
Electrochemical and Mechanistic Study of Superoxide Scavenging by Pyrogallol in N,N-Dimethylformamide through Proton-Coupled Electron Transfer



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Electrochemical and In Situ Electrolytic ESR/UV-vis Spectrum Measurements
2.3. Calculation
3. Results
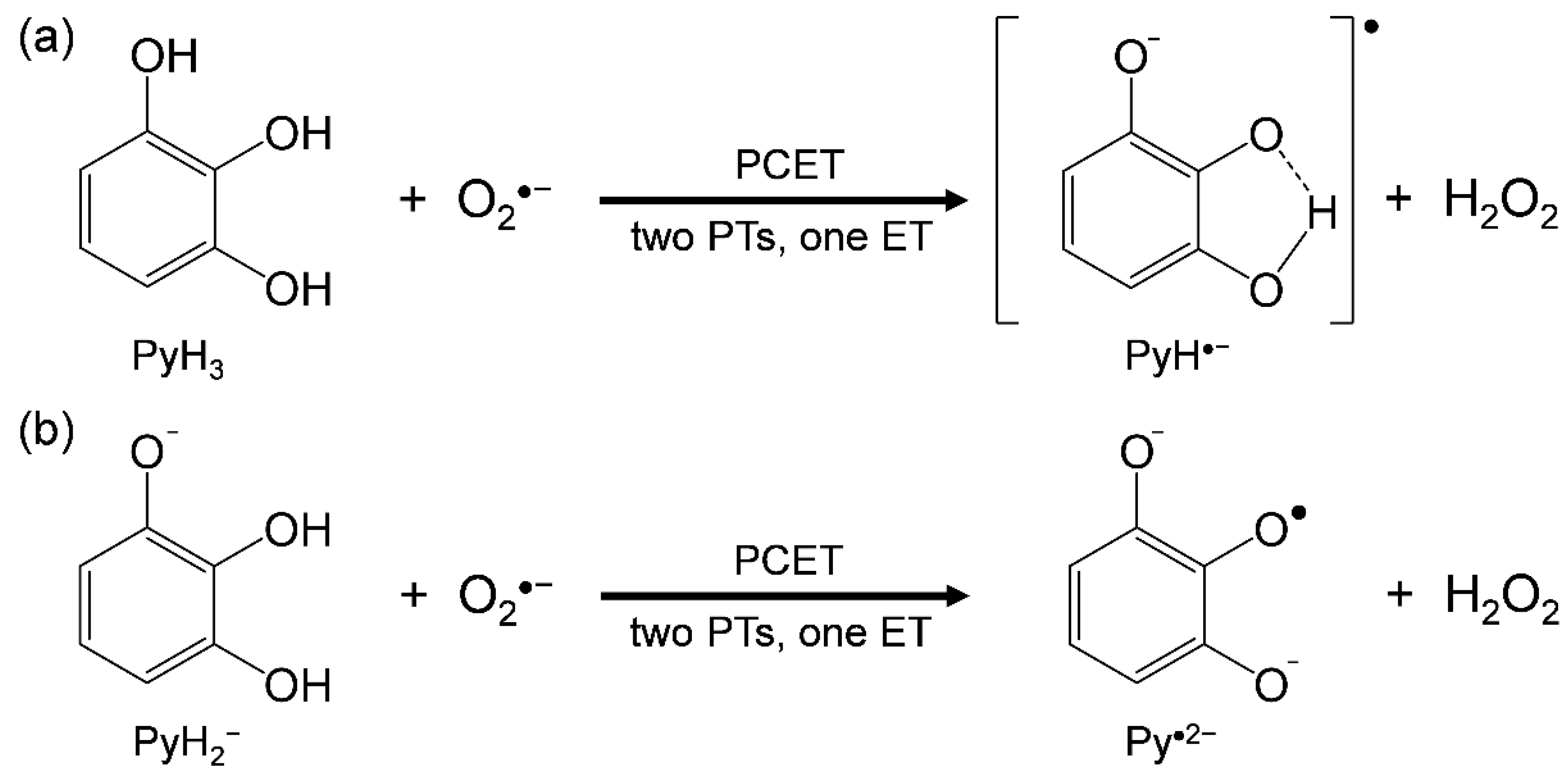
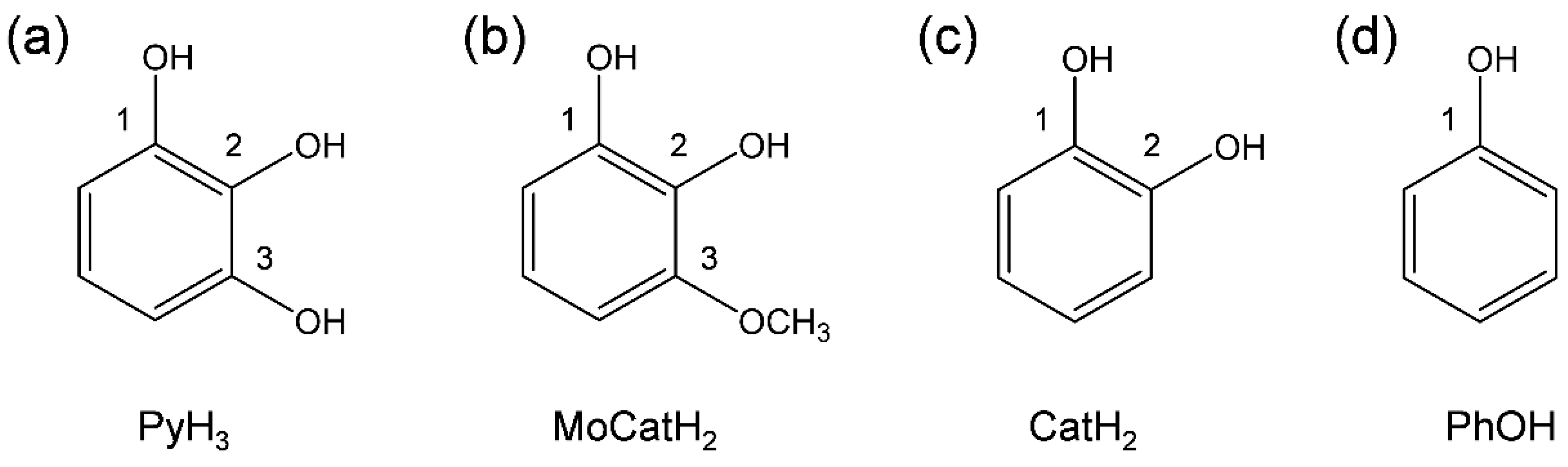
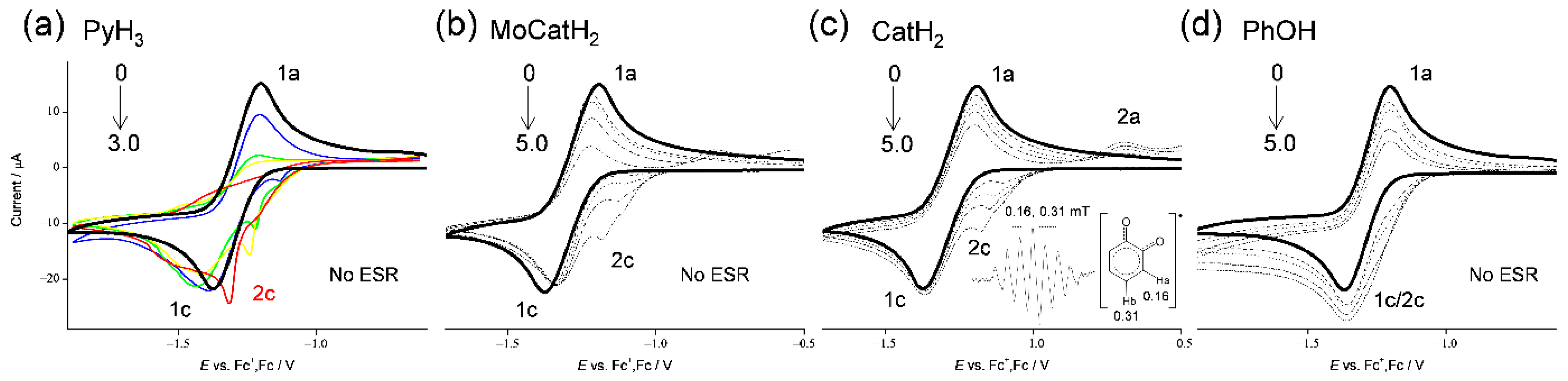
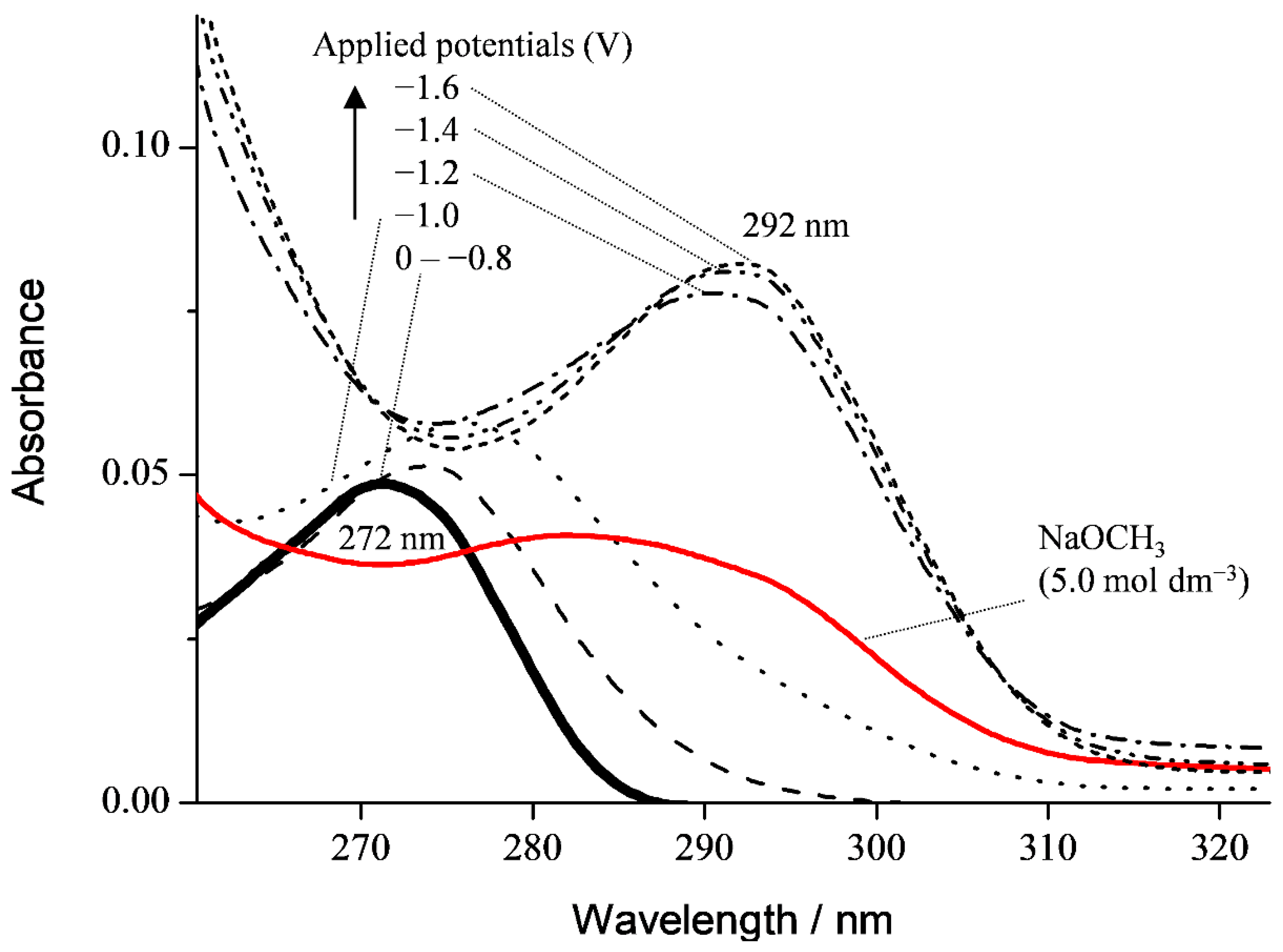
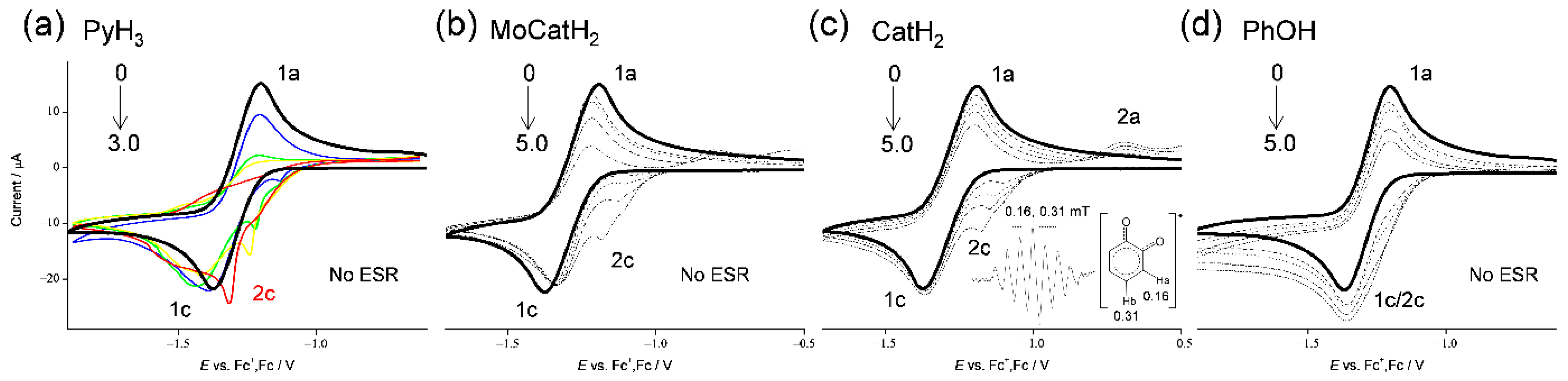
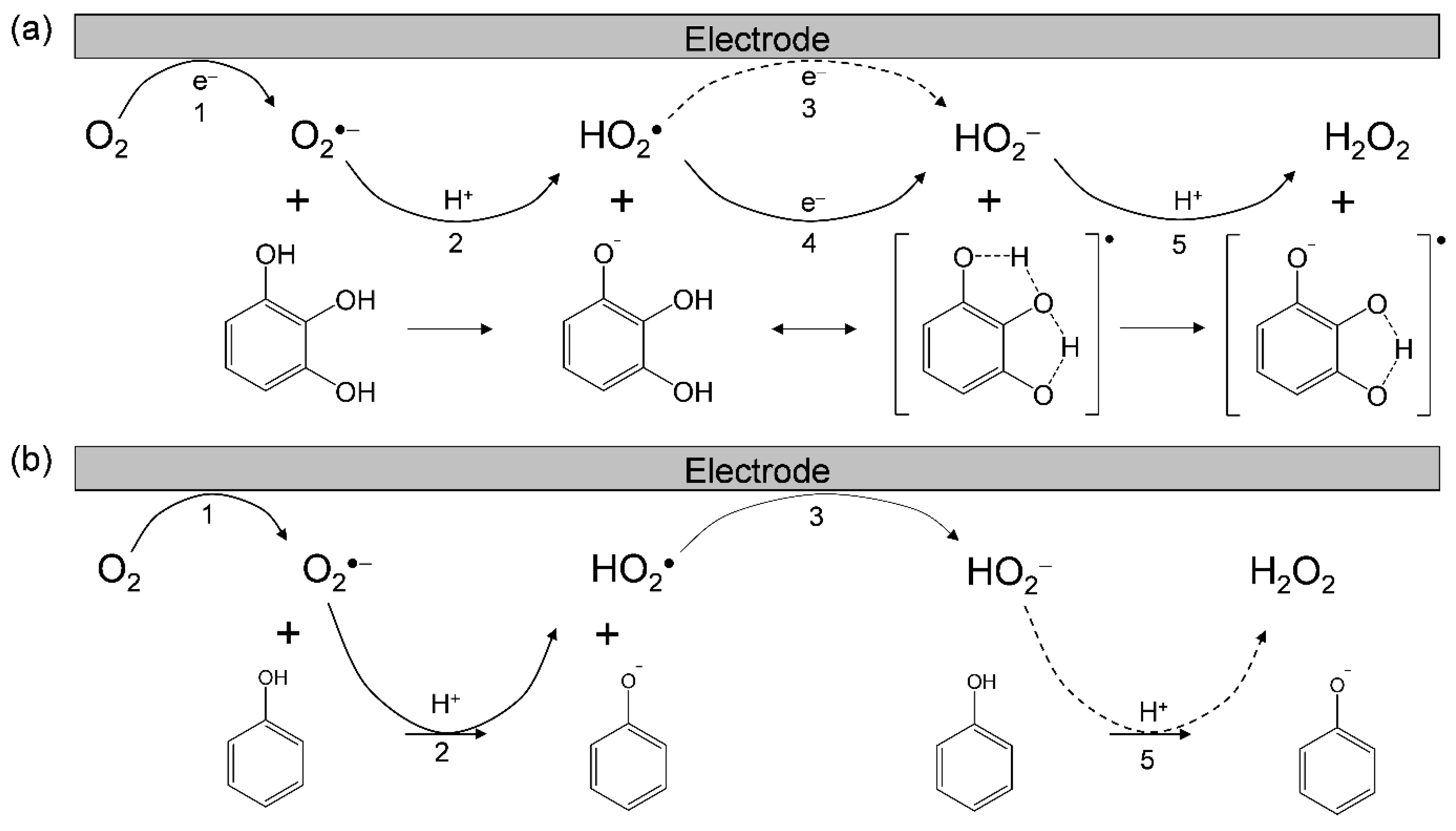
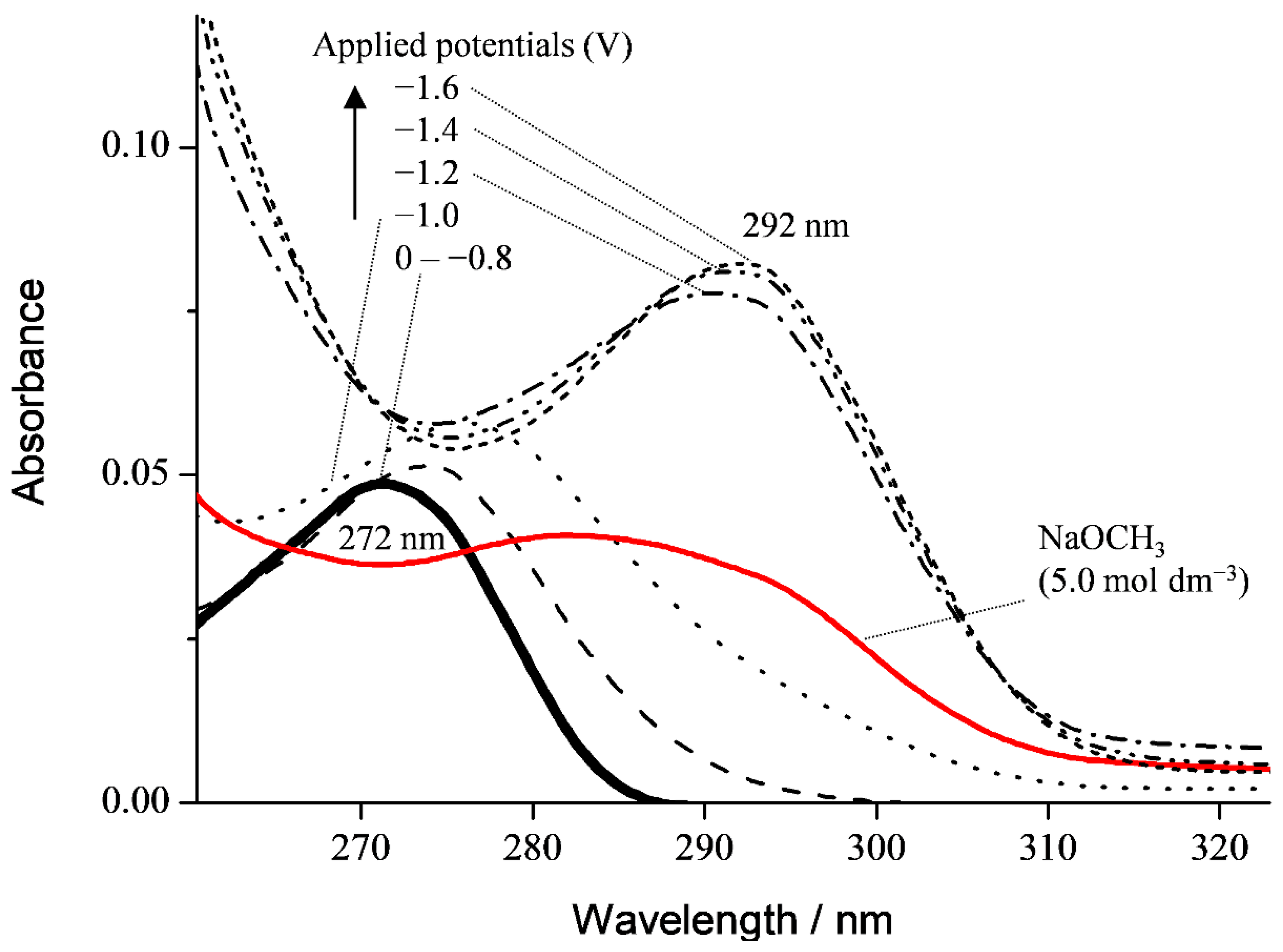
3.1. Cyclic Voltammetry and ESR Analysis of O2/O2•− in the Presence of PyH3
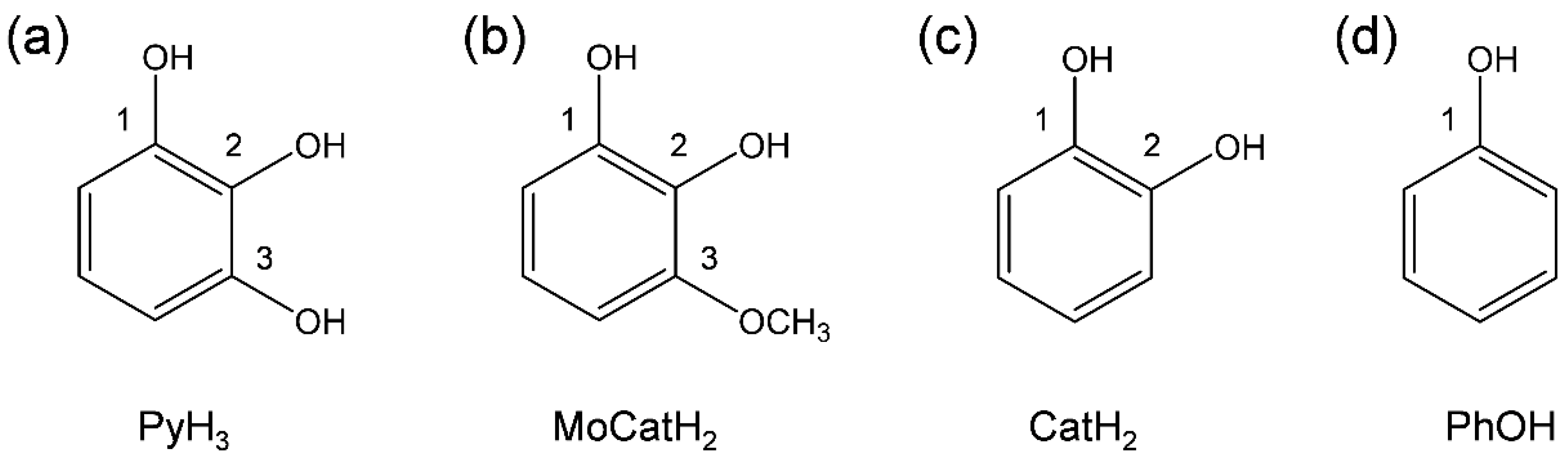
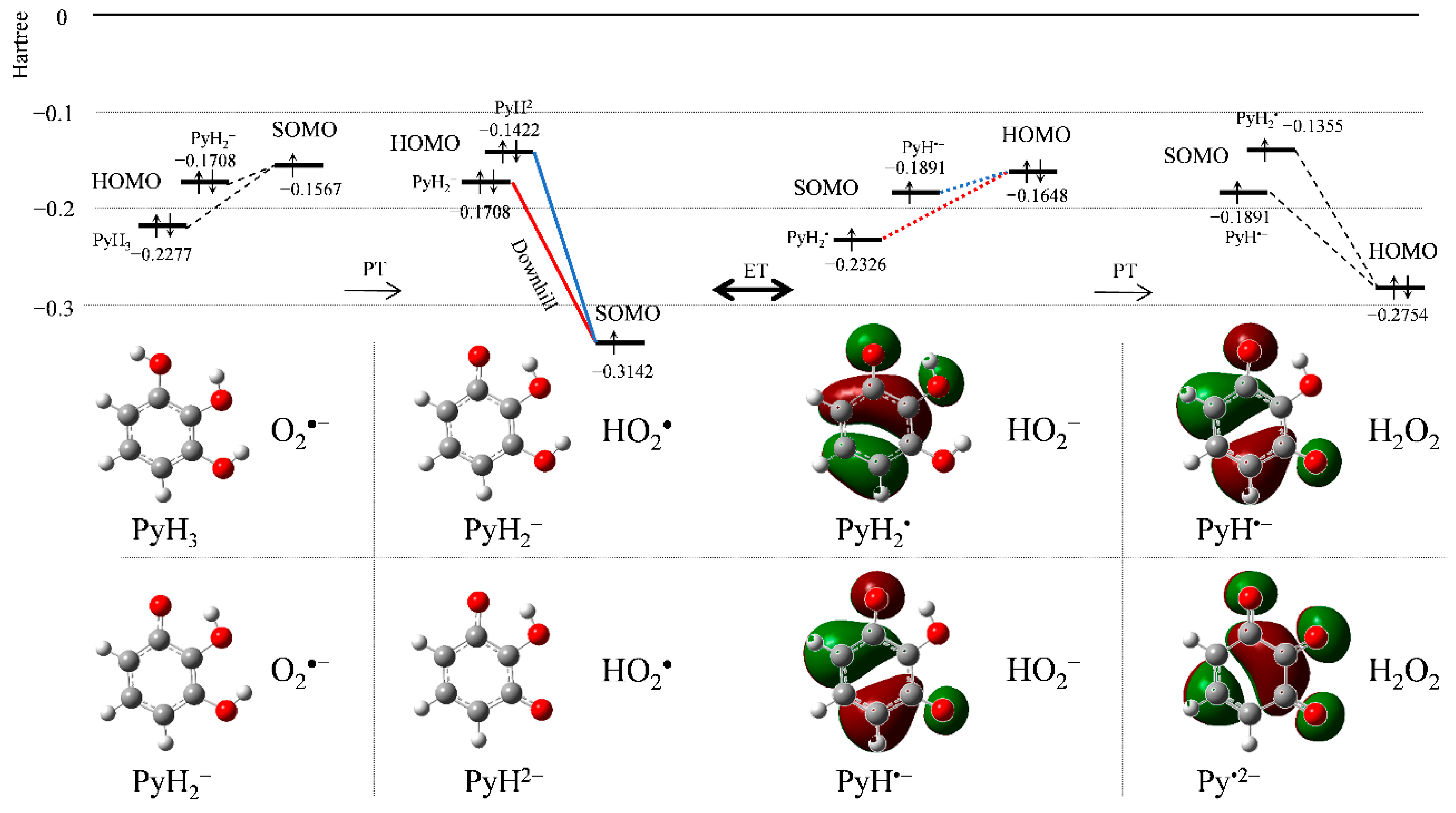
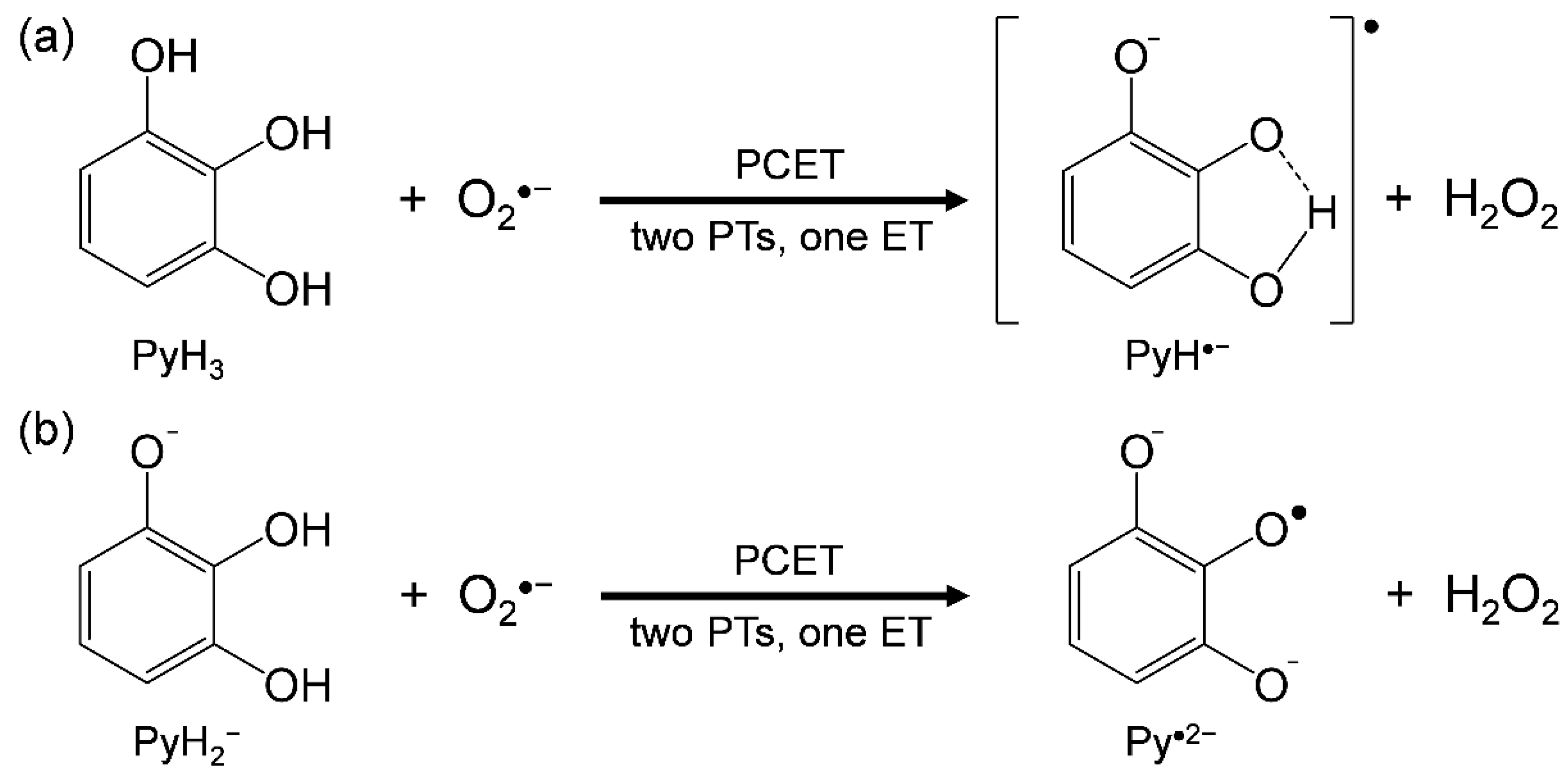
3.2. Change in HOMO–LUMO Energies upon PCET between PyH3 and O2•− in DFT Analyses
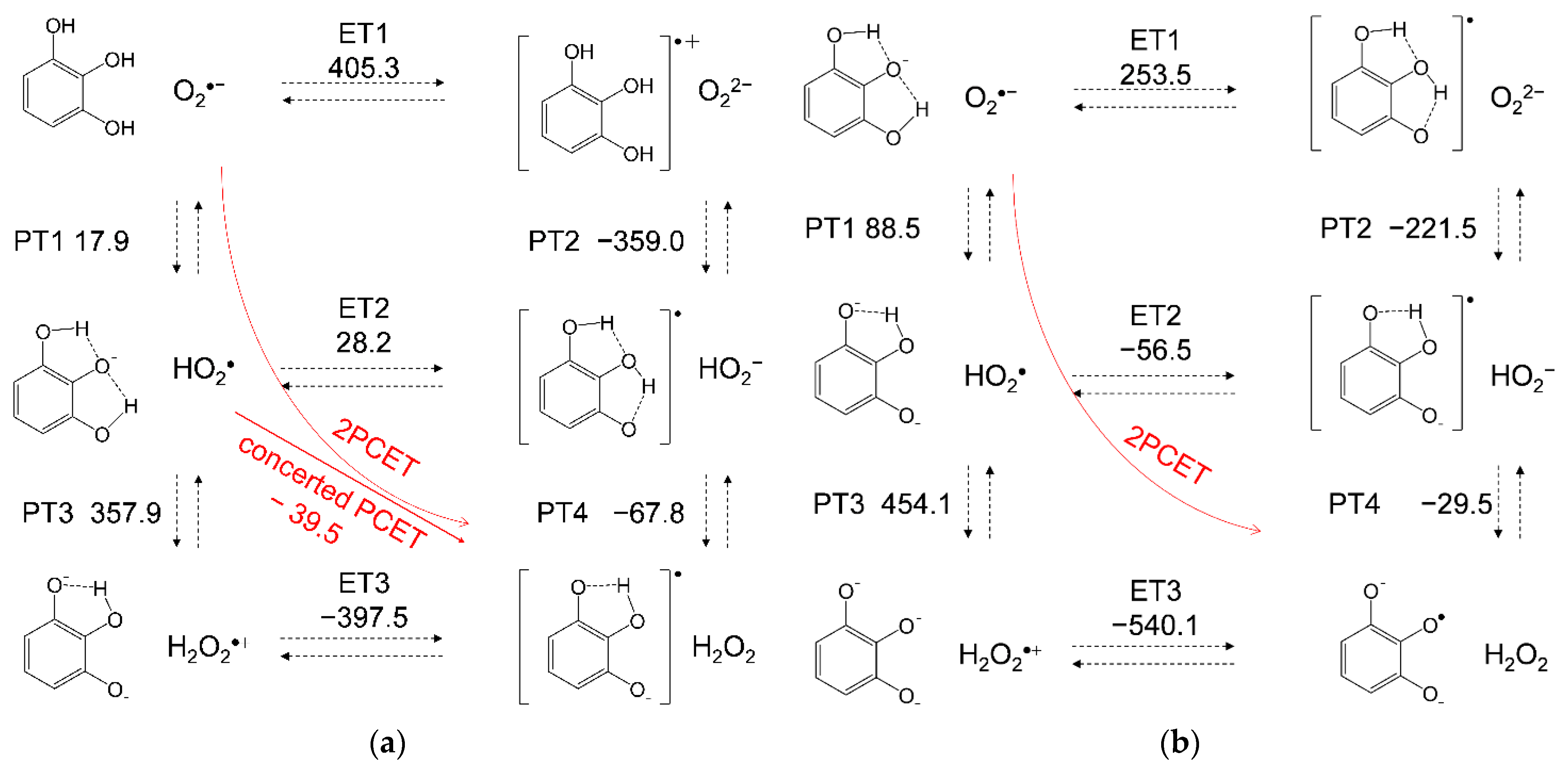
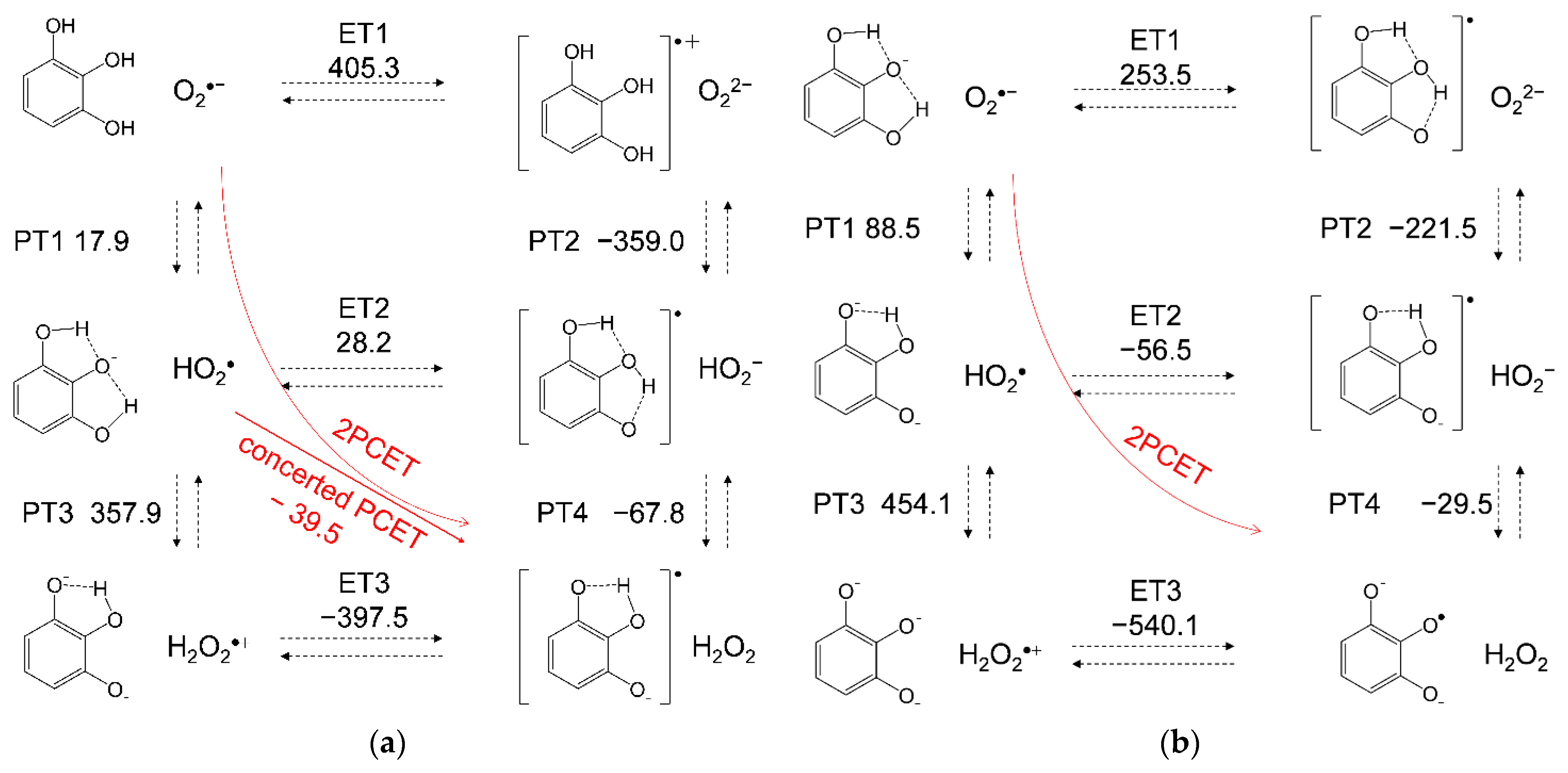
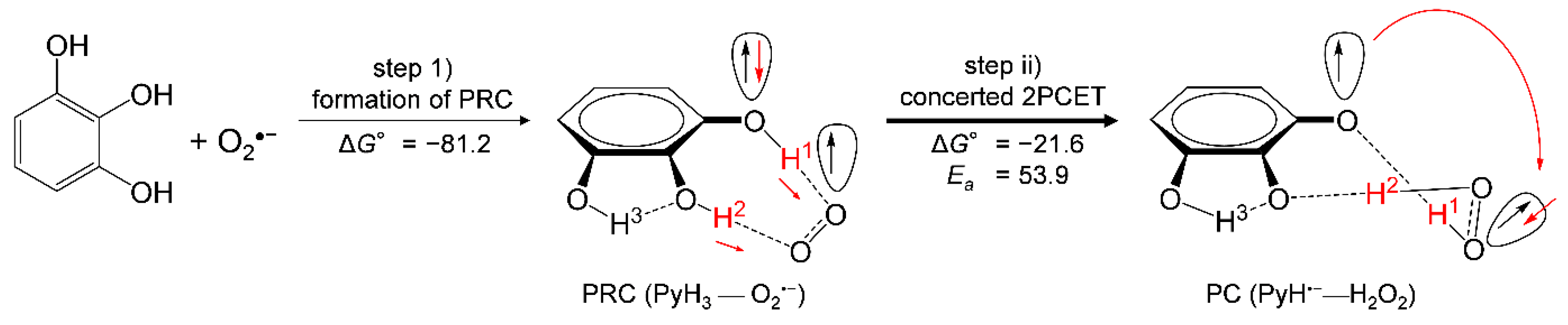
3.3. Free Energy Calculations of PCET between PyH3 and O2•−
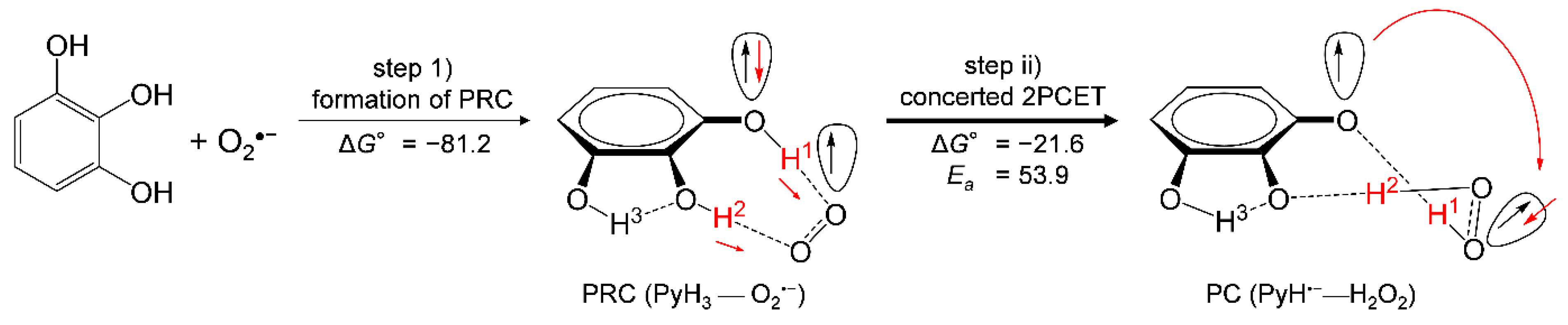
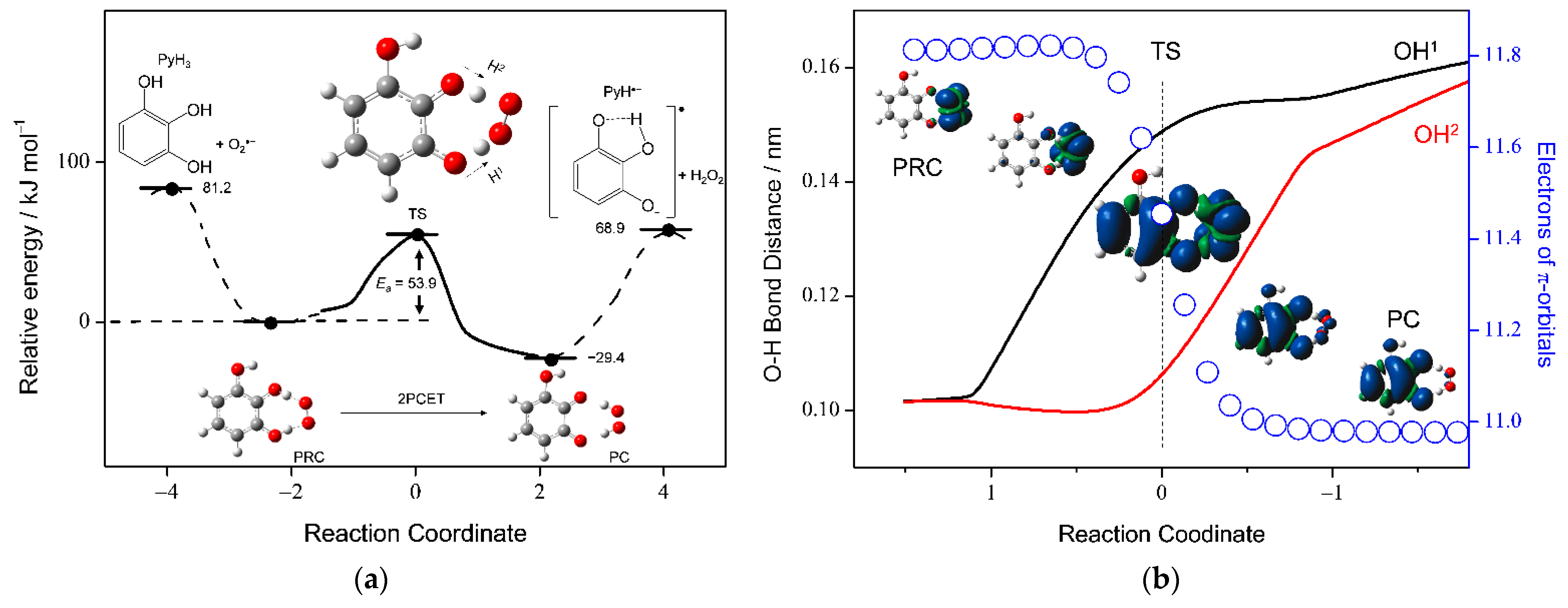
3.4. Potential Energy Surfaces of the PCET between PyH3 and O2•−
