1. Introduction
Ovarian cancer is the most lethal gynecological cancer, with a 5 year survival rate of 50%. In 2020, ovarian cancer was one of the five leading causes of death from all cancers in females aged 40–79 years [
1]. Ovarian cancer has been named a “silent killer” due to the lack of effective diagnostic methods available at the early stages when the disease is treatable. If diagnosed in the first stage (localized), the 5 year survival rate can be as high as 92.4%, while in the regional and distant stages, it drops to 72.9 and 31.5%, respectively [
1,
2]. Cancer antigen 125 (CA125) is the only clinically approved biomarker for ovarian cancer diagnosis; however, it is neither specific nor effective enough for early screening. In its early stages, CA125 was found to be elevated in only 50% of patients with a confirmed diagnosis. Moreover, false-positive results were observed in cases such as pregnancy [
2].
Lysophosphatidic acid (LPA) is a promising biomarker for ovarian cancer. LPA is a phospholipid involved in a number of cellular processes including cellular proliferation, prevention of apoptosis, cell migration, and more [
3,
4]. In ovarian cancer cases, malignant ovarian epithelium produces LPA to stimulate growth of cancer cells [
5,
6]. There has been an increasing interest in LPA as a prospective ovarian cancer biomarker. Not only is it significantly elevated in the first stage of the disease, but it is also more specific and selective than CA125 [
6,
7,
8]. Studies have shown LPA elevation in 90% of patients at stage I of ovarian cancer, as well as the dependency of its concentration on disease progression [
3,
8]. Notably, several studies found that LPA was not elevated in the cases of breast cancer and leukemia, suggesting its specificity in regard to ovarian cancer [
5,
6]. While LPA levels do not necessarily differ significantly between patients with benign and malignant tumors, with a few contradicting studies, it holds great potential as an effective biomarker for detecting the presence of ovarian cancer tumors and predicting the disease stage [
3,
7]. However, the lack of cost-effective and reliable standard techniques for LPA detection has prevented mass clinical trials and clinical approval of LPA as an ovarian cancer biomarker. Traditional analytical techniques like gas chromatography–mass spectrometry have been employed to quantify LPA levels in serum. However, these methods have not been widely used for large-scale screening due to their time-consuming nature, high cost, and the need for specialized technicians [
9,
10,
11].
Electrochemical biosensors have emerged as powerful tools for detecting biological molecules, offering advantages in terms of selectivity, sensitivity, and specificity [
9,
12,
13,
14,
15,
16,
17,
18]. Their affordability and ease of use make them appealing in the context of point-of-care testing (POCT) devices [
19]. The main challenge in biosensor technology is the issue of non-specific adsorption (NSA) when interacting with biological fluids [
20,
21]. This issue becomes especially pronounced on gold surfaces. Thiol chemistry, which is used to create a self-assembled monolayer (SAM) for bio-functionalization, can also contribute to NSA, as any biomolecules containing thiol groups in biofluids may bind to the gold electrode, causing false results [
22]. To address the issue of NSA, we incorporated a newly synthesized linker, 3-dithiothreitol propanoic acid (DTT
COOH), which our research team recently introduced, demonstrating its promising antifouling properties using quartz crystal microbalance (QCM) [
22]. The formation of the SAM of this linker establishes a “water barrier”, as the dithiol structure enhances the hydration network within the SAM [
23]. In this study, we examined the antifouling properties of DTT
COOH in developing electrochemical biosensors. We employed the gelsolin–actin system, which was introduced by our group, as the biorecognition element for detecting LPA [
24]. This system was used to develop a method based on fluorescence spectroscopy capable of detecting LPA with a limit of detection (LOD) of 5 μM [
24,
25]. We also used this system to develop an affinity-based electrochemical biosensor for LPA detection, achieving an LOD of 0.7 μM [
26]. The biosensor fabrication involved the use of medical-grade stainless steel as the working electrode, an unconventional yet effective material choice, coupled with silane-based interfacial chemistry [
26].
In this study, we introduced a cost-effective strategy for immobilizing the gelsolin–actin system through thiol-based interfacial chemistry using screen-printed electrodes (SPE). The synthesis of the thiol-based linkers is simpler, and does not require specific precautions compared to silane-based linkers. Silane-based linkers are sensitive to oxygen and water; therefore, they require a glovebox to synthesize them. Using thiol-based chemistry has the advantages of providing a more robust synthesis method, thereby reducing the cost and time for developing the biosensor.
2. Materials and Methods
2.1. Materials and Reagents
All materials were purchased from Sigma-Aldrich, unless otherwise specified. Gelsolin plasmids were provided by Professor Robert Robinson of the University of Singapore, and the first three domains of gelsolin (gelsolin) were synthesized from plasmids, as reported by Franier, D.L. et al. [
24]. Actin from rabbit muscle was purchased from Alfa Aesar by Thermo Fisher Scientific, United States. All the aqueous solutions were prepared using Milli-Q water.
