundan, and Rajesh Ahluwalia. 2025. "Chemical Stability of PFSA Membranes in Heavy-Duty Fuel Cells: Fluoride Emission Rate Model" Electrochem 6, no. 3: 25. https://doi.org/10.3390/electrochem6030025
APA StyleJohnson, L. R., Wang, X., Quesada, C., Wang, X., Mukundan, R., & Ahluwalia, R. (2025). Chemical Stability of PFSA Membranes in Heavy-Duty Fuel Cells: Fluoride Emission Rate Model. Electrochem, 6(3), 25. https://doi.org/10.3390/electrochem6030025