4. Conclusions
- PyH3 scavenges O2•− through the 2PCET involving concerted two PTs and one ET, in a similar mechanism for CatH2;
- the net mechanism involves the initial formation of PRC followed by concerted 2PCET;
- the 3OH group thermodynamically promotes the formation of PRC via two HBs but does not promote the latter 2PCET, resulting in an effective O2•− scavenging ability of PyH3.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furuno, K.; Akasako, T.; Sugihara, N. The contribution of the pyrogallol moiety to the superoxide radical scavenging activity of flavonoids. Biol. Pharm. Bull. 2002, 25, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X. Improved pyrogallol autoxidation method: A reliable and cheap superoxide-scavenging assay suitable for all antioxidants. J. Agric. Food Chem. 2012, 60, 6418–6424. [Google Scholar] [CrossRef] [PubMed]
- Ramasarma, T.; Rao, A.V.S.; Devi, M.M.; Omkumar, R.V.; Bhagyashree, K.S.; Bhat, S.V. New insights of superoxide dismutase inhibition of pyrogallol autoxidation. Mol. Cell. Biochem. 2015, 400, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Doona, C.J.; Kustin, K. Kinetics and mechanism of pyrogallol autoxidation: Calibration of the dynamic response of an oxygen electrode. Int. J. Chem. Kinet. 1993, 25, 239–247. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef]