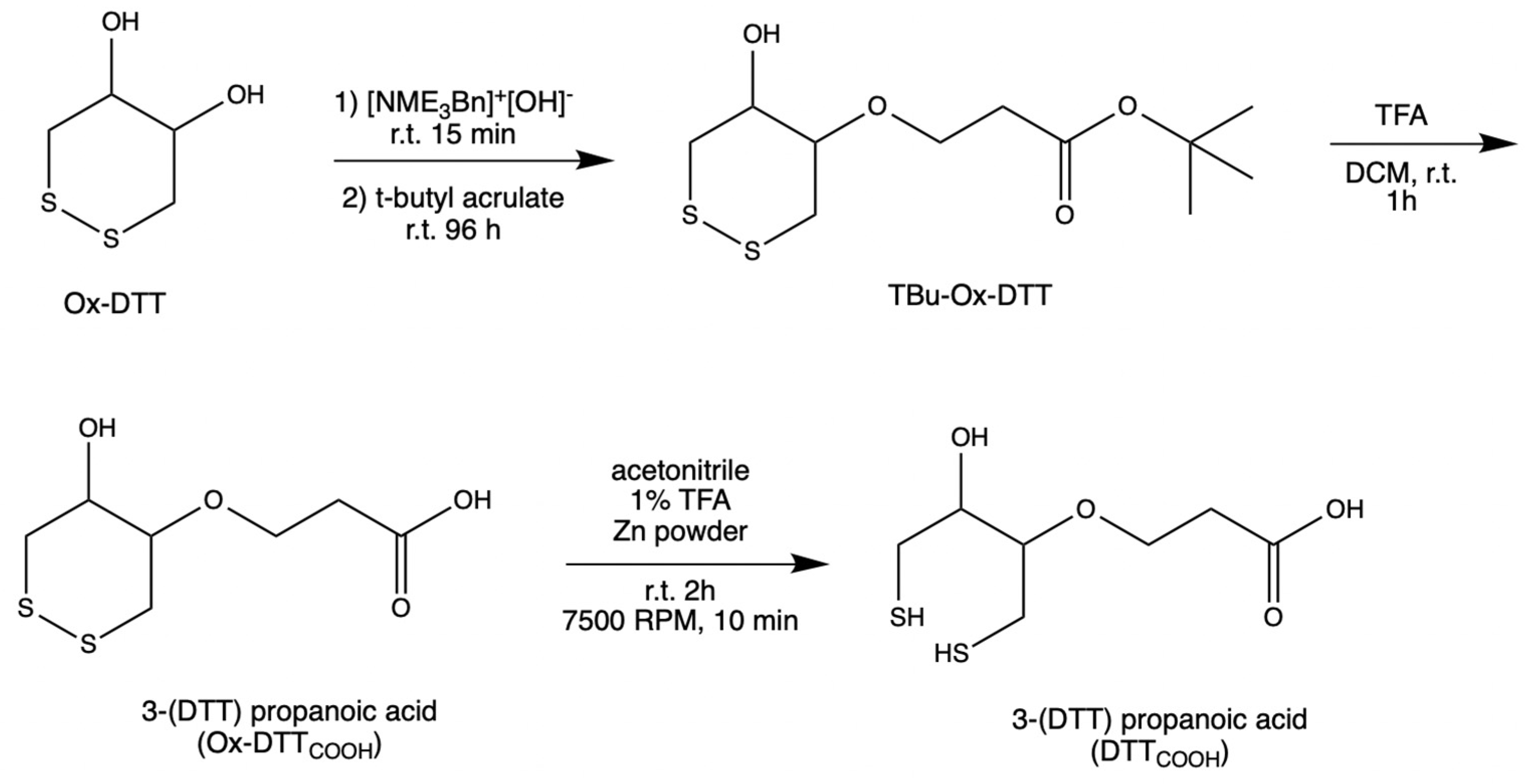
2.2. Synthesis of the Linker
DTT
COOH was synthesized (
Figure 1) by optimizing the procedure that was reported by Spagnolo et al. [
22]. To provide a mild chemical reaction instead of applying a temperature of 58–60 °C, the reaction was performed at room temperature for 96 h.
An oven-dried flask was used to concentrate N-benzyltrimethylammonium hydroxide (40% w/w in methanol, 600 μL, 3 mmol) under vacuum. After cooling to room temperature, a solution of ox–DTT (200 mg, 1.3 mmol) in acetonitrile (60 mL) was added to the flask. The reaction mixture was stirred at room temperature for 15–30 min. Then, tert-butyl acrylate (287 μL, 1.9 mmol) was added to the solution and stirred for 96 h at room temperature under an inert atmosphere, and then was monitored with TLC (3:1 and 1:1 v/v hexane/ethyl acetate). TLC plates were run with various ratios of hexane and ethyl acetate to choose the right conditions for subsequent flash chromatography analysis. It was determined that the 3:1 v/v hexane/ethyl acetate was the optimal condition. Flash chromatography was performed using high-grade silica gel (60 Å pore size, 200–400 mesh) and yielding 63.5% tert-butyl 3(DTT)propanoate (TBu–Ox–DTT) (127 mg, 453 μM). TBu–Ox–DTT (127 mg, 453 μmol) was then mixed with trifluoroacetic acid (316.4 μL) and dissolved dichloromethane (9 mL). The solution was then stirred for 60–90 min at room temperature. The product was obtained by evaporating trifluoroacetic acid under reduced pressure yielding 51% 3–(DTT) propanoic acid (Ox–DTTCOOH) (65 mg, 289.8 μmol).
Ox–DTT
COOH (32.5 mg, 144.9 μmol) was dissolved in a solution of 1% trifluoroacetic acid in acetonitrile (12 mL) and mixed with zinc powder. The solution was stirred at room temperature for 2 h, then centrifuged at 7500 RPM for 10 min. The supernatant was concentrated under reduced pressure, yielding 81.8% 3–(DTT) propanoic acid (DTT
COOH) (26.6 mg, 111.75 μmol).
13C NMR (
Figure S1) and
1H NMR spectra were obtained to confirm the structure of the final product.
2.3. Electrochemical Measurements
All electrochemical experiments were conducted using a CHI440A electrochemical workstation (CH Instruments, Austin, TX, USA) and disposable screen-printed gold electrodes (SPEs). The SPEs (220 AT), which employed gold nanotube inks and underwent high-temperature curing, were purchased from DropSense Metrohm, Llanera (Asturias), Spain. The electrode consisted of a gold working electrode (4 mm diameter), a gold auxiliary electrode, and a silver reference electrode. The electrochemical measurements were performed by immersing the electrodes in 10 mL aqueous solutions of 10 mM K4[Fe(CN)6]/K3[Fe(CN)6] as redox probes containing 0.5 M KCl as a supporting electrolyte. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were employed to monitor the current response as a function of applied potential. CV measurements were started at the open circuit potential (OCP) with positive initial scan polarity and a 5 s quiet time. A scan rate of 0.3 V/s was applied, unless otherwise mentioned. The DPV measurements were performed with the increment potential of 0.004 V, amplitude of 0.05 V, pulse width of 0.05 s, and pulse period 0.5 s. DPV measurements started with negative or positive initial scan polarity and 2 s quiet time with sensitivity of 5 × 10−4 A/V.
2.4. Surface Modification of Screen-Printed Electrodes
SPE modification was conducted by directly applying 10 μL of various reagents onto the working electrode surface through an incubation process. Two different linkers, 11-mercaptoundecanoic acid (MUA) and 3-dithiothreitol propanoic acid (DTTCOOH), were used to immobilize the gelsolin–actin system on the SPE. The SPEs were modified by either a 2 mM MUA aqueous solution containing 30% ethanol or a 3 mM DTTCOOH aqueous solution containing 13% EtOH and 7 mM KOH. The SPEs were then allowed to incubate overnight to form a SAM composed either of MUA or DTTCOOH, which functioned as linkers. Then, electrodes were modified with an aqueous solution of N-hydroxysuccinimide (NHS) and 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (NHS/EDC: 20 mM NHS and 50 mM EDC) for 35 min. Following this, electrodes were incubated in Nα,Nα-bis (carboxymethyl)-L-lysine and NiCl2 (Ni-NTA) (2 mg/mL) in phosphate-buffered saline buffer (PBS pH 7.4) overnight. Then, the modified SPEs were introduced to the gelsolin–actin complex (0.01 mg/mL in PBS pH 7.4) for 1 h. Prior to this, the gelsolin–actin complex was prepared by preincubating actin and gelsolin in a fridge for 1 hr. We use a polyhistidine-tagged gelsolin that effectively binds to Ni-NTA via the imidazole rings of histidine residues. This binding occurs due to the additional free coordination sites on the Ni2+ in Ni-NTA, which are occupied by the imidazole ring of histidine acting as a ligand.
After this, the modified SPEs were exposed to LPA solution with concentration between 0.01 and 10 μM LPA in either PBS or goat serum. To remove all the unbounded and loosely bound molecules from the surface after each step of the modification, SPEs were first rinsed thoroughly with the compatible solvent. Then, they were rinsed with copious amounts of Milli-Q water to remove any residual solvent. Finally, they were dried under a nitrogen stream to ensure that they were completely dry before being used for further modification or analysis.
2.5. Surface Characterization
Each step of SPE modification was monitored by performing CV and DPV using 10 mM K4[Fe(CN)6]/K3[Fe(CN)6] as an external redox probe containing 0.5 M KCl as a supporting electrolyte. Successfully modified electrodes were then used to measure LPA in either buffer or goat serum. In addition to electrochemical measurements, contact angle goniometry was used for surface characterization. The contact angle goniometry experiments were conducted to assess the hydrophobicity of the electrode surface following each modification step. A KSV CAM 101 contact angle goniometer (KSV Instruments Ltd., Helsinki, Finland) was employed, with deionized water as the testing liquid in standard atmospheric conditions. To prevent water evaporation, the angle between the droplet and the electrode was measured immediately.
3. Results and Discussion
To fabricate the electrochemical biosensor, we used DTT
COOH, which was recently introduced by our group and showed promising antifouling properties [
23]. This novel thiol linker is a derivative of DTT, with a modification that replaces one hydroxyl group with propanoic acid. This alteration enables the immobilization of probes through NHS/EDC reactions. The formation of SAM facilitates the creation of a “water barrier”, as the dithiol structure enhances the hydration network by providing distance between the propanoic acid chains within the SAM. As a result, the surface area available for interfacial water molecules increases. The antifouling properties of DTT
COOH against untreated human serum were validated using the quartz crystal microbalance (QCM) [
22]. In this study, we compared the effectiveness of this new linker with the MUA, a linear thiol molecule, for developing a label-free electrochemical biosensor using gold SPEs. We showed that this linker provides a comparable antifouling property compared to the silane-based linker that we recently used to develop an affinity-based electrochemical biosensor for LPA detection [
26]. The simplicity of synthesis and reduced requirements for thiol-based linkers compared to silane-based linkers led to improvements in cost and time for biosensor development. By taking an advantage of the affinity-based gelsolin–actin system, we engineered a label-free electrochemical biosensor for precise LPA detection [
23,
25]. Unlike the fluorescence methods that were reported before [
24], this biosensor exhibits higher sensitivity and does not require complex instrumentation. SPEs offer a unique set of features that make them suitable for biosensor applications, including their affordability, user-friendliness, and minimal sample volume requirements. Moreover, using SPE offers a miniaturized solution, suitable for the fabrication of compact POCT devices [
9,
19,
26]. These features position SPE as an ideal candidate for mass production and commercialization [
12,
13,
14,
15,
27].