- Marklund, S.; Marklund, G. Involvement of the Superoxide Anion Radical in the Autoxidation of Pyrogallol and a Convenient Assay for Superoxide Dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Alavi Rafiee, S.; Farhoosh, R.; Sharif, A. Antioxidant Activity of Gallic Acid as Affected by an Extra Carboxyl Group than Pyrogallol in Various Oxidative Environments. Eur. J. Lipid Sci. Technol. 2018, 120, 1800319. [Google Scholar] [CrossRef]
- Sutanto, H.; Susanto, B.H.; Nasikin, M. Solubility and antioxidant potential of a pyrogallol derivative for biodiesel additive. Molecules 2019, 24, 2439. [Google Scholar] [CrossRef] [Green Version]
- Romero, M.; Rojano, B.; Mella-Raipán, J.; Pessoa-Mahana, C.D.; Lissi, E.; López-Alarcón, C. Antioxidant Capacity of pure compounds and complex mixtures evaluated by the ORAC-pyrogallol red assay in the presence of Triton X-100 micelles. Molecules 2010, 15, 6152–6167. [Google Scholar] [CrossRef] [Green Version]
- Mu, S.; Chen, C. Electrochemical Oxidation of Pyrogallol: Formation and Characterization of Long-Lived Oxygen Radicals and Application To Assess the Radical Scavenging Abilities of Antioxidants. J. Phys. Chem. B 2012, 116, 12567–12573. [Google Scholar] [CrossRef]
- Zhang, Q.A.; Wang, X.; Song, Y.; Fan, X.H.; García-Martín, J.F. Optimization of pyrogallol autoxidation conditions and its application in evaluation of superoxide anion radical scavenging capacity for four antioxidants. J. AOAC Int. 2016, 99, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Nasr, B.; Hsen, T.; Abdellatif, G. Electrochemical treatment of aqueous wastes containing pyrogallol by BDD-anodic oxidation. J. Environ. Manag. 2009, 90, 523–530. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridovich, I. Superoxide dismutase. In Encyclopedia of Biological Chemistry, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 352–354. ISBN 9780123786319. [Google Scholar]
- Costentin, C.; Robert, M.; Savéant, J.M. Concerted proton-electron transfers: Electrochemical and related approaches. Acc. Chem. Res. 2010, 43, 1019–1029. [Google Scholar] [CrossRef]