3.1. Characterization of Biorecognition Surface
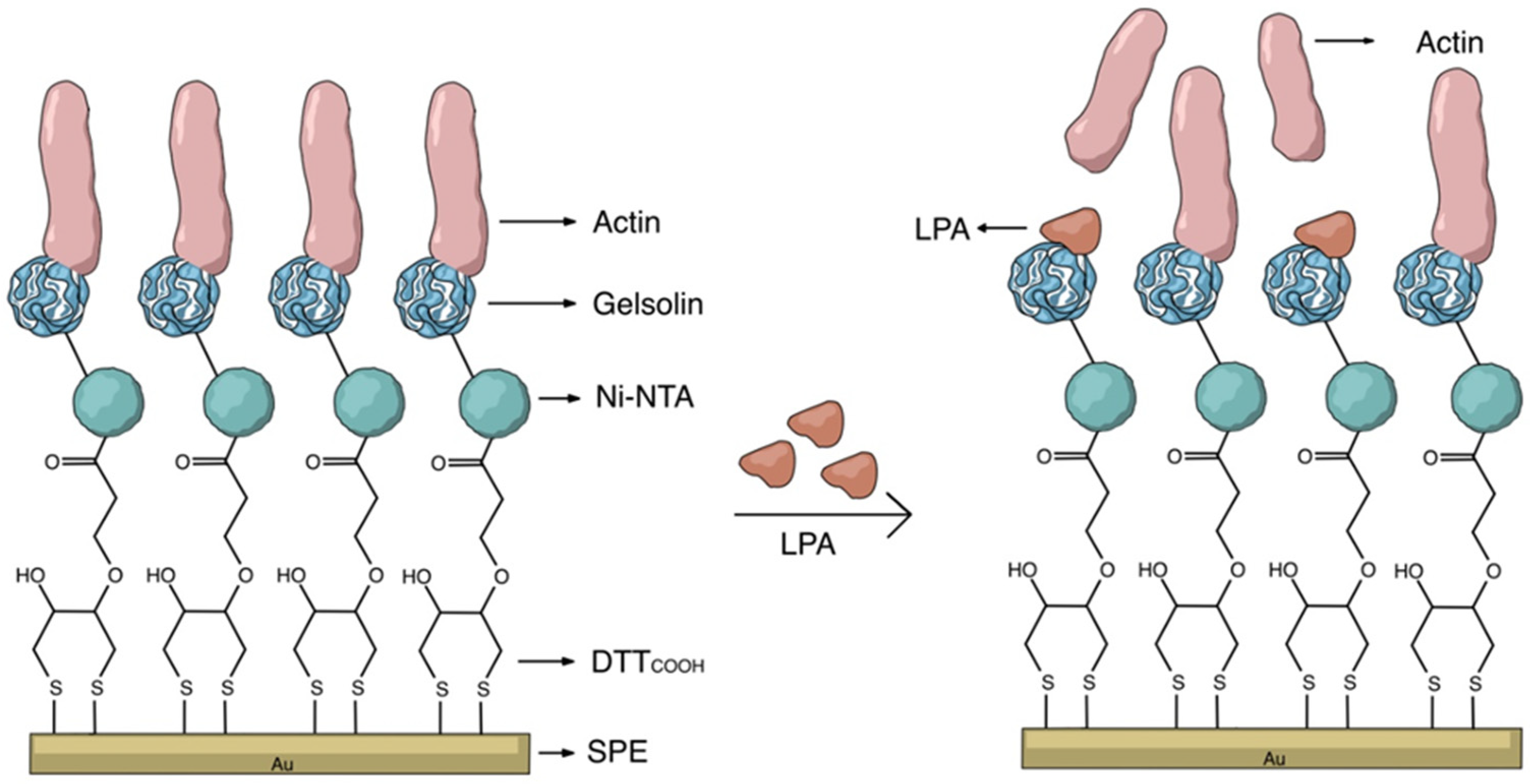
The key to constructing electrochemical biosensors lies in developing an effective biorecognition surface.
Figure 2 illustrates the layers of the surface modification to create an effective biorecognition surface on the SPEs for LPA detection. To characterize the surface at each stage of SPEs modification, we employed CV and DPV.
Figure S2 illustrates the scan rate study of the unmodified gold SPE which employed [Fe(CN)
6]
3−/4− as the redox probe. Although the CN
− may react with gold, forming Au(CN)
2− and causing some drift from an ideal reversible redox reaction, this study indicates that the redox process is diffusion controlled, confirming that the electrode’s surface is not involved in electron transfer.
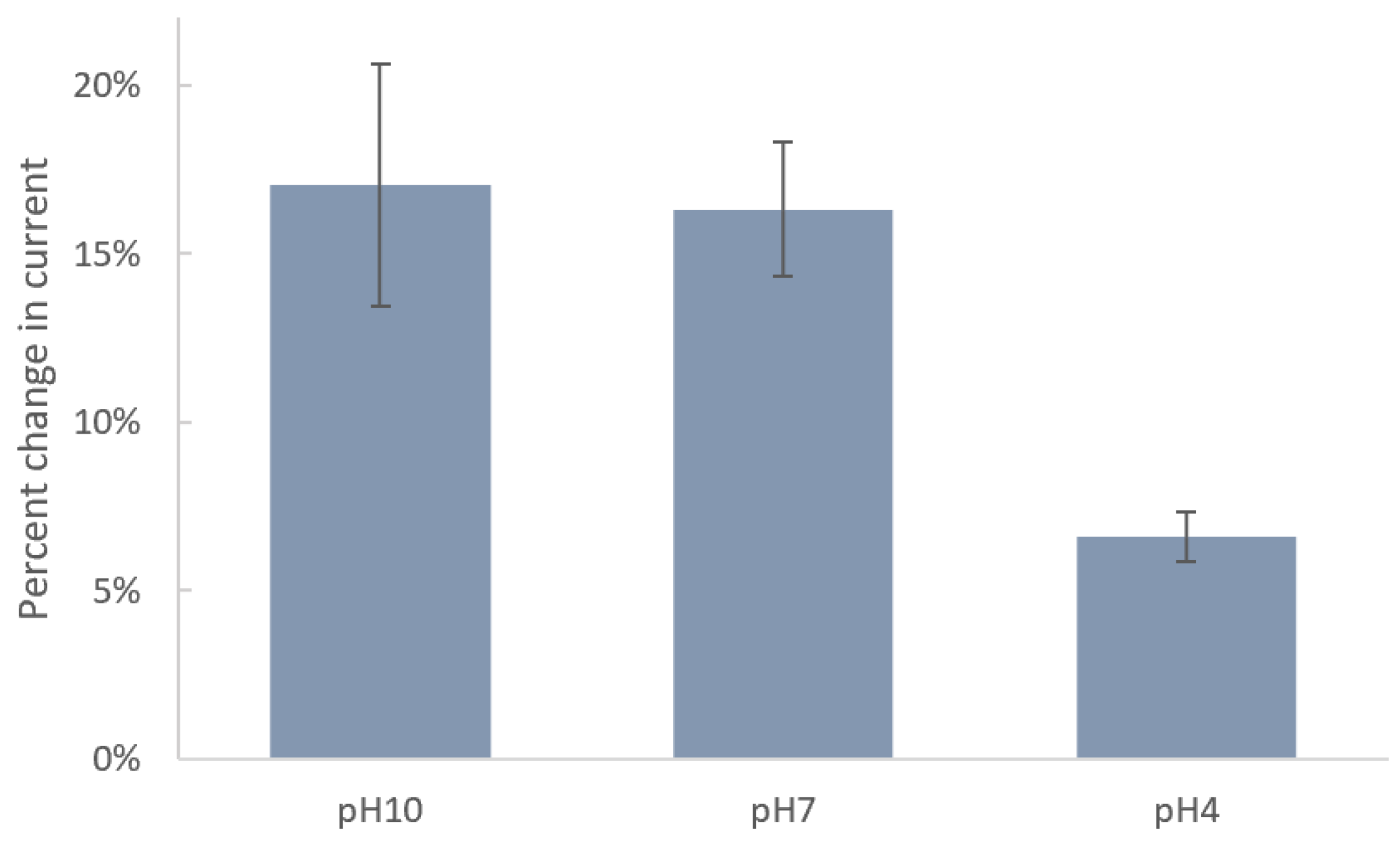
3.1.1. Study of the Effect of pH
Since the acidity of solution can significantly impact the molecular–surface interaction, we studied the effect of pH on immobilization of DTT
COOH on the SPE surface.
Figure 3 shows the changes in current after incubation of SPEs in DTT
COOH at pH 4, 7, and 10. Carboxyl and thiol groups in DTT
COOH have pKa values of 4.8 and 8.6, respectively. Therefore, at pH 10, both are deprotonated, forming carboxylate (COO
−) and thiolate (S
−) ions, respectively. At pH 7, the carboxyl group is fully deprotonated, while the thiol group may partially exist in its deprotonated form due to its higher pKa value. At pH 4, both groups are fully protonated. The stronger binding affinity of thiolate ions (-S
−) to the gold surface, in comparison to that of protonated thiols (-SH), explains the minimal changes in current observed after an overnight incubation at pH 4, indicating ineffective binding under acidic conditions due to protonation of the thiol group. Consequently, the greater changes in current at pH 10 and, to a slightly lesser extent, at pH 7 can be attributed to the deprotonation of the thiol group under these conditions, resulting in a more robust covalent bond between the linker and the gold surface. The fact that deprotonated thiolate ions form stronger and more stable bonds with the gold surface justify the preference for pH 7, which is closer to physiological levels and exhibits a lower standard deviation (1.98%) compared to pH 10 (3.60%), which indicates a more consistent interaction between the linker and the surface. Therefore, we chose pH 7 for subsequent experiments. These results align with those reported previously by Xue, Y. et al. [
28].
3.1.2. Development of the Biorecognition Surface
The SPEs were initially modified with either MUA or DTT
COOH to create a SAM, acting as the linker to immobilize NHS/EDC, followed by subsequent modifications with Ni-NTA, and gelsolin–actin complex. The NHS/EDC are commonly used as cross-linking agents in surface modification. EDC is a crosslinker that activates the terminal carboxyl groups of the thiol linker to form reactive O-acylisourea intermediates. This intermediate is susceptive to nucleophilic attack by NHS, and forms a stable amide bond with the original carboxyl groups. The EDC by-product is a soluble urea derivative that can be easily removed from the surface. The resulting semi-stable NHS ester facilities the immobilization of Ni-NTA on the surface.
Figure 4C shows the changes in current after modification with MUA, which is a linear thiol-based linker. In contrast, the incubation with DTT
COOH (
Figure 4 and
Figure S4) displays a more reproducible surface modification. Our previous study, employing a quartz crystal microbalance, demonstrated that DTT
COOH provides superior surface coverage and antifouling properties compared to MUA, possibly due to the additional thiol group [
23]. This structure could potentially prevent surface crowding when subsequent modifications are introduced with larger molecules of gelsolin and actin.