- Wenger, O.S. Proton-coupled electron transfer with photoexcited metal complexes. Acc. Chem. Res. 2013, 46, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Hammes-Schiffer, S. Theory of proton-coupled electron transfer in energy conversion processes. Acc. Chem. Res. 2009, 42, 1881–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.S.; Evans, D.H. Study of the electrochemical reduction of dioxygen in acetonitrile in the presence of weak acids. J. Phys. Chem. B 2006, 110, 637–644. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Importance of proton-coupled electron transfer from natural phenolic compounds in superoxide scavenging. Chem. Pharm. Bull. Tokyo 2015, 63, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Biela, M.; Rimarčík, J.; Senajová, E.; Kleinová, A.; Klein, E. Antioxidant action of deprotonated flavonoids: Thermodynamics of sequential proton-loss electron-transfer. Phytochemistry 2020, 180, 112528. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Quinone-hydroquinone π-conjugated redox reaction involving proton-coupled electron transfer plays an important role in scavenging superoxide by polyphenolic antioxidants. Chem. Lett. 2010, 39, 162–164. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Concerted two-proton-coupled electron transfer from catechols to superoxide via hydrogen bonds. Electrochim. Acta 2016, 208, 304–309. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Structural properties of 4-substituted phenols capable of proton-coupled electron transfer to superoxide. Int. J. Adv. Res. Chem. Sci. 2016, 3, 11–19. [Google Scholar] [CrossRef]
- Nakayama, T.; Honda, R.; Kuwata, K.; Usui, S.; Uno, B. Electrochemical and Mechanistic Study of Reactivities of α-, β-, γ-, and δ-Tocopherol toward Electrogenerated Superoxide in N, N-Dimethylformamide through Proton-Coupled Electron Transfer. Antioxidants 2021, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Uno, B. Electronic spectra of the electrogenerated 1,4-benzoquinone π-dianion and the strongly hydrogen-bonded charge-transfer complex with methanol. Bull. Chem. Soc. Jpn. 1999, 72, 1213–1217. [Google Scholar] [CrossRef]
- Frisch, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; ISBN 9781935522027. [Google Scholar]
- Quintero-Saumeth, J.; Rincón, D.A.; Doerr, M.; Daza, M.C. Concerted double proton-transfer electron-transfer between catechol and superoxide radical anion. Phys. Chem. Chem. Phys. 2017, 19, 26179–26190. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Nakayama, T.; Honda, R. Electrochemical and mechanistic study of superoxide elimination by mesalazine through proton-coupled electron transfer. Pharmaceuticals 2021, 14, 120. [Google Scholar] [CrossRef]
- Nakayama, T.; Honda, R. Electrochemical and Mechanistic Study of Oxidative Degradation of Favipiravir by Electrogenerated Superoxide through Proton-Coupled Electron Transfer. ACS Omega 2021, 6, 21730–21740. [Google Scholar] [CrossRef]
- Hammett, L.P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | PT1 | PT2 | PT3 | PT4 | ET1 | ET2 | ET3 | Concerted | Total 1 |
|---|---|---|---|---|---|---|---|---|---|
| PyH3 | 17.9 | −359.0 | 357.9 | −67.8 | 405.3 | 28.2 | −397.5 | −39.6 | −21.6 |
| PyH2− | 88.5 | −221.5 | 454.1 | −29.5 | 253.5 | −56.5 | −540.1 | - | 2.4 |
| CatH2 | 19.4 | −364.2 | 390.6 | −78.7 | 406.8 | 23.0 | −446.3 | −55.7 | −36.2 |
| MoCatH2 | 46.0 | −335.4 | 379.5 | −92.4 | 394.8 | 13.3 | −458.6 | −79.1 | −32.9 |
| Reactants | 1 FR | TS (Ea) | PC | FP |
|---|---|---|---|---|
| PyH3 (+O2•−) | 81.2 | 53.9 | −29.4 | 68.9 |
| CatH2 (+O2•−) | 71.6 | 52.5 | −20.9 | 45.3 |