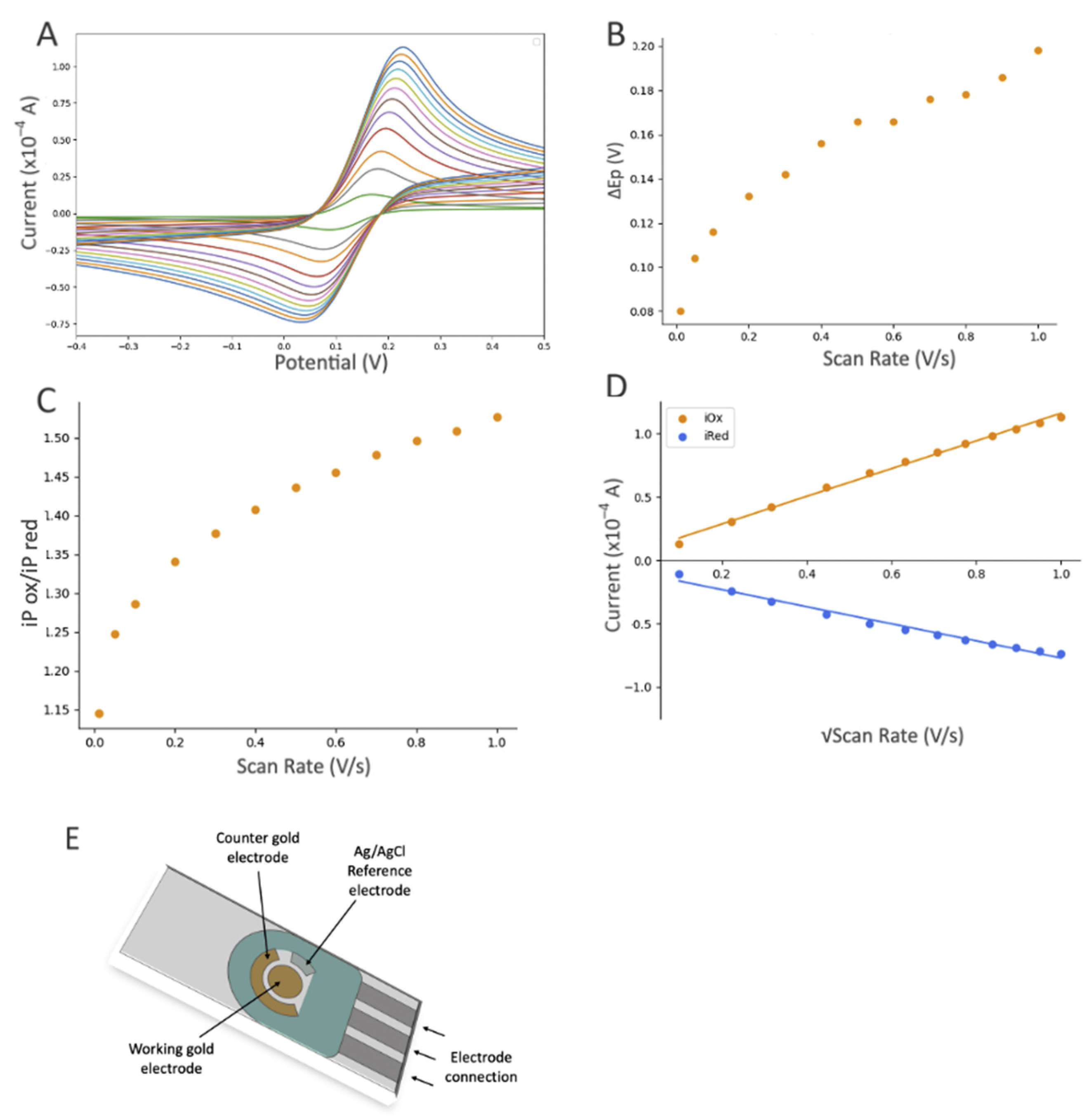
The decrease in the current after linker modification provided substantial evidence for the successful immobilization of the linker on the gold surface. Furthermore, using a Randles–Sevcik equation (assuming all parameters in the equation except current and surface area remain the same after modification) and the average oxidation peaks from
Figure 4A,C, we were able to estimate the surface coverage of DTT
COOH and MUA to be 18.06% and 8.16%, respectively. As such, DTTCOOH has a better estimated surface coverage by more than 10%, supporting the previous statement. Further modification with Ni-NTA resulted in a slight decrease in the current, which is believed to be associated with Ni-NTA’s potential participation in the redox process, and was previously observed on the stainless steel electrode [
25]. This hypothesis was confirmed though a CV scan rate investigation (
Figure S3), which showed a linear correlation between the current and both the scan rate and the square root of the scan rate at scan rates above 0.2 V/s. This suggests that both diffusion and adsorption (see
Figure S3B) are involved in the electron transfer process.
Modification with the gelsolin–actin complex resulted in a slight current increase (
Figure 4A). Although both gelsolin and actin are large proteins and have a higher coverage on the electrode surface, this increase is due to the redox activity of actin that has been previously observed in physiological and pathological conditions [
29,
30,
31]. This redox activity of actin might arise from the direct oxidation or reduction in its amino acid constituents, such as tyrosine, histidine, cysteine, tryptophan, and methionine [
32,
33]. The scan rate study showed that adsorption is also involved in electron transfer, suggesting the participation of actin on electron transfer (
Figure 5 and
Figure S3C). This redox behavior on the modified electrode was also reported before [
26].
3.1.3. Study of the Hydrophobicity of the Biorecognition Surface
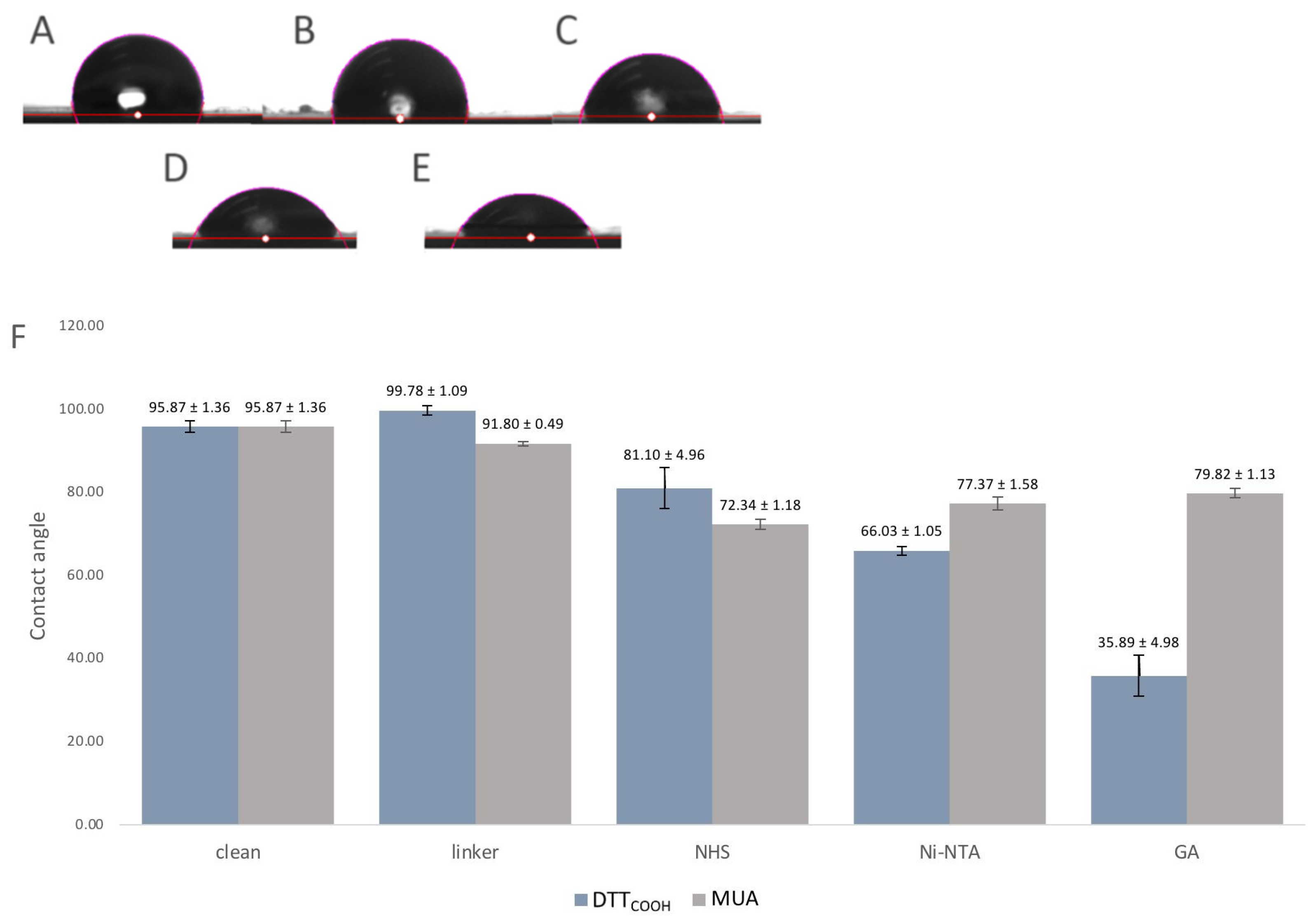
Surface hydrophobicity often changes through surface modifications, indicating how much this surface repels water molecules, and can be used to observe the changes to the surface chemistry. For that reason, the contact angles between a water droplet and a solid surface were used as an indicator of surface hydrophobicity and modifications. The contact angle measurement is a widely accepted method for studying surface wettability and hydrophobicity. A higher contact angle indicates enhanced hydrophobicity, implying that the surface is less wettable by water.
Figure 6 illustrates the contact angles of the SPEs following each modification step. Introduction of DTT
COOH to the surface resulted in a slightly increased contact angle compared to the bare gold surface, which may be due to the hydrophobic nature of the molecule. In contrast, the SPE modified with MUA exhibited less hydrophobicity, indicating the linear immobilization of MUA and the exposure of carboxylic acid groups on the surface (
Figure 6F). Subsequent modification with NHS/EDC and Ni-NTA led to a further reduction in the contact angle, likely due to the polarity of NHS and the ionic charges on Ni-NTA. The gelsolin–actin complex on the surface significantly increased its hydrophilicity, as evidenced by the decrease in contact angle (35.89 ± 4.98°), confirming successful surface modification. Notably, the SPE surface exhibited less hydrophilicity (
Figure 3SB, contact angle 79.8 ± 1.13°) when MUA was employed as the linker for immobilizing gelsolin–actin. The higher contact angle observed when using MUA compared to DTT
COOH may indicate lesser immobilization of gelsolin–actin, leading to less hydrophilicity. This observation provides further confirmation of the superior effectiveness of DTT
COOH in developing the biosensor compared to MUA.
3.2. Evaluation of the Electrochemical Biosensor Performance
The performance of the developed biosensor was evaluated by detecting different concentrations of LPA in the buffer solution and goat serum.