| MoCatH2 (+O2•−) | 61.4 | 50.7 | −28.4 | 38.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, T.; Honda, R.; Kuwata, K.; Usui, S.; Uno, B. Electrochemical and Mechanistic Study of Superoxide Scavenging by Pyrogallol in N,N-Dimethylformamide through Proton-Coupled Electron Transfer. Electrochem 2022, 3, 115-128. https://doi.org/10.3390/electrochem3010008
Nakayama T, Honda R, Kuwata K, Usui S, Uno B. Electrochemical and Mechanistic Study of Superoxide Scavenging by Pyrogallol in N,N-Dimethylformamide through Proton-Coupled Electron Transfer. Electrochem. 2022; 3(1):115-128. https://doi.org/10.3390/electrochem3010008
Chicago/Turabian StyleNakayama, Tatsushi, Ryo Honda, Kazuo Kuwata, Shigeyuki Usui, and Bunji Uno. 2022. "Electrochemical and Mechanistic Study of Superoxide Scavenging by Pyrogallol in N,N-Dimethylformamide through Proton-Coupled Electron Transfer" Electrochem 3, no. 1: 115-128. https://doi.org/10.3390/electrochem3010008
APA StyleNakayama, T., Honda, R., Kuwata, K., Usui, S., & Uno, B. (2022). Electrochemical and Mechanistic Study of Superoxide Scavenging by Pyrogallol in N,N-Dimethylformamide through Proton-Coupled Electron Transfer. Electrochem, 3(1), 115-128. https://doi.org/10.3390/electrochem3010008