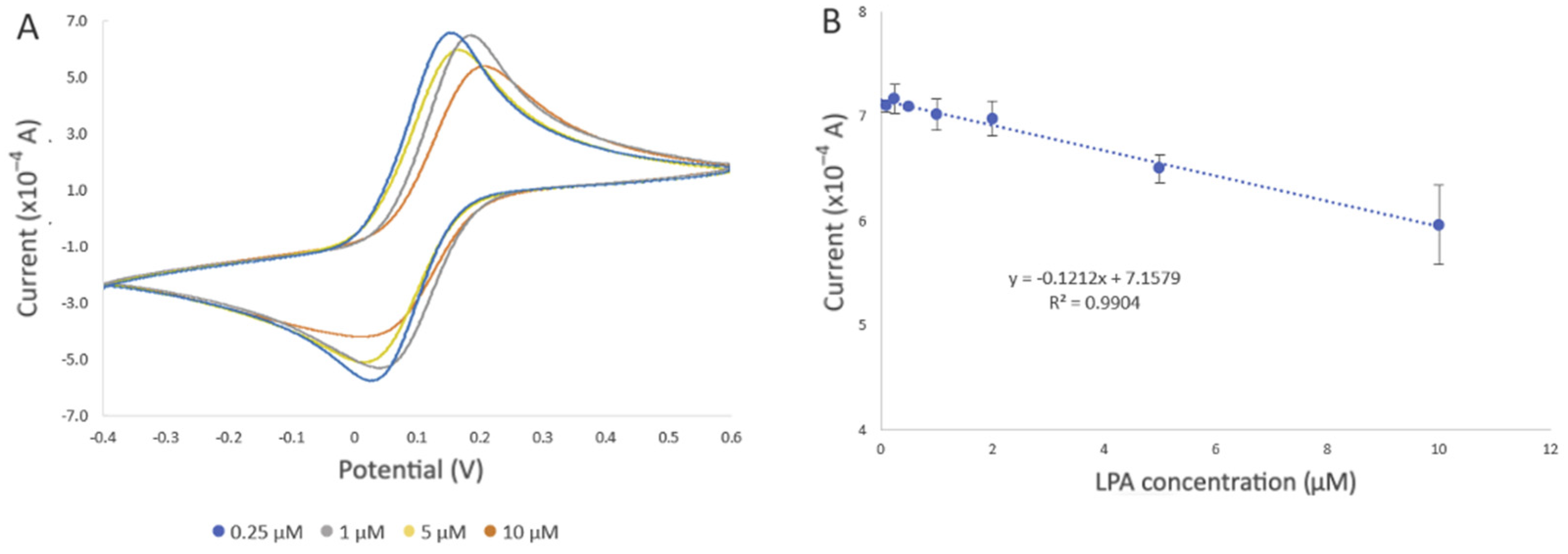
Figure 7A displays the cyclic voltammograms of the developed biosensor upon exposure to LPA concentrations ranging from 0.1 to 10 µM, under optimal experimental conditions in a PBS solution. After incubation of the developed biosensor in the LPA solution, LPA displaces actin in the gelsolin–actin complex due to its higher affinity toward gelsolin. Given that the molecular weight of LPA (436 Da) is smaller than that of actin (42 kDa), it was expected that binding LPA would result in a more efficient electron transfer between the solution and the modified electrode. In CV and DPV, this would appear as an increase in the current. Contrary to our belief, the results showed a decrease in the current after exposing the developed biosensor to LPA. As was previously reported, actin can exhibit some redox activity [
26]. As such, participation of redox-active amino acids within the actin structure in the electron transfer process at the electrode surface was validated through a scan rate study (
Figure 5 and
Figure S3C), as discussed in
Section 3.1.2. It is also probable that LPA molecules, upon binding to the biorecognition surface, may create a barrier or hinder access to active sites essential for electron transfer. This interference could disrupt the efficient flow of electrons between the solution and the electrode, thereby contributing to the observed decrease in current intensity. This observation confirmed that the replacement of actin by LPA, which lacks redox activity, could result in the decreased electroactivity and a subsequent reduction in current intensity.
In addition to the change in the current intensity, we also observed variations in peak potentials (
Figure 7A), which became more pronounced when the biosensor was exposed to 10 µM LPA. This could be due to the displacement of actin by LPA, altering the electrochemical behavior of the SPE. Such changes affect the electron transfer kinetics and, consequently, the redox reaction of [Fe(CN)
6]
3−/4− occurring at the electrode interface, leading to shifts in the peak potentials [
33].
The decrease in current was also observed with increased LPA concentration, which further confirmed the involvement of actin in the electron transfer process. Evaluation of the biosensor with different concentrations of LPA revealed a negative correlation between current intensity and LPA concentration (R
2 = 0.9904) (
Figure 7B). The estimated limit of detection (LOD) was 0.99 µM, along with a limit of quantification (LOQ) of 3.0 µM (refer to the SI for LOD and LOQ calculation), and a standard error of the predicted y-value for each x in a regression (STEYX) of 0.3899. These data were determined through Regression Analysis, with a maintained a 98% confidence level (
Figure 7B).
4. Conclusions
Electrochemical biosensors have a great potential to be used as effective clinical diagnosis tools due to their exceptional sensitivity, rapid response, and potential for miniaturization. In this study, we developed a sensitive label-free electrochemical biosensor while addressing the current challenges of NSA associated with gold surfaces. By using a new linker, DTTCOOH, we successfully engineered a SAM on the gold surface less prone to fouling compared to MUA, a linear thiol linker. Using an affinity-based gelsolin–actin system, we developed a biorecognition surface tailored for LPA detection. Our developed biosensor showed a higher sensitivity compared to the previously reported fluorescence method, achieving the LOD and LOQ of 0.9 µM and LOQ of 2.76 µM in goat serum, which is within the threshold of ovarian cancer diagnosis at early stages. Furthermore, the utilization of thiol chemistry is less challenging compared to silane chemistry, which we previously used to develop an electrochemical biosensor for LPA detection. Unlike silane-based linkers, thiols are not sensitive to oxygen and water, presenting a more practical approach to surface modification. Without the need of complex instrumentation and time-consuming sample preparation, the miniaturized design of the developed biosensor further demonstrates its potential for compact POCT device fabrication. This study presents a proof of concept of using SPE to detect LPA. Future work will evaluate the selectivity and sensitivity of the developed biosensor using human serum. It is crucial to examine the stability and shelf-life of the developed biosensor in order to use it for clinical trials. While stability testing to determine the shelf life was not conducted in this proof-of-concept work, the stability of the modified SPE in an electrochemical environment was assessed by conducting at least seven CV cycles after each modification step. Furthermore, prior research has suggested that surfaces with a thiolate SAM bound to NTA can exhibit stability. This suggests that we could pre-functionalize these surfaces in advance and then further modify them with proteins just before detection, potentially reducing the overall preparation time. It is worth mentioning that understanding the surface chemistry of modified electrodes can open a new direction for designing new electrochemical biosensors with better performance.